 Nicholas Landini1
Nicholas Landini1 Pierluigi Ciet2,3,4*Hettie M. Janssens4Silvia Bertolo5Mirco Ros6
Pierluigi Ciet2,3,4*Hettie M. Janssens4Silvia Bertolo5Mirco Ros6 Monica Mattone1Carlo Catalano1
Monica Mattone1Carlo Catalano1 Fabio Majo7
Fabio Majo7 Stefano Costa8
Stefano Costa8 Andrea Gramegna9,10
Andrea Gramegna9,10 Francesca Lucca11
Francesca Lucca11 Giuseppe Fabio Parisi12Luca Saba3Harm A. W. M. Tiddens2,4
Giuseppe Fabio Parisi12Luca Saba3Harm A. W. M. Tiddens2,4 Giovanni Morana5
Giovanni Morana5
- 1Department of Radiological, Oncological and Pathological Sciences, Policlinico Umberto I Hospital, “Sapienza” Rome University, Rome, Italy
- 2Department of Radiology and Nuclear Medicine, Erasmus MC – Sophia, Rotterdam, Netherlands
- 3Department of Radiology, University Cagliari, Cagliari, Italy
- 4Department of Pediatrics, division of Respiratory Medicine and Allergology, Erasmus MC – Sophia Children’s Hospital, University Medical Center Rotterdam, Rotterdam, Netherlands
- 5Department of Radiology, S. Maria Ca’Foncello Regional Hospital, Treviso, Italy
- 6Department of Pediatrics, Ca’Foncello S. Maria Hospital, Treviso, Italy
- 7Pediatric Pulmonology & Cystic Fibrosis Unit Bambino Gesú Children's Hospital, IRCCS Rome, Rome, Italy
- 8Department of Pediatrics, Gaetano Martino Hospital, Messina, Italy
- 9Department of Pathophisiology and Transplantation, University of Milan, Milan, Italy
- 10Respiratory Disease and Adult Cystic Fibrosis Centre, Internal Medicine Department, IRCCS Ca’ Granda, Milan, Italy
- 11Regional Reference Cystic Fibrosis Center, University Hospital of Verona, Verona, Italy
- 12Pediatric Pulmonology Unit, Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy
Respiratory tract exacerbations play a crucial role in progressive lung damage of people with cystic fibrosis, representing a major determinant in the loss of functional lung tissue, quality of life and patient survival. Detection and monitoring of respiratory tract exacerbations are challenging for clinicians, since under- and over-treatment convey several risks for the patient. Although various diagnostic and monitoring tools are available, their implementation is hampered by the current definition of respiratory tract exacerbation, which lacks objective “cut-offs” for clinical and lung function parameters. In particular, the latter shows a large variability, making the current 10% change in spirometry outcomes an unreliable threshold to detect exacerbation. Moreover, spirometry cannot be reliably performed in preschool children and new emerging tools, such as the forced oscillation technique, are still complementary and need more validation. Therefore, lung imaging is a key in providing respiratory tract exacerbation-related structural and functional information. However, imaging encompasses several diagnostic options, each with different advantages and limitations; for instance, conventional chest radiography, the most used radiological technique, may lack sensitivity and specificity in respiratory tract exacerbations diagnosis. Other methods, including computed tomography, positron emission tomography and magnetic resonance imaging, are limited by either radiation safety issues or the need for anesthesia in uncooperative patients. Finally, lung ultrasound has been proposed as a safe bedside option but it is highly operator-dependent and there is no strong evidence of its possible use during respiratory tract exacerbation. This review summarizes the clinical challenges of respiratory tract exacerbations in patients with cystic fibrosis with a special focus on imaging. Firstly, the definition of respiratory tract exacerbation is examined, while diagnostic and monitoring tools are briefly described to set the scene. This is followed by advantages and disadvantages of each imaging technique, concluding with a diagnostic imaging algorithm for disease monitoring during respiratory tract exacerbation in the cystic fibrosis patient.
Introduction
Cystic fibrosis (CF) is a systemic disease in which lung involvement is the major cause of morbidity and mortality (1). Although improvements in patients' management and therapeutic strategies have increased the life expectancy of people with CF (PCF), it remains lower than in the general population (1). Respiratory tract exacerbations (RTE) contribute significantly to lung morbidity and disease progression in PCF; recurrent RTE during childhood are associated with lung function deterioration, analysed, for example, via spirometry, and structural abnormalities, such as bronchiectasis, detected by computed tomography (CT) (2). The frequency of RTE increases with patient age. Moreover, older PCF require longer antibiotic treatment, which impacts adversely on both patient quality of life and costs for the healthcare system (3). Moreover, most PCF who experienced RTE do not recover their pre-exacerbation lung function (3).
The etiology of RTE is not completely understood. Infections by new pathogens (e.g., viruses) and overgrowth of preexisting airway flora (mostly bacteria) can determine the nature of acute infection and, since pathogen determination is not always possible via sputum sample, treatment may be complex. While invasive approaches, such as bronchoalveolar lavage (BAL), provide higher sensitivity and specificity (4), they are not routinely performed in all CF centers.
The treatment of RTE is not standardized either (2) and there is no international consensus on its optimal duration. Current RTE treatment, especially antibiotic therapy, lasts on average 14 days, with conflicting evidence for the use of prolonged treatment intervention (2). Thus, clinicians have no objective tools or guidelines to support clinical decision making, relying, instead, on improvement of pulmonary function tests (PFTs), e.g. forced expiratory volume in the first second (FEV1), and relief of symptoms (i.e., cough and sputum production) (5).
For all these reasons, early diagnosis of RTE is crucial for timely initiation of therapy, as well as optimizing treatment efficacy. As recently highlighted in national guidelines (1), imaging techniques can improve both RTE detection and monitoring of therapy, providing information that may support clinicians with patient care.
Although evidence for its use remains controversial, chest radiography (CR) is the most frequently used imaging technique for RTE detection (6). The gold standard for the assessment of structural lung abnormalities in PCF, however, is computed tomography (CT), especially after the introduction of the low and ultra-low dose CT protocols (7). Despite its advantages, the use of CT remains limited for short-term follow-up serial imaging—as used in RTE—largely due to concern regarding excessive radiation exposure.
Conversely, magnetic resonance imaging (MRI) offers a useful and safe alternative, especially for both inflammation and functional assessment (1), while lung ultrasound (LUS) has been recently proposed as a point-of-care diagnostic tool for repeatable bedside RTE monitoring (8).
This state-of-the-art review summarizes the clinical challenges of RTE in PCF with special focus on imaging. A short description of the definition of RTE, available diagnostic and monitoring tools, and current treatment options are firstly given to illustrate the clinical context. The review then discusses the advantages and disadvantages of respective imaging techniques, which incorporates a diagnostic imaging algorithm for monitoring PCF during RTE.
Respiratory tract exacerbations definition
The Cystic Fibrosis Foundation defines RTE as “acute worsening of respiratory symptoms” requiring medical treatment, mainly represented by antimicrobial therapy and airway clearance through physiotherapy, often under hospitalization (9). The occurrence of RTE is a risk factor for decreased life quality (10, 11), increased medical resource utilization (12), and mortality (13, 14). Therefore, reduction in RTE rate and/or risk has become a robust efficacy measure for trials of chronic CF therapies (15).
Nonetheless, a shared and objective definition of RTE, exploitable as an outcome in clinical trials, is still lacking.
The easiest way to define RTE is to consider the beginning and end dates of antimicrobial treatment on the patient medical record, assuming that the treatment correlated with the onset and resolution of RTE. The choice of drug administration route (intravenous vs. oral) and intervention type (hospital at home vs. in-hospital stay) depend on the RTE severity (16, 17).
The need for more objective criteria for identifying RTE has been driven by the need to avoid variability in clinician assessment.
The dornase alfa Phase 3 study (16) defined RTE as a respiratory event treated with intravenous (IV) antimicrobials, where at least 4 out of 12 specific signs and symptoms were present. Following this publication, these criteria are referred to as the “Fuchs criteria” and are frequently used in both studies and clinical practice (Table 1). Other groups have proposed similar criteria to define RTE, such as the Rosenfeld criteria (18). Since the probability of requiring IV antimicrobial treatment for exacerbation is age- and lung-function-dependent (19) [unlike the overall rate of RTE subjectively assessed by clinicians (20)], the RTE definition has been expanded to include any antimicrobial treatment. This kind of definition has been recently used to explore the rate/risk of exacerbation in clinical trials on CF transmembrane conductance regulator (CFTR) modulators (21).
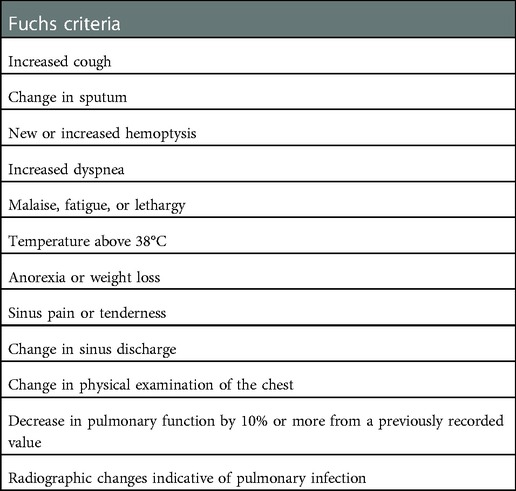
Table 1. Original fuchs criteria: at least 4 out of 12 of the following signs and symptoms must be present for respiratory tract exacerbation diagnosis.
Clinical scenario and diagnosis
A challenging aspect of using the Fuchs criteria in clinical practice is that prescribed antibiotic therapy forms part of the diagnostic criteria for RTE as well as its treatment.
The selection of antibiotic therapy is governed by strong subjective influence, which greatly limits the applicability of the Fuchs criteria in daily practice. For example, in a clinical trial using hypertonic saline, Elkins et al. (22) compared a version of the Fuchs criteria based on symptoms (regardless of the prescribed treatment) with classic criteria, confirming the strong variability introduced by clinician opinion. This is unsurprising, since some of the original criteria, such as physical examination, may be nonspecific or present with various clinical scenarios, that can lead to different therapeutic approaches.
Therefore, an international group of experts (23) has suggested a modified version of the Fuchs criteria that involves the presence of two out of six signs or symptoms (Table 2). The following original criteria were removed: new or increased hemoptysis, temperature over 38°C, sinus pain or tenderness, change in sinus discharge and change in chest findings on physical examination; the latter was excluded because it was too difficult to define and standardize. However, although physical examination has low sensitivity and is difficult to standardize, it remains the first and most used method to diagnose RTE.
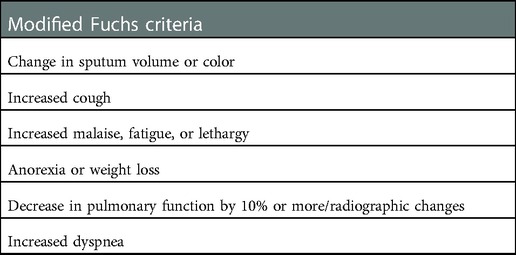
Table 2. Modified Fuchs criteria according to the European Consensus Group: at least 2 out of 6 of the following signs and symptoms must be present for respiratory tract exacerbation diagnosis.
Many gaps in our knowledge about the clinical management of the acute phase of RTE remain (3). Firstly, standardized diagnostic criteria might help define RTE in both daily practice and clinical research; possible nonbacterial and noninfectious etiologies, uncontrolled comorbidities and poor treatment adherence should also be carefully considered. Secondly, predictive biomarkers must be identified and implemented to confirm the RTE diagnosis and monitor treatment response. Thirdly, the establishment of agreement on standardized antibacterial therapy and its duration for RTE are areas of new research, which offer many implications for clinical practice (24). Then, although RTE increase in frequency and severity based on the degree of lung damage, there is still no means of structured assessment that can be used in the decision-making process for treatment selection. Finally, the therapeutic landscape of CF lung disease is being significantly altered following the arrival of new classes of CFTR modulators (25), where their long-term impact on CF progression and RTE frequency is still unknown. Whether RTE will play a different role in CF progression in the near future is a key topic to be addressed by further research.
Physical examination
During RTE, findings of the physical examination are part of the objective measures that most scoring tools use combined with (subjective) symptoms. However, these findings are frequently subject to low interobserver repeatability, e.g., changes on auscultation (26). The odds of a clinician-diagnosed RTE have recently been shown to increase when associated with reported symptoms and a drop in forced expiratory volume (FEV1). However, evaluating FEV1 decline and subsequent RTE management in non-cooperative children can be challenging (27).
Physical examination of the chest is a Fuchs criterion for RTE diagnosis (16) and part of the scoring system in the multivariate model developed by Rosenfeld; however, in the study by Rosenfeld et al., the most important features in determining the presence of RTE were symptoms and clinical history rather than physical examination and laboratory results (18).
Most scoring tools are barely used in a clinical setting, while lung examination findings remain central to the clinical practice of any clinician. However, the last two years of the COVID-19 pandemic have reduced reliance on traditional diagnostic criteria, especially the physical examination. The impossibility of visiting patients made telehealth more prevalent, and RTE had to be defined without examination or spirometry (28).
In the absence of any physical examination, symptoms recalled by PCF or caregivers are a direct and fundamental source of information. Patient-reported outcomes represent the first attempt at standardizing subjective symptoms and their incorporation as additional endpoints in scoring tools and clinical practice is becoming a developing area in RTE definition and monitoring (29).
Pulmonary function tests and functional biomarkers
Spirometry has always been considered the main pulmonary function test (PFT) in PCF. In particular, the forced expiratory volume in the first second (FEV1) is the most used parameter to assess the severity of lung disease, measure outcomes in clinical trials, evaluate therapeutic response and refer patients for lung transplantation (16, 30, 32). The measure can also be combined with clinical criteria to identify RTE and assess its trend over time or evaluate therapy duration and efficacy (16, 32, 33). Most authors recognize an FEV1 decline >10% as an indicator of RTE, and it is used as a criterion for initiating oral or IV antibiotic therapy (16, 34). This explains the need for spirometry as a cornerstone of the annual check-up performed at CF centers (35, 36).
On the other hand, PFTs parameters assessed by spirometry are also used to determine the duration of antibiotic therapy during RTE, but data from the literature are controversial.
Redding et al. monitored the lung function of 17 children with CF every day during a 14-day hospitalization to undergo IV antibiotic therapy, but failed to demonstrate an optimal duration of treatment because the FEV1 response was highly variable (36); similar results were observed in the retrospective study by Rosenberg and Schramm (37).
Conversely, a retrospective study by Collaco et al. suggested that the duration of antibiotic therapy during RTE can be reduced to 7–10 days when a stable improvement in FEV1 is evident within this period (38). More recently, Stephen et al. assessed daily FEV1 in an adult CF population with RTE, demonstrating that achieving an FEV1 increase of >10% takes an average of 6 days; however, the result variability was so large that the authors could not assess the optimal duration of antibiotic therapy. Therefore, other independent variables of PFT have been suggested to determine the therapy duration (40).
Moreover, RTE may result in an irreversible loss of lung function, with an FEV1 that does not always return to pre-exacerbation values (39, 40), although the reason that lies behind this eventuality remains unclear.
Besides, PFTs have some further intrinsic limitations, since there may be a big discrepancy between structure and function (41), e.g., preserved lung function may disguise extensive bronchiectasis. Furthermore, young children (below 6 years) cannot perform the forced maneuver for traditional PFTs. These observations emphasize the need for new tools to ensure a more sensitive assessment of RTE, beyond clinical and spirometric analyses (42).
New PFTs correlated with structural changes have been recently investigated, including the multiple breath washout test, which allows the assessment of the lung clearance index (LCI). The LCI is a parameter of ventilatory inhomogeneity which is currently recognized as the most sensitive index for identifying small airway obstruction (43–45). Despite intra-individual variability of up to 15%, higher values of LCI may be a telltale sign of RTE (46, 47). Walicka et al. proposed a lower cut-off to define RTE based on LCI, where the LCI threshold should be reduced to 10%, as this would increase its sensitivity compared with conventional spirometry and facilitate calculations in clinical practice (48). They also showed that RTE are associated with a 9% worsening of LCI even at the end of treatment (48), a percentage that is reduced to 3% according to Perrem et al. (46). Furthermore, more recent studies indicate the LCI to be a far more sensitive parameter than FEV1, with rapid LCI changes being seen at the onset of RTE. The combined assessment via spirometry and LCI is superior to diagnose and monitor PCF during RTE (47–49). However, LCI is not currently used as a standard of care because it requires specialized personnel and equipment and is rather time-consuming.
Other biomarkers
Infection
Respiratory tract exacerbations in CF are treated with antibiotics directed against microorganisms found in the airways. Because infections by microorganisms, such as Pseudomonas aeruginosa, accelerate lung function decline in PCF, eradication therapy must be quickly initiated.
Microbiological cultures from airways are therefore taken regularly and represent an important biomarker for the management of CF lung disease; samples can be collected from BAL, sputum or the upper airways, with decreasing reliability of accuracy for finding microorganisms in the lower airways (50, 51). Bronchoalveolar lavage is considered the gold standard for assessing the presence of microorganisms in the lungs. However, the nature of the procedure—which is invasive, requiring bronchoscopy under anesthesia—means that it is not routinely done in all CF centers. Sputum sampling is a good alternative, but young children cannot always cough up enough sputum for cultures. Furthermore, cultures obtained from the upper airways often do not reflect the microorganisms residing in the lungs.
As a result of these limitations, research has been focused on finding other ways to detect microorganisms in the lungs. For example, the analysis of volatile organic compounds in exhaled breath is a promising tool; this technique can discriminate between positive and negative Pseudomonas cultures in PCF (52). However, most research on other biomarkers is still in the developmental phase.
Inflammation
There has been an increase in our understanding of the role of inflammation in the pathophysiology of CF lung disease. Although it is known that a vicious circle of infection and neutrophilic inflammation causes structural damage to the lungs, a debate exists regarding which comes first. By 1995 it was shown that, despite the absence of microorganisms in BAL cultures, there were signs of inflammation in the lungs in patients as young as 4 weeks. It has also been demonstrated that increased neutrophil elastase (NE) and increased neutrophils in BAL infants are risk factors for developing bronchiectasis at a later age (53, 54). Research on RTE biomarkers—for example, as found in sputum, serum and exhaled breath condensate (EBC)—in CF has therefore focused on assessing inflammation to predict exacerbations (55).
Numerous studies have investigated sputum as a source of biomarkers in CF. Existing sputum biomarkers with higher scientific reliability include interleukin (IL)-8 and NE, since their levels increase during RTE and decrease after treatment (56–58). A recent extensive review by the biomarker Special Interest Working Group of the European Cystic Fibrosis Society–Clinical Trials Network Standardization Committee stated that sputum biomarkers hold promise as a direct measure of inflammation, with NE, IL-8, tumor necrosis factor alpha, and IL-1β showing that validity and responsiveness should be considered as outcome measures in clinical trials. However, variability in study design and sampling methods has limited their use in clinical practice. Future focus should therefore be on creating standards for the collection, storage, and analysis of sputum biomarkers (59).
EBC analysis represents an innovation that has great potential for understanding the biochemical and metabolic mechanisms of respiratory disease. The EBC is the fluid obtained in a condenser by cooling exhaled air during current volume breathing. In PCF, an important contribution to pulmonary damage originates from an increasing imbalance between oxidant and antioxidant molecules in the airways. For example, extracellular glutathione transport may be altered in PCF, contributing to the loss of an important airway antioxidant defense mechanism. Furthermore, high levels of malondialdehyde and 8-isoprostane, two biomarkers of oxidative stress, have been found during RTE in the EBC of PCF (60, 61). However, in another study, 8-isoprostane and nitrite in EBC failed to show sufficient sensitivity for RTE prediction (62). A further study has reported increased NE in the EBC of children with CF. However, nitric oxide, IL-17, IL-8, e-cadherin, NE, or leukotriene B4 levels in the EBC of CF children were not related to P. Aeruginosa infection, FEV1 levels, or hospital admission in the last year (63). So far, sampling difficulties and variable results have failed to prove the usefulness of EBC in clinical practice.
Finally, metabolomic studies provide potentially interesting data. In this regard, a recent systematic review identified two potential metabolites (4-hydroxycyclohexylcarboxylic acid and lactic acid) that could predict RTE when measured in EBC and four other metabolites (sputum lactic acid, nitrate, plasma arginine, and methionine) with a high sensitivity for RTE diagnosis (Figure 1) (64).
Regarding blood biomarkers, a recent meta-analysis concluded that there is reasonable evidence to support their application in RTE diagnosis but further studies are needed to demonstrate the usefulness of this approach (65). Although the most used serum marker in CF is C-reactive protein, there is insufficient evidence that levels correlate with RTE severity (66). More interestingly, levels of calprotectin ̶ a neutrophil-derived protein ̶ increase during RTE and decrease after treatment, and apparently correlate with RTE prediction more sensitively than C-reactive protein (67, 68). Other biomarkers under study include serum amyloid A, IL-1Ra, and some cleavage products of the endogenous respiratory tract proteins produced during episodes of infection, e.g., secretory leukocyte protease inhibitor, elafin, and LL-37 (69, 70).
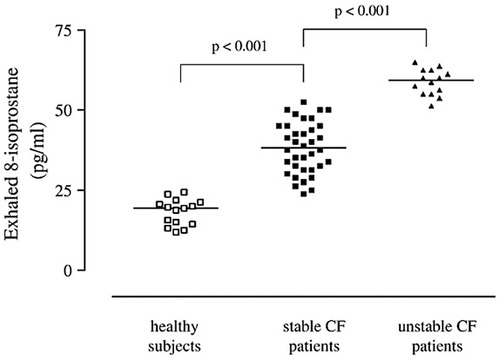
Figure 1. Adapted from “Exhaled 8-isoprostane and prostaglandin E(2) in patients with stable and unstable cystic fibrosis”: 8-Isoprostane concentrations in exhaled breath condensate in healthy subjects (open squares) and patients with stable (filled squares) and unstable (filled triangles)cystic fibrosis. Horizontal bars represent median values (61).
In conclusion, the most promising predictive inflammatory biomarkers for RTE in PCF appear to be NE, IL-8, tumor necrosis factor alpha, and IL-1 β in sputum and calprotectin in serum. The metabolomic analysis of EBC, sputum, and serum has shown interesting results but needs further investigation.
Imaging techniques
Imaging plays a key role in RTE management by its ability to provide structural and functional information. The advantages and disadvantages of the imaging techniques currently available are discussed in the following paragraphs and summarized, along with the other diagnostic methods, in Table 3.
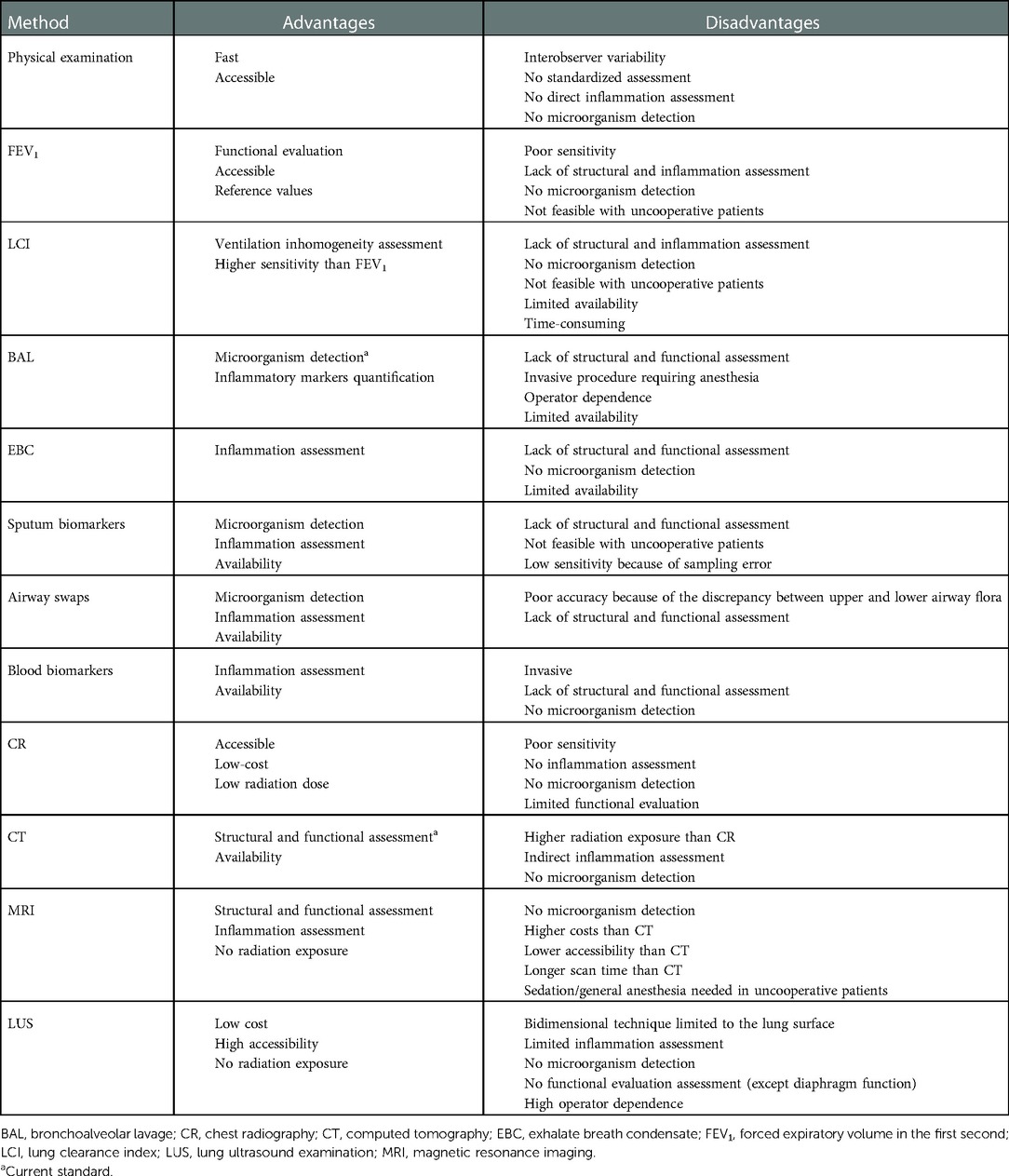
Table 3. Advantages and disadvantages for each respiratory tract exacerbation evaluation method.
Chest radiography
CR is usually the first imaging tool utilized for detecting RTE due to its ready availability, low cost, and lowest radiation exposure compared with CT (1). A recent review has given dose ranges for CR and CT imaging in PCF (7). When comparing the effective dose of current low-dose CT protocols for PCF to a single projection CR (either anterior-posterior or posterior-anterior) the dose range of CT is in the order of 15–30 CRs (7). This range is expected to further decrease by the introduction of ultra-low-dose protocols (7).
However, CR has low sensitivity and specificity in both detecting and following up on new lung alterations. Since lung abnormalities may not differ between patients with and without RTE, nor before and after therapy (6), because of differing disease severities, new CR abnormalities can be very difficult to detect among severe parenchymal changes (70) (Figure 2). Moreover, some alterations are focal in nature and not directly visible in a single projection, being hidden in blind spots, e.g., lung apices and retrocardiac regions (71). Thus, the use of CR is mainly supported by the need to exclude major complications, such as pneumothorax (6), or for assessment of gross abnormalities (Figure 3).
More recently, dynamic CR has been proposed for RTE monitoring. FitzMaurice et al. demonstrated improving speed and range of the diaphragmatic movement in PCF during RTE following treatment (72). Dynamic CR data were acquired over 10 seconds; with machine learning software, diaphragm motion and lung density were extracted during tidal breathing acquisition and from full inspiration to passive end-expiration. The results of this single-center study, however, need further confirmations to assess the possible role of this technique in RTE evaluation.
Computed tomography
Given its ability to provide a detailed view of regional airway and parenchymal changes, CT remains the tool of choice for assessing structural changes in CF lung disease alongside CR. The technique currently offers the best spatial resolution of all imaging modalities. Common CT scanners have a resolution in the order of one millimeter (mm), which allows detection of airways as small as 2 mm in diameter. For an adult without lung disease, this degree of resolution means that during an inspiratory CT scan airways up to the 13th generation can be detected. This is less for preschool children when only more central airways up to the 9th generation can be seen (73, 74). However, inspiratory and expiratory CT scans reveal data on ventilatory alterations and show higher sensitivity than spirometry in detecting early small airways disease (70). Therefore, well-established scores of lung involvement in PCF can be useful during RTE.
Davis et al., using the modified Brody score, demonstrated that CT can reveal areas of active inflammation during RTE (which is sometimes used to guide sampling) which improves after treatment (75). Moreover, CT may be helpful in the stratification of risk for RTE. For example, a higher modified Bhalla score or a higher bronchiectasis score at baseline can be used to identify patients with higher RTE frequency (76, 77). Nonetheless, although CT findings can demonstrate the reversibility of RTE-induced lung abnormalities, CT-detected abnormalities may not differ between RTE and asymptomatic patients (78), that may show a large variability in the resolution of structural abnormalities. This indicates that CT is a reliable tool to monitor new structural changes related to RTE but not to diagnose RTE itself (Figure 4).
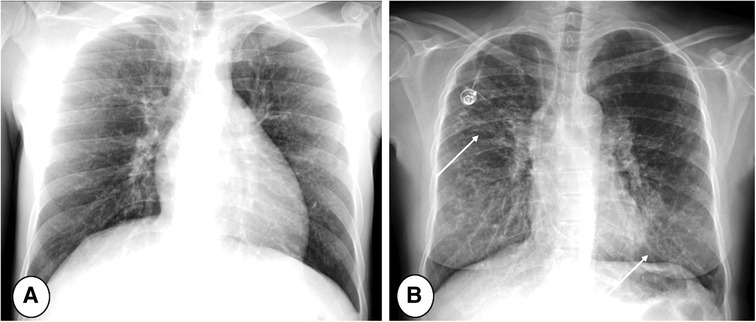
Figure 2. Posterior/anterior chest radiographs in (A) mild and (B) severe cystic fibrosis lung disease. Note the progressive increase of bronchial pathology in the subject with severe disease (B, arrows). The more severe the disease, the more difficult the detection of new abnormalities, especially during respiratory tract exacerbation.
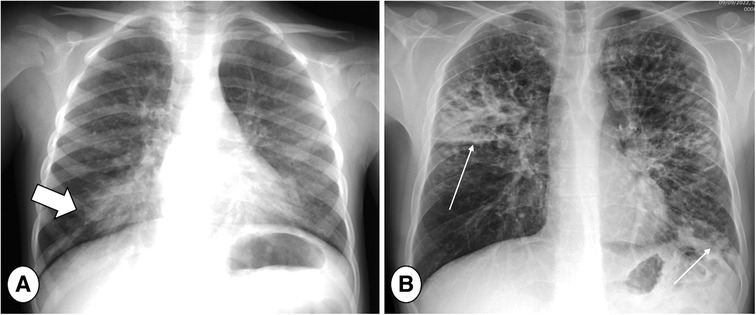
Figure 3. Posterior-Anterior (PA) chest radiograph in mild (A) and severe (B) cystic fibrosis lung disease during respiratory tract exacerbation. Note large consolidation in the right lung base in image A indicating respiratory tract exacerbation (thick arrow) and bilateral consolidations in the right upper lobe and left lung base (B, thin arrows).
Current low and ultra-low CT protocols have further reduced the risk of radiation exposure (7), especially during long-term monitoring of PCF. However, clinicians remain reluctant to use CT for short-term follow-up of RTE treatment, especially in children, who are more sensitive to radiation (79). Further dose reduction could be achieved following the recent introduction of the photon-counting detector CT, a new technology which enables higher resolution at lower dose range (7, 80). Therefore, concerns about radiation exposure may become a minor issue for short-term RTE monitoring.
Another major advantage of CT is its scan speed: high image-quality ̶ thanks to fast acquisition strategies and iterative reconstructions ̶ can also be achieved in non-collaborative patients, such as infants and young children (81), i.e., adopting high-pitch with dual source acquisitions (Figure 5). This facility reduces the need for sedation or general anesthesia compared with MRI, which requires longer acquisition time. Conversely, CT is more limited than MRI in providing functional information such as that which may relate to perfusion and inflammation. For instance, the use of iodine contrast is needed to obtain perfusion mapping with CT, while non-contrast techniques, described below, are available for MRI.
Finally, CT imaging is far beyond the capability of MRI in terms of automatic quantification of lung abnormality. Automatic scoring systems for CT in PCF have been proposed and tested in large cohorts (82, 83). These tools eliminate the variability related to inter-observer assessment and increase robustness of quantitative data. Other software now focuses on the assessment of airway-artery ratio, the most sensitive outcome parameter of early airways disease in PCF (73, 74).
Magnetic resonance imaging
Magnetic Resonance Imaging might become the “one-stop-shop” for diagnosing and monitoring RTE in PCF. In facts, thanks to several MRI techniques, information on structure, inflammation and function may be collected during a single examination and without radiation exposure.
Morphological changes during RTE can be evaluated using conventional sequences such as T1-, T2- and proton density-weighted images (84, 85). According to the patient's age, the protocol can be adapted in terms acquisition type, using end-expiratory triggered sequences in non-cooperative PCF, and breath-hold scans in older patients (Figure 6). Dedicated chest MRI scoring allows a more objective assessment of disease burden, even if the lower resolution of MRI compared with CT remains a limitation. For example, an inferior performance of MRI compared with CT was observed in the morphologic assessment of peripheral alteration, e.g., bronchiectasis and tree in bud, which may be underestimated by MRI sequences (Figure 7) (85).
Nevertheless, recently developed zero echo time or ultrashort echo time (ZTE and UTE) sequences, which are currently the MRI sequences with the best resolution and signal-to-noise ratio (SNR) (Figure 8), were tested with promising results in diffuse lung disease and CF (84, 86, 87). While these sequences allow sub-millimetric isotropic voxel resolution (88), they are limited by a long acquisition time, which are usually around 10 min (86), compared with the few seconds or less of the CT scan. Moreover, in terms of spatial resolution, MRI still does not match CT performance, with best voxel size for MRI of 0.86 mm3 against the current 0.2 mm3 capability of new Photon Counting CTs (7, 88). Further improvement in image quality is expected from low-field MRI (i.e., 0.55 T) scanners, which should provide higher SNR, as recently shown in diffuse lung diseases (89, 90). However, to date, there are no articles showing the use of low-field MRI scanner for PCF.
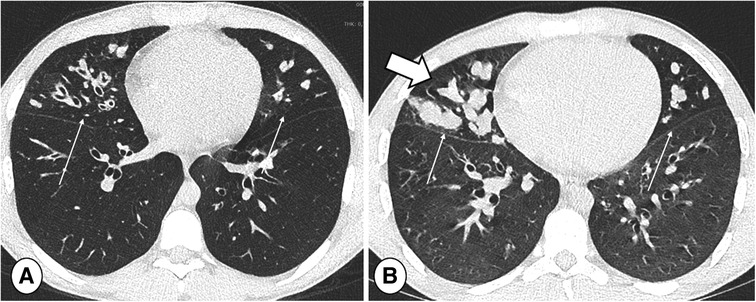
Figure 4. End-inspiratory CT of a cystic fibrosis patient (A) without and (B) with respiratory tract exacerbation, lung window. Note bronchiectasis and mucus plugs, especially in the middle lobe and lingula (thin arrows). The only difference indicating RTE is an increase in central mucus plugs (thick arrow).
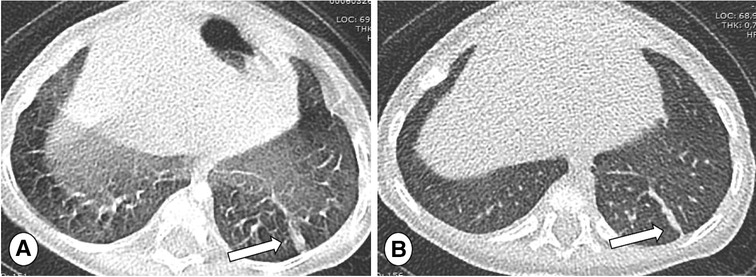
Figure 5. Axial CT of 5 months old patients with CF, thickness 0.7 mm, scanned in single source-mode with pitch 1.2 (A) and repeated in double source-mode with pitch 3.2 (B). Note that respiratory artifacts are absent using an higher pitch acquisition (B), providing a high image quality and facilitating the detection of atelectasis in the left lower lobe (arrows).
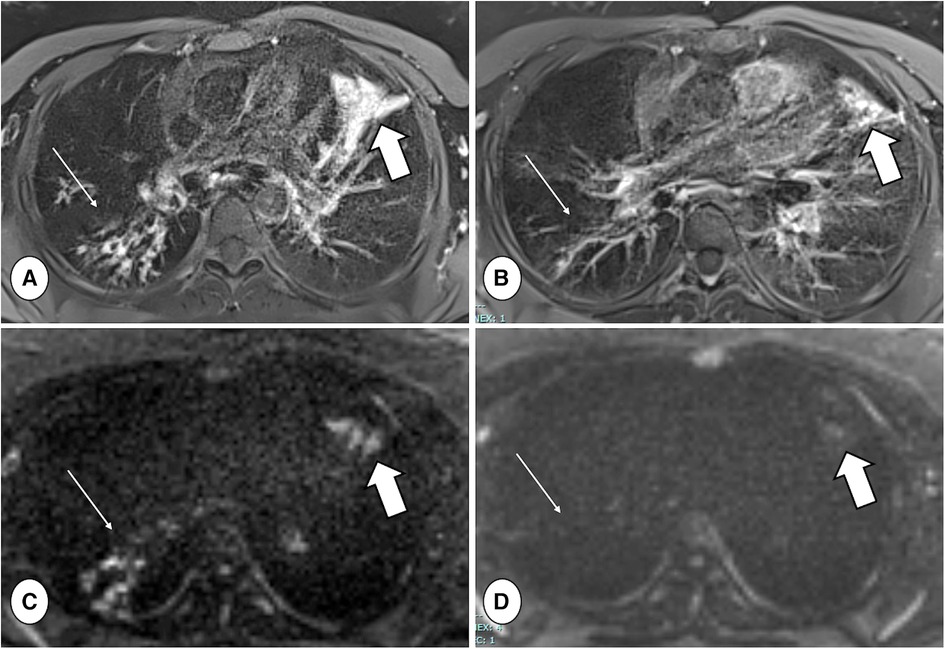
Figure 6. Axial proton density-weighted PROPELLER free-breathing scan (A,B) and diffusion-weighted imaging (DWI) (b = 800 mm/s2) acquisition (C,D), both performed with a 1.5 T MRI system (MAGNETOM avanto, siemens healthineers, enlargen, Germany), in a patient with CF during respiratory tract exacerbation at baseline (A,C) and after treatment (B,D). Note the reduction in mucus plugs and DWI signal in the right lower lobe between baseline and follow-up (thin arrows), as well as the reduced consolidation and DWI signal in the lingula (thick arrows).
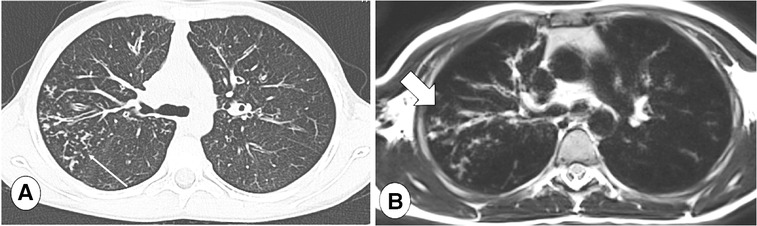
Figure 7. Axial CT (A), lung window, and axial T2-weighted MR image (B) in a CF patient with tree in bud in the posterior segment of the right upper lobe (thin arrow). Note loss in definition of lung abnormalities in the MR image, where peripheral abnormalities may be not be visible in the MRI sequence (B, thick arrow).
Inflammation has been studied through diffusion-weighted imaging (DWI) and T2-weighted scans (91–93). Ciet et al., by using a semi-quantitative DWI score on high b-value images, analyzed PCF with and without RTE and those with RTE before and after treatment, interestingly showing significant differences between the groups (91) (Figure 6). Similar results were reported by Benlala et al. when quantitatively assessing T2 signal abnormalities during RTE (93). These studies demonstrate the ability of chest MRI to track RTE changes and monitor treatment response. Contrast-enhanced MRI can also be used to evaluate the effects of therapy during RTE. Wielputz et al. reported that contrast-enhanced MRI is the most sensitive imaging biomarker of bronchial inflammation during RTE in preschool children with CF (94). Furthermore, using contrast-enhanced MRI, bronchial wall inflammation can be better seen and discriminated from mucus plugs (Figure 9) (94).
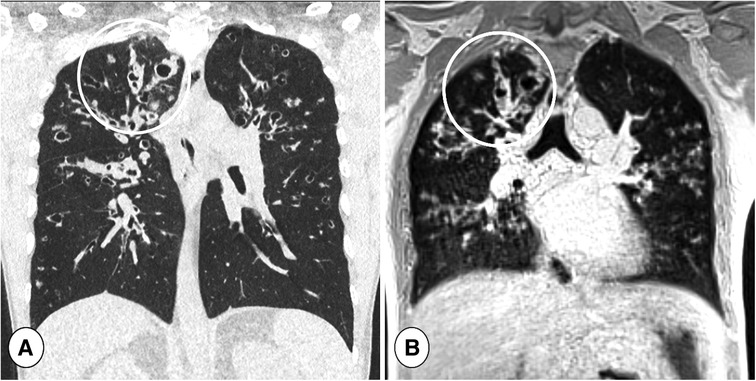
Figure 8. Coronal reformatted CT (A) and coronal ultra-short TE (UTE) MR scan (B) in a cystic fibrosis patient during respiratory tract exacerbation. Note the high definition of lung abnormalities in both CT and MRI in the right lung (circles).
Although data on inflammatory activity can be also obtained by positron emission tomography (PET)–CT, the radiation exposure is even higher than with standard CT. As an alternative, PET–MRI may offer the same information at a much lower radiation dose, but there is a lack of RTE data using this technique (1).
Functional information on ventilation are obtained using 3D spoiled-gradient echo, which may show areas of air trapping at end-expiration. In common with CT, this technique can be only used in cooperative patients as it necessitates the ability to breath hold for 5–10 s at both end-inspiration and, especially, end-expiration (95).
Moreover, Fourier decomposition (FD) MRI and derivations such as Phase-Resolved FUnctional Lung (PREFUL) MRI sequences, both ventilation and perfusion can be assessed without intravenous or gaseous inhaled contrast administration (96). PREFUL MRI is also feasible in uncooperative patients, since it is performed in free-breathing conditions (Figure 10). This technique avoids the disadvantages of hyperpolarized-gas MRI in terms of the need for dedicated hardware and the gases costs and storage. Moreover, PREFUL-MRI does not use gadolinium, the use of which is debated because of concerns regarding potential tissue deposition (97). Munisada et al. demonstrated that, during RTE, the ventilation distribution obtained with FD is correlated with hyperpolarized gas imaging and both can monitor functional improvement after therapy (96).
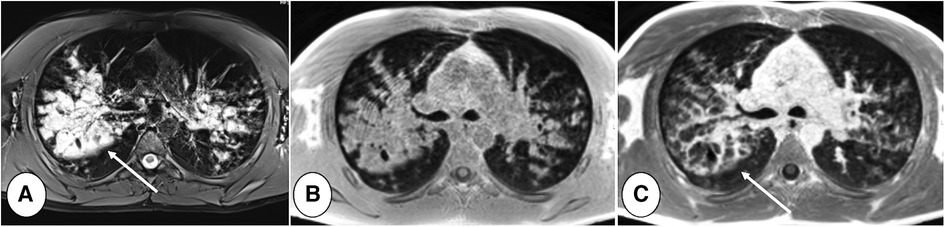
Figure 9. 16 years old boy with cystic fibrosis during respiratory tract exacerbation. T2-weighted PROPELLER image (A), T1-weighted (B) pre and post-contrast (C) gradient-echo acquisitions, performed on 1.5 T MRI system (MAGNETOM Avanto, Siemens Healthineers, Enlargen, Germany). Note that in T2-weighted scan bronchial wall cannot be identified in the large bronchiectasis with mucus plugging in the right upper lobe (A, arrow). Gadolinium injection allows identification of bronchial wall thickening and to distinguish between bronchial wall and mucus plugs (C, arrow).
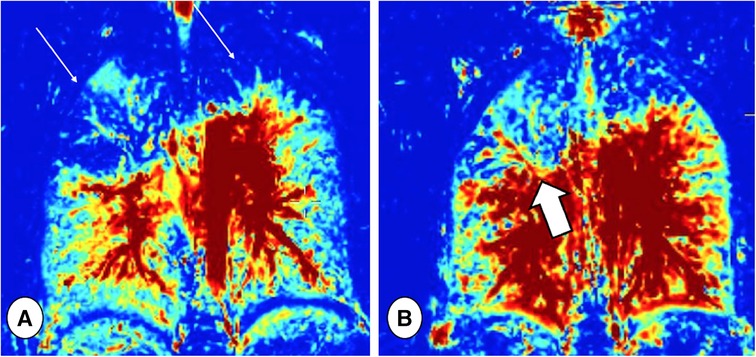
Figure 10. Ventilation maps taken before (A) and after (B) respiratory tract exacerbation treatment in a patient with CF, obtained by Fourier decomposition MRI. Note the ventilation defects in the upper lobes at baseline (thin arrows), which improved after treatment, especially in the right lung (thick arrow).
Thus, the ventilation–inflammation–perfusion–structure (VIPS) MRI protocol proposed by Tiddens et al. (98) might be a game changer in the diagnosis and management of RTE. However, l studies on chest MRI during RTE are needed though to define a standard MRI protocol for RTE.
In fact, the main factor limiting the use of chest MRI in clinical practice and for RTE monitoring is the need for sedation or general anesthesia in uncooperative patients, both of which require specialized personnel, while the general anesthesia conveys some risks for brain development (99, 100). Higher costs and lower availability of MRI compared with CT, the lack of standardized MRI protocols, and large variability in image quality among MRI vendors may represent further limitations. Finally, only few studies have provided a fully automatic assessment of parenchymal abnormalities using MRI (93), therefore full automation of validated MRI outcomes are needed to facilitate implementation in clinical practice.
Lung ultrasound
Lung ultrasound is a low-cost, bedside and safe imaging technique used both in pediatric and adult patients with pulmonary diseases. In recent years, interest in this technique has increased, making LUS a valuable point-of-care option.
Lung ultrasound is the method of choice for the diagnosis of pleural diseases, such as pleural effusion and pneumothorax. The technique offers relatively easy access to the costal and diaphragmatic pleura, being also capable of detecting small volumes of pleural effusion. Moreover, despite its use is limited to the lung surface, interest in using LUS to assess diffuse lung disease is increasing, for example, in adult respiratory distress syndrome in intensive care units, Covid-19 pneumonia and interstitial lung diseases (101–105). Unfortunately, limited data are available about the use of LUS to monitor lung status in PCF or evaluate RTE. Nonetheless, preliminary results have shown a good relationship between LUS and CT in assessing structural changes in CF (106, 107). Furthermore, Hassanzad et al. demonstrated that LUS is comparable with CT in detecting some pulmonary findings during RTE, especially consolidations and air bronchogram (8). Hence, LUS may be adopted as a complementary imaging tool, as already proposed for CF follow-up, allowing repeatable and easily available examination without any risk to the patient during RTE. However, further multicenter validation studies are needed to evaluate its use in the assessment of CF lung disease.
Imaging algorithm and recommendations for respiratory tract exacerbations
Based on this literature review and the experience of authors from CF centers, an imaging algorithm for assessing RTE in PCF is proposed (Figure 11).
Given its large availability and low dose-exposure, once the clinical diagnosis is made CR can be performed to look for gross lung abnormalities that may explain the patient's symptoms during RTE and exclude chest complications such as pneumothorax. Comparison with previous CR images is always recommended since it aids the detection of new radiographic abnormalities. In RTE with moderate or severe symptoms (e.g., increased sputum and cough, hemoptysis, dyspnea, chest pain or fever), if the CR outcome is negative or showing no improvement under therapy, further examination is warranted, especially if a recent (<2 years) CT or MRI scan is not available. The choice of the imaging technique depends upon the patient age and compliance. In uncooperative patients or children under 5 years old, a free-breathing CT may be suitable since it is usually conducted without sedation. Fast-acquisition strategies, such as high-pitch with dual source scanning, or restraining tools like vacuum mattresses, often provide sharp image quality without needing sedation or anesthesia (Figure 5). Since free-breathing acquisitions do not allow end-inspiratory and end-expiratory scans to assess air trapping, indirect signs of airway obstruction can be presumed when mosaic attenuation is observed. CT is mainly indicated to exclude major complications, such as empyema or necrotic pneumonia, and is frequently performed with iodine contrast. Computed Tomography is rarely repeated to monitor RTE treatment effects because of concerns about radiation and is thus replaced by CR. In specialized centers with long experience of chest MRI, the latter can also be used in young children under sedation or anesthesia.
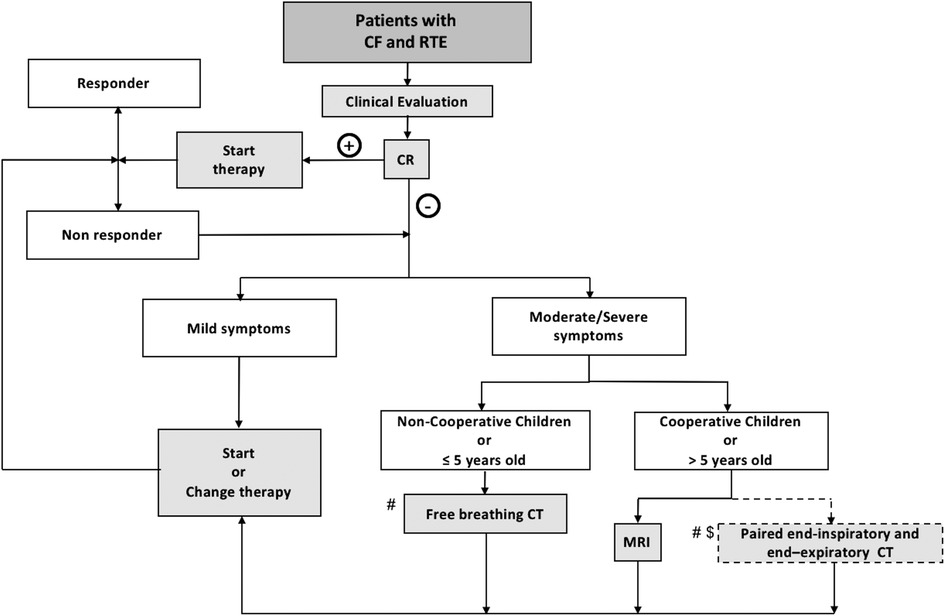
Figure 11. Workflow of the proposed imaging algorithm. CF, Cystic fibrosis; CR, Chest Radiography; CT, computed tomography; MRI, magnetic resonance imaging; RTE: respiratory tract exacerbation; # recent CT or MRI not available (<2 years); $, center with no MRI or no MRI experience; +, positive CR findings; −, negative CR findings.
In cooperative PCF, generally more than 5 years old, MRI can offer a valid alternative to assess both structural and functional information related to RTE. Chest MRI provides relevant information about ventilation and perfusion, both of which are important in the assessment of RTE. Moreover, MRI can be used repeatedly to monitor the effects of treatment without any limitation in terms of radiation exposure. In centers with no experience of chest MRI, a paired non-contrast-enhanced end-expiratory and contrast-enhanced end-inspiratory CT can be considered.
Conclusions
Physical examinations and PFTs have several limitations in respect to RTE diagnosis and monitoring. Current sputum and blood biomarkers of infection and inflammation during RTE are not sensitive nor robust enough for the reliable diagnosis of RTE. Therefore, lung imaging is still crucial for diagnosing RTE and monitoring treatment response. While chest radiography can exclude gross abnormalities or complications, it cannot sensitively diagnose new structural changes induced by RTE, especially in PCF with severe lung disease. Computed tomography allows a detailed assessment of RTE-related structural changes and represent the current standard for CF lung disease evaluation. However, short-term CT serial imaging during RTE is limited by the risk of radiation exposure, and it might be only advised with the introduction of ultralow-dose CT protocols. On the other hand, magnetic resonance imaging (MRI) remains behind CT in terms of morphologic assessment capability but, thanks also to recently developed morphological sequences (i.e., zero echo time or ultrashort echo time sequences), could become a one-stop-shop for obtaining both structural and functional information in a single examination. Current limitations of MRI include the need for sedation or anesthesia in uncooperative PCF, the lack of image-quality standardization and the necessity of developing dedicated post-processing tools.
Author contributions
NL and PC wrote the draft of the article, critically reviewed literature articles, critically read the paper and approved the final version to be published. HMJ, SB, MR, MM, CC, FM, SC, AG, FL, GFP, LS, HAWMT and GM critically read the article and approved the final version to be published. All authors contributed to the article and approved the submitted version.
Acknowledgments
We'd like to thank Silvia Maina, Editamed srl, for coordination of the project, and Michele Fusaro, for providing Figure 5.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BAL, bronchoalveolar lavage; CF, cystic fibrosis; CFTR, CF transmembrane conductance regulator; CR, chest radiography; CT, computed tomography; DWI, diffusion-weighted imaging; EBC, exhaled breath condensate; FD, Fourier decomposition; FEV1, forced expiratory volume in the first second; IL, interleukin; IV, intravenous; LCI, lung clearance index; MRI, magnetic resonance imaging; NE, neutrophil elastase; RTE, respiratory tract exacerbations; PET, positron emission tomography; PFT, pulmonary function test; PCF, people with CF; PREFUL, Phase-Resolved FUnctional Lung.
References
1. Ciet P, Bertolo S, Ros M, Casciaro R, Cipolli M, Colagrande S, et al. State-of-the-art review of lung imaging in cystic fibrosis with recommendations for pulmonologists and radiologists from the “iMAging managEment of cySTic fibROsis” (MAESTRO) consortium. Eur Respir Rev. (2022) 31(163):210173. doi: 10.1183/16000617.0173-2021
2. Goss CH. Acute pulmonary exacerbations in cystic fibrosis. Semin Respir Crit Care Med. (2019) 40(6):792–803. doi: 10.1055/s-0039-1697975
3. Stanford GE, Dave K, Simmonds NJ. Pulmonary exacerbations in adults with cystic fibrosis. Chest. (2021) 159(1):93–102. doi: 10.1016/j.chest.2020.09.084
4. Ishak A, Stick SM, Turkovic L, Ranganathan SC, King L, Jarrison J, et al. BAL Inflammatory markers can predict pulmonary exacerbations in children with cystic fibrosis. Chest. (2020) 158(6):2314–22. doi: 10.1016/j.chest.2020.06.044
5. West NE, Beckett VV, Jain R, Sanders DB, Nick JA, Heltshe SL, et al. Standardized treatment of pulmonary exacerbations (STOP) study: physician treatment practices and outcomes for individuals with cystic fibrosis with pulmonary exacerbations. J Cyst Fibros. (2017) 16(5):600–6. doi: 10.1016/j.jcf.2017.04.003
6. Greene KE, Takasugi JE, Godwin JD, Richardson ML, Burke W, Aitken ML. Radiographic changes in acute exacerbations of cystic fibrosis in adults: a pilot study. Am J Roentgenol. (1994) 163(3):557–62. doi: 10.2214/ajr.163.3.8079843
7. Ciet P, Booij R, Dijkshoorn M, van Straten M, Tiddens HAWM. Chest radiography and computed tomography imaging in cystic fibrosis: current challenges and new perspectives [published online ahead of print, 2022 Oct 29]. Pediatr Radiol. (2022) :10.1007/s00247-022-05522-4. doi: 10.1007/s00247-022-05522-4
8. Hassanzad M, Kiani A, Abedini A, Ghaffaripour H, Emami H, Alizadeh N, et al. Lung ultrasound for the diagnosis of cystic fibrosis pulmonary exacerbation. BMC Pulm Med. (2021) 21(1):353. doi: 10.1186/s12890-021-01728-8
9. Flume PA, Mogayzel PJ Jr, Robinson KA, Goss CH, Rosenblatt RL, Kuhn RJ, et al. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med. (2009) 180(9):802–8. doi: 10.1164/rccm.200812-1845PP
10. Orenstein DM, Pattishall EN, Nixon PA, Ross EA, Kaplan RM. Quality of well-being before and after antibiotic treatment of pulmonary exacerbation in patients with cystic fibrosis. Chest. (1990) 98(5):1081–4. doi: 10.1378/chest.98.5.1081
11. Britto MT, Kotagal UR, Hornung RW, Atherton HD, Tsevat J, Wilmott RW. Impact of recent pulmonary exacerbations on quality of life in patients with cystic fibrosis. Chest. (2002) 121(1):64–72. doi: 10.1378/chest.121.1.64
12. Ouyang L, Grosse SD, Amendah DD, Schechter MS. Healthcare expenditures for privately insured people with cystic fibrosis. Pediatr Pulmonol. (2009) 44(10):989–96. doi: 10.1002/ppul.21090
13. Liou TG, Adler FR, Fitzsimmons SC, Cahill BC, Hibbs JR, Marshall BC. Predictive 5-year survivorship model of cystic fibrosis. Am J Epidemiol. (2001) 153(4):345–52. doi: 10.1093/aje/153.4.345
14. Mayer-Hamblett N, Rosenfeld M, Emerson J, Goss CH, Aitken ML. Developing cystic fibrosis lung transplant referral criteria using predictors of 2-year mortality. Am J Respir Crit Care Med. (2002) 166(12 Pt 1):1550–5. doi: 10.1164/rccm.200202-087OC
15. VanDevanter DR, Konstan MW. Outcome measures for clinical trials assessing treatment of cystic fibrosis lung disease. Clin Investig (Lond). (2012) 2(2):163–75. doi: 10.4155/cli.11.174
16. Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. N Engl J Med. (1994) 331(10):637–42. doi: 10.1056/NEJM199409083311003
17. Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. N Engl J Med. (1999) 340(1):23–30. doi: 10.1056/NEJM199901073400104
18. Rosenfeld M, Emerson J, Williams-Warren J, Pepe M, Smith A, Montgomery AB, et al. Defining a pulmonary exacerbation in cystic fibrosis. J Pediatr. (2001) 139(3):359–65. doi: 10.1067/mpd.2001.117288
19. Goss CH, Burns JL. Exacerbations in cystic fibrosis–1: epidemiology and pathogenesis. Thorax. (2007) 62(4):360–7. doi: 10.1136/thx.2006.060889
20. Wagener JS, Rasouliyan L, VanDevanter DR, Pasta DJ, Regelmann WE, Morgan WJ, et al. Oral, inhaled, and intravenous antibiotic choice for treating pulmonary exacerbations in cystic fibrosis. Pediatr Pulmonol. (2013) 48(7):666–73. doi: 10.1002/ppul.22652
21. Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. (2011) 365(18):1663–72. doi: 10.1056/NEJMoa1105185
22. Elkins MR, Robinson M, Rose BR, Harbour C, Moriarty CP, Marks GB, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. (2006) 354(3):229–40. doi: 10.1056/NEJMoa043900
23. Bilton D, Canny G, Conway S, Dumcius S, Hjelte L, Proesmans M, et al. Pulmonary exacerbation: towards a definition for use in clinical trials. Report from the EuroCareCF Working Group on outcome parameters in clinical trials. J Cyst Fibros. (2011) 10(Suppl 2):S79–S81. doi: 10.1016/S1569-1993(11)60012-X
24. Goss CH, Heltshe SL, West NE, Skalland M, Sanders DB, Jain R, et al. A randomized clinical trial of antimicrobial duration for cystic fibrosis pulmonary exacerbation treatment. Am J Respir Crit Care Med. (2021) 204(11):1295–305. doi: 10.1164/rccm.202102-0461OC
25. Gramegna A, Contarini M, Aliberti S, Casciaro R, Blasi F, Castellani C. From ivacaftor to triple combination: a systematic review of efficacy and safety of CFTR modulators in people with cystic fibrosis. Int J Mol Sci. (2020) 21(16):5882. doi: 10.3390/ijms21165882
26. Spiteri MA, Cook DG, Clarke SW. Reliability of eliciting physical signs in examination of the chest. Lancet. (1988) 1(8590):873–5. doi: 10.1016/s0140-6736(88)91613-3
27. Bouzek DC, Ren CL, Thompson M, Slaven JE, Sanders DB. Evaluating FEV1 decline in diagnosis and management of pulmonary exacerbations in children with cystic fibrosis. Pediatr Pulmonol. (2022) 57(7):1709–16. doi: 10.1002/ppul.25925
28. Patel S, Thompson MD, Slaven JE, Sanders DB, Ren CL. Reduction of pulmonary exacerbations in young children with cystic fibrosis during the COVID-19 pandemic. Pediatr Pulmonol. (2021) 56(5):1271–3. doi: 10.1002/ppul.25250
29. Perrem L, Stanojevic S, Shaw M, Pornillos M, Guido J, Sanders DB, et al. Comparative analysis of respiratory symptom scores to detect acute respiratory events in children with cystic fibrosis [published online ahead of print, 2022 Jun 23]. J Cyst Fibros. (2022):S1569-1993(22)00594-X. doi: 10.1016/j.jcf.2022.06.007
30. Mogