 Xingxing Qiao
Xingxing Qiao Liping Wu2,3
Liping Wu2,3 Yaqin Chen
Yaqin Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 18 January 2023
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1089194
Background: Campomelic dysplasia (CD) is an autosomal dominant skeletal dysplasia syndrome characterized by shortness and bowing of lower extremities, and often accompanied by XY sex reversal. Heterozygous pathogenic variants of SOX9 or rearrangement involving the long arm of chromosome 17 are the causes of disease. However, evidence for pathogenesis of SOX9 haploinsufficiency is insufficient.
Methods: We enrolled a Chinese family where the fetus was diagnosed with CD. The affected fetus was selected for whole-exome sequencing to identify the pathogenic mutations in this family.
Results: After data filtering, a novel non-sense SOX9 variant (NM_000346.3; c.1249C > T; p.Q417*) was identified as the pathogenic lesion in the fetus. Further co-segregation analysis using Sanger sequencing confirmed that this novel SOX9 mutation (c.1249C > T; p.Q417*) was a de novo mutation in the affected fetus. This terminated codon mutation identified by bioinformatics was located at an evolutionarily conserved site of SOX9. The bioinformatics-based analysis predicted this variant was pathogenic and affected SOX9 transactivation activity.
Conclusion: CD is a rare condition, which connected with SOX9 tightly. We identified a novel heterozygous SOX9 variant (p.Q417*) in a Chinese CD family. Our study supports the putative reduced transactivation of SOX9 variants in the pathogenicity of CD.
Campomelic dysplasia (CD, OMIM: #114290) is a rare autosomal dominant skeletal dysplasia syndrome caused by rearrangement of 17 chromosome and mutations in SOX9 gene. The diagnosis is usually made after 20 weeks of pregnancy by ultrasonography characterized by malformation of skeletal including bowing of the legs, especially the tibias, small scapular bones, and cleft palate (1). Otherwise, a small chest, eleven pairs of ribs, micrognathia, flat face, and hypertelorism are also featured. CD is a lethal syndrome due to respiratory distress related to small chest and tracheobronchial hypoplasia (2). In two-thirds of reported 46, XY karyotype affected individuals existed male to female sex reversal or had ambiguous genitalia because of the essential role of SOX9 in sex determination (3, 4).
SOX9 (SRY-related HMG-box gene 9, OMIM: 608160), located in chromosome 17q24.3, is an essential transcription factor for both sex and skeletal development. Anomalies of SOX9 are the main causes of CD, and the rearrangement involving the long arm of chromosome 17 will interrupt the upstream of SOX9 (5, 6). Most reported cases of CD are caused by intragenic heterozygous mutations in SOX9 gene, and the deletions of SOX9 represent strong evidence for the dosage-dependent action of SOX9 protein in normal chondrogenesis (7, 8).
Heterozygous de novo mutations are the main cause of CD and sex reversal. The detected shortness and bowing of tubular lower bones by ultrasonography could lead to the suspect of CD. Here we reported on novel heterozygous terminated mutation in SXO9 gene (c.1249C > T; p.Q417*) in a CD-affected individual in a Chinese family, and we present new evidence for the pathogenesis of SOX9 in CD.
The present study was approved by the Review Board of Shenzhen Longgang District Maternity and Child Healthcare Hospital and was performed in accordance with the principles outlined and enshrined in the Declaration of Helsinki. Written informed consent was obtained from the parents for the publication of any potentially identifiable images or data.
The affected fetus and parents were investigated in this study. Amniocentesis was performed to collected the sample of fetus and peripheral blood samples were collected from the parents of fetus. Clinical data are mainly collected from ultrasound measurements and are carefully recorded.
Genomic DNA was extracted using the DNeasy blood and tissue kit (Qiagen, Valencia, CA, United States). The main part of WES was performed at Guangdong Women’s and Children’s Hospital. The filter strategies used are consistent with those outlined in our previous research (9). Briefly, after initial quality control of the data, introns, intergenic and untranslated regions (UTRs), homogeneous single nucleotide variants (SNVs), and variants with a frequency of substitution alleles exceeding 1% in the public databases [1,000 Genomes, dbSNP144, YH database, and Genome Aggregation database (gnomAD)] are first removed and then continue further analyzed. Then, the variants predicted by SIFTm PolyPhen-2, and Mutation Taster as “Disease-causing,” were retained.
A 30-year-old, nulliparous woman (G0P0A0) presented in Shenzhen Longgang District Maternity and Child Healthcare Hospital for routine pregnancy prenatal Doppler ultrasound following up and care. The couple has no history of consanguinity, but the gravida has a history of intellectual disability. The initial fetal ultrasonography revealed abnormal long bones in both lower extremities, mild hydronephrosis with detached right renal pelvis, and varus in both feet (Figures 1A–C). Thus, they received further detailed anomaly scan of the fetus. A detailed fetal examination revealed severe malformation of bones, including shortened spine and ribs, leading to a small chest, and the long bones of the fetal limbs are distinctly short and curved, especially the femur. The face of the fetus is seen with micrognathia and significant thickening of the neck fold of 1.3 cm (>6 mm). The amniotic fluid index is 21.7 cm, beyond the normal range of 14.3–15.9 cm. Typical pot-shaped external genitalia was observed in the fetus (Figures 1E–G). After ultrasonography examination, the diagnosis of thanatophoric dysplasia or CD was suspected, and WES was requested for the pathogenesis of the affected fetus.
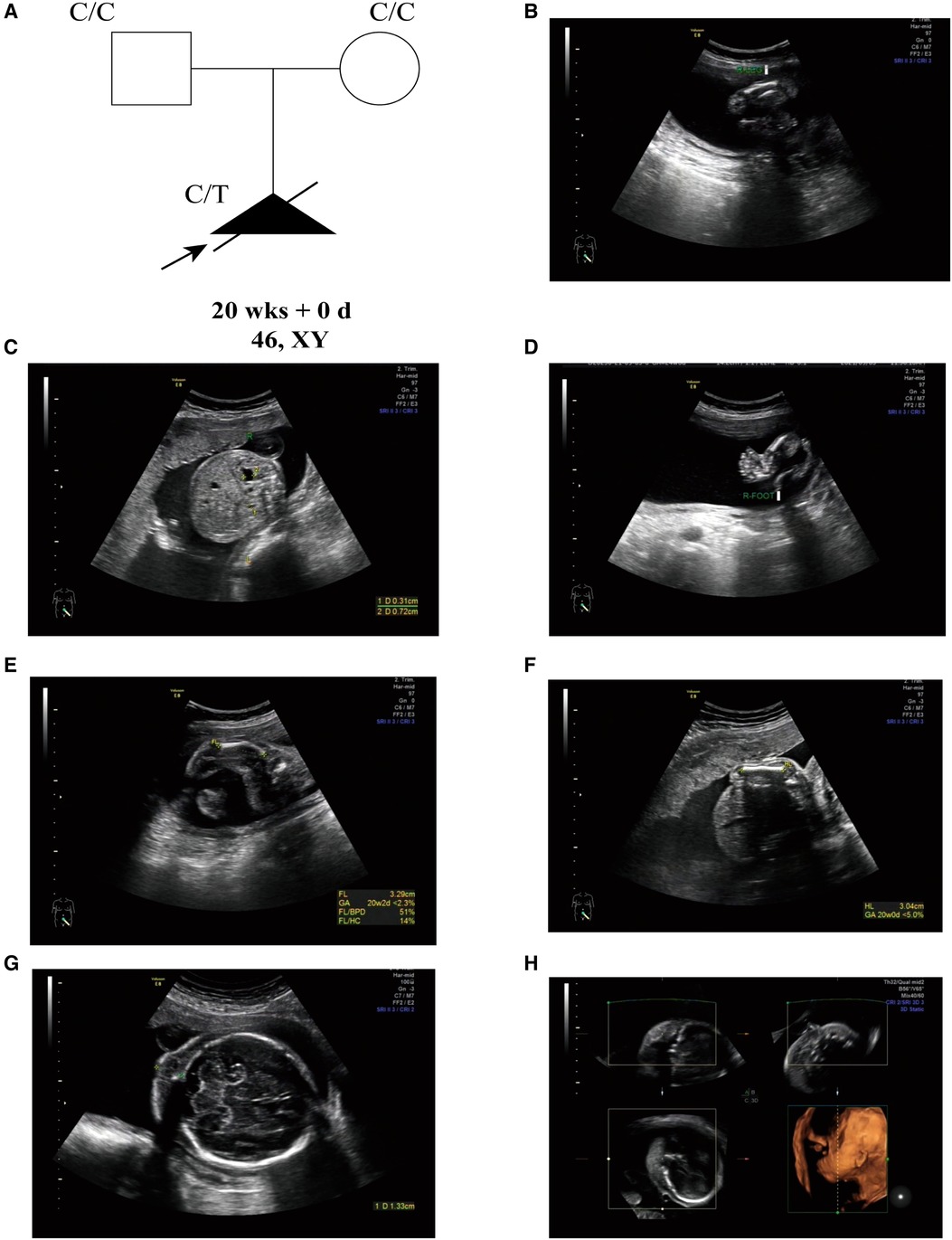
Figure 1. (A) the pedigree of this family. Black indicates the affected fetus with CD. White circles are unaffected. Arrow indicates the proband. (B) Short femurs. (C) Detached right renal pelvis. (D) Foot varus. (E) Short and curved femurs (<2.5%). (F) Shorted humerus (<5%). (G) Thickened neck fold (>6 mm). (H) Micrognathia.
After data filtering, a novel mutation of SOX9 (NM_000346.3; c.1249C > T; p.Q417*) was highly suspected to be the genetic lesion in the fetus (Figure 2A). No other potential pathogenic mutations were found to cause the malformation of bones. Further co-segregation analysis revealed that the novel SOX9 mutation did not exist in the parents. This mutation is a de novo mutation in this family. Bioinformatics-based prediction revealed that this termination mutation is pathogenic and may affect transactivation of SOX9. Otherwise, this novel mutation (c.1249C > T; p.Q417*) was located at a highly evolutionarily conserved site of the SOX9 protein (Figure 2B).
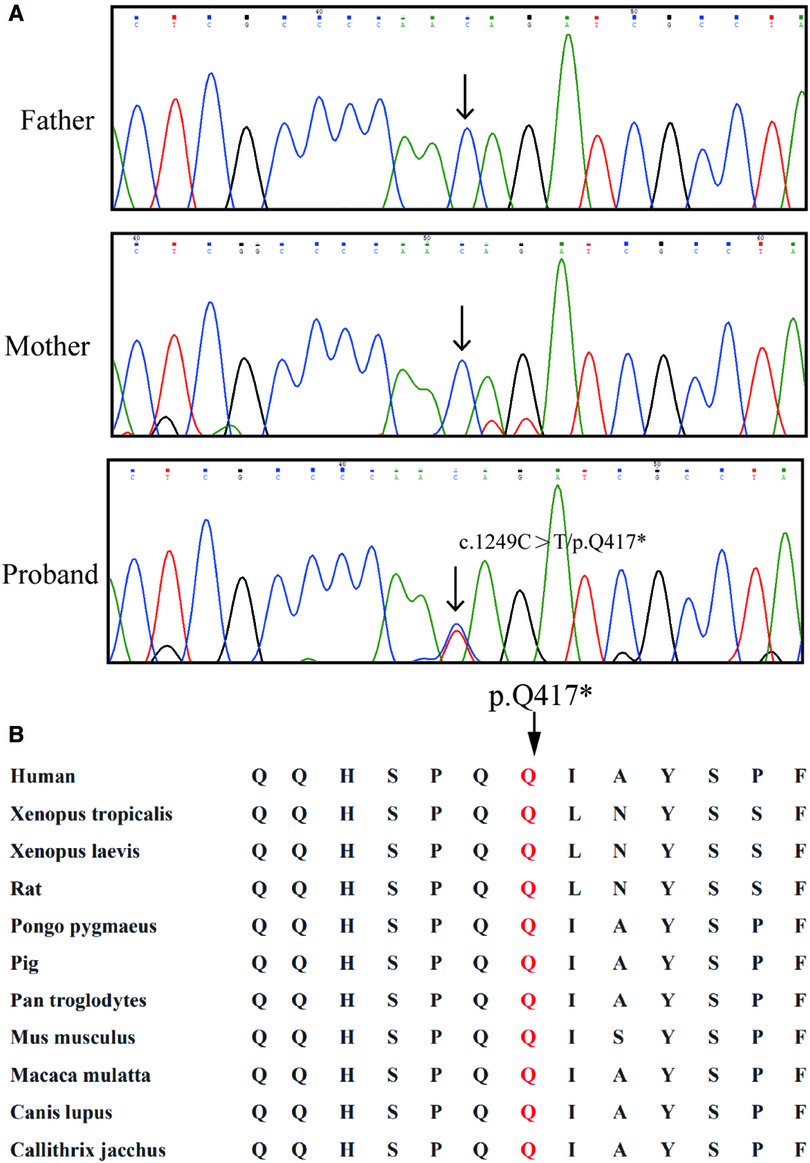
Figure 2. Genetic description of the Chinses family. (A) Sequencing results of the SOX9 mutation. Sequence chromatograms indicate the heterozygosity of the SOX9 non-sense mutation (NM_000346.3; c.1249C > T; p.Q417*) in the fetus and in the normal parents. Black arrow indicates the mutation site. (B) Alignment analysis of this site (p.G285) in COL4A4 amino acid sequences shows that the site (p.417) is highly conserved.
In this study, a novel mutation (c.1249C > T; p.Q417*) was identified in the affected fetus of a Chinese family by WES and Sanger sequencing. Our results were consistent with previous studies in humans and animals that heterozygous mutations in SOX9 may lead to CD (10, 11). This mutation occurred within the C-terminus of SOX9 and was presumably thought to led to a truncated SOX9 protein and affected the transactivation of SOX9. Our study further supports the recommendations released by the International Society for Prenatal Diagnosis, which highlight the application of WES when a fetal anomaly was observed (12).
CD is a lethal, autosomal dominant skeletal dysplasia usually caused by a heterozygous mutation of SOX9 and often accompanied by sex reversal (4). The SOX9 protein mainly consists of several domains, including a dimerization domain (DIM), a DNA-binding domain (high-mobility group, HMG), two transactivation domains, and a proline/glutamine/alanine (PQA)-rich domain (13). The missense, nonsense, frameshift, consensus splice site, and loss of function mutations located on one of the three exons of SOX9 are the main causes of CD (14). Otherwise, the reciprocal translocation involving SOX9 or its regulatory region also led to CD (15). The reported cases of CD that SOX9 being completely deletion over the past few decades were thought to represent evidence for the dosage-dependent action of SOX9 protein in normal chondrogenesis (8, 16). However, Csukasi et al. (14) revealed dominant-negative mutations of SOX9 in CD-affected individuals, and the reported deletion of SOX9 also overlapped the upstream enhancer region (7, 17, 18). Thus, it is difficult to confirm the haploinsufficiency of SOX9 in CD.
The correlation between the type and position of mutations of SOX9 with resulting phenotype is also lacking. Kwok et al. (19) reported two patients with the same frameshift mutation, but one presented with male phenotype and the other displayed a female phenotype with XY sex reversal. The reported Y440X mutation that presumably resulted in a truncated protein but retains some transactivation function has been described as a less lethal form of CD. The patient carrying the nonsense mutation Q117X has been reported performing well over a 10-year period. However, other reported patients with missense mutations such as Q375X and E400X died in the neonatal period (20).
The CD-causing mutations are usually located in the HGM domain and transactivation domain. SOX9 gene has two transactivation domains located in the middle (TAM) and the C-terminus (TAC), interacting with transcriptional co-activators or basal transcriptional machinery components. SOX9 transactivation domains work synergistically or independently of each other to activate chondrocyte-specific genes (13). A recent study revealed an evolutionarily conserved EΦ[D/E]QYΦ motif in TAM, playing a critical role in the transactivation function of SOX9 protein (21). The missense mutation in EΦ[D/E]QYΦ motif of SOX18 showed impaired transactivation (22). In addition, the interaction of SOX9 with β-catenin and TRAP230 required TAC, and the reported mutation of TAC (R394G and R437C) which retained normal transactivation of Col2a1 caused testicular dysgenesis without CD and sex reversal, a less lethal result (23, 24). However, the terminated mutation (Q412X) causes a severe phenotype and has a dramatically reduced effect on the SOX9 activation of Col2a1 (14). In our case, we reported a novel terminated mutation (p.Q417*) of SOX9, causing severe and lethal clinical phenotypes, including the leading causes of fetal mortality such as small thoracic, micrognathia, and cleft palate. This mutation is located in the TAC transactivation domain and the terminated mutation p.Q417* presumably resulted in a truncated SOX9 protein and impaired transactivation. The phenotype caused by mutation p.Q417X located in TAC of SOX9 was consistent with CD, providing increasing evidence for the correlation between the clinical and radiographic phenotype and the extent to SOX9 mutations affect transactivation.
In conclusion, there is growing evidence to support the role of SOX9 in the pathogenesis of CD, but the link between mutations and clinical phenotypes has not been established. Recent studies have shown that the pathogenicity of SOX9 is related to the activity of transactivation of mutant SOX9 protein. Severe clinical phenotype is closely related to the reduced activation of Col2a1. Mutations in the two transactivation domains of SOX9 have received additional attention, and this study provides a truncated mutation located in the TAC domain of SOX9, which may affect the transactivation activity of SOX9. Our study confirmed the clinical phenotype of this variant. Future research on SOX9 mutations should pay more attention to the correlation between SOX9 mutation and transactivation activity.
The results of genetic and ultrasound reports supported the diagnosis of CD, and the couple decided to terminate the pregnancy. The parents of the fetus provided written informed consent to participate in this study. Written informed consent was obtained from the couple for the publication of potentially identifiable images or data included in this article.
'The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Review Board of Shenzhen Longgang District Maternity and Child Healthcare Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
YC designed the research and collected the data of the Chinese family. LW performed the WES experiment and ultrasound examination. JT, RX and FL collected the data and information. HH and XQ analyzed the WES data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Fundamental Research Funds for the Central Universities of Central South University (grant No. 2021zzts1051), the Research project of Hunan Provincial Health Commission (grant No. 20200768), Chinese Cardiovascular Association- Access fund (grant No. 2020-CCA-ACCESS-115).
We thank the family for their participation in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tongsong T, Wanapirak C, Pongsatha S. Prenatal diagnosis of campomelic dysplasia. Ultrasound Obstet Gynecol. (2000) 15(5):428–30. doi: 10.1046/j.1469-0705.2000.00126.x
2. Mansour S, Hall CM, Pembrey ME, Young ID. A clinical and genetic study of campomelic dysplasia. J Med Genet. (1995) 32(6):415–20. doi: 10.1136/jmg.32.6.415
3. Cox JJ, Willatt L, Homfray T, Woods CG. A SOX9 duplication and familial 46,XX developmental testicular disorder. N Engl J Med. (2011) 364(1):91–3. doi: 10.1056/NEJMc1010311
4. Cameron FJ, Sinclair AH. Mutations in SRY and SOX9: testis-determining genes. Hum Mutat. (1997) 9(5):388–95. doi: 10.1002/(SICI)1098-1004(1997)9:5%3C388::AID-HUMU2%3E3.0.CO;2-0
5. Pfeifer D, Kist R, Dewar K, Devon K, Lander ES, Birren B, et al. Campomelic dysplasia translocation breakpoints are scattered over 1 mb proximal to SOX9: evidence for an extended control region. Am J Hum Genet. (1999) 65(1):111–24. doi: 10.1086/302455
6. Leipoldt M, Erdel M, Bien-Willner GA, Smyk M, Theurl M, Yatsenko SA, et al. Two novel translocation breakpoints upstream of SOX9 define borders of the proximal and distal breakpoint cluster region in campomelic dysplasia. Clin Genet. (2007) 71(1):67–75. doi: 10.1111/j.1399-0004.2007.00736.x
7. Olney PN, Kean LS, Graham D, Elsas LJ, May KM. Campomelic syndrome and deletion of SOX9. Am J Med Genet. (1999) 84(1):20–4. doi: 10.1002/(SICI)1096-8628(19990507)84:1%3C20::AID-AJMG5%3E3.0.CO;2-N
8. Kayhan G, Calis P, Karcaaltincaba D, Tug E. Prenatal diagnosis of campomelic dysplasia due to deletion. J Obstet Gynaecol. (2019) 39(8):1175–6. doi: 10.1080/01443615.2019.1601165
9. Huang H, Chen Y, Jin J, Du R, Tang K, Fan L, et al. CSRP3, P.Arg122*, is responsible for hypertrophic cardiomyopathy in a Chinese family. J Gene Med. (2022) 24(1):e3390. doi: 10.1002/jgm.3390
10. Bi W, Huang W, Whitworth DJ, Deng JM, Zhang Z, Behringer RR, et al. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci U S A. (2001) 98(12):6698–703. doi: 10.1073/pnas.111092198
11. Foster JW, Dominguez-Steglich MA, Guioli S, Kwok C, Weller PA, Stevanović M, et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature. (1994) 372(6506):525–30. doi: 10.1038/372525a0
12. Van den Veyver IB, Chandler N, Wilkins-Haug LE, Wapner RJ, Chitty LS. International Society for Prenatal Diagnosis Updated Position Statement on the use of genome-wide sequencing for prenatal diagnosis. Prenat Diagn. (2022) 42(6):796–803. doi: 10.1002/pd.6157
13. Ming Z, Vining B, Bagheri-Fam S, Harley V. SOX9 In organogenesis: shared and unique transcriptional functions. Cell Mol Life Sci. (2022) 79(10):522. doi: 10.1007/s00018-022-04543-4
14. Csukasi F, Duran I, Zhang W, Martin JH, Barad M, Bamshad M, et al. Dominant-negative SOX9 mutations in campomelic dysplasia. Hum Mutat. (2019) 40(12):2344–52. doi: 10.1002/humu.23888
15. Cooke CT, Mulcahy MT, Cullity GJ, Watson M, Srague P. Campomelic dysplasia with sex reversal: morphological and cytogenetic studies of a case. Pathology. (1985) 17(3):526–9. doi: 10.3109/00313028509105515
16. Liu X, Wang J, Yang M, Tian T, Hu T. Case report: cystic hygroma accompanied with campomelic dysplasia in the first trimester caused by haploinsufficiency with deletion. Front Genet. (2022) 13:950271. doi: 10.3389/fgene.2022.950271
17. Pop R, Conz C, Lindenberg KS, Blesson S, Schmalenberger B, Briault S, et al. Screening of the 1 mb SOX9 5’ control region by array CGH identifies a large deletion in a case of campomelic dysplasia with XY sex reversal. J Med Genet. (2004) 41(4):e47. doi: 10.1136/jmg.2003.013185
18. Smyk M, Obersztyn E, Nowakowska B, Bocian E, Cheung SW, Mazurczak T, et al. Recurrent SOX9 deletion campomelic dysplasia due to somatic mosaicism in the father. Am J Med Genet A. (2007) 143A(8):866–70. doi: 10.1002/ajmg.a.31631
19. Kwok C, Weller PA, Guioli S, Foster JW, Mansour S, Zuffardi O, et al. Mutations in SOX9, the gene responsible for campomelic dysplasia and autosomal sex reversal. Am J Hum Genet. (1995) 57(5):1028–36.7485151
20. Meyer J, Südbeck P, Held M, Wagner T, Schmitz ML, Bricarelli FD, et al. Mutational analysis of the SOX9 gene in campomelic dysplasia and autosomal sex reversal: lack of genotype/phenotype correlations. Hum Mol Genet. (1997) 6(1):91–8. doi: 10.1093/hmg/6.1.91
21. Haseeb A, Lefebvre V. The SOXE transcription factors-SOX8, SOX9 and SOX10-share a bi-partite transactivation mechanism. Nucleic Acids Res. (2019) 47(13):6917–31. doi: 10.1093/nar/gkz523
22. Sandholzer J, Hoeth M, Piskacek M, Mayer H, de Martin R. A novel 9-amino-acid transactivation domain in the C-terminal part of Sox18. Biochem Biophys Res Commun. (2007) 360(2):370–4. doi: 10.1016/j.bbrc.2007.06.095
23. Katoh-Fukui Y, Igarashi M, Nagasaki K, Horikawa R, Nagai T, Tsuchiya T, et al. Testicular dysgenesis/regression without campomelic dysplasia in patients carrying missense mutations and upstream deletion of SOX9. Mol Genet Genomic Med. (2015) 3(6):550–7. doi: 10.1002/mgg3.165
Keywords: SOX9, transactivation, campomelic dysplasia, terminated mutation, non-sense
Citation: Qiao X, Wu L, Tang J, Xiang R, Fan L, Huang H and Chen Y (2023) Case report: A de novo Non-sense SOX9 mutation (p.Q417*) located in transactivation domain is Responsible for Campomelic Dysplasia. Front. Pediatr. 10:1089194. doi: 10.3389/fped.2022.1089194
Received: 4 November 2022; Accepted: 28 December 2022;
Published: 18 January 2023.
Edited by:
Paul Lasko, McGill University, CanadaReviewed by:
Hala El-Bassyouni, National Research Centre, Egypt© 2023 Qiao, Wu, Tang, Xiang, Fan, Huang, Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaqin Chen YXZpdmE5OTAzQGNzdS5lZHUuY24=
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.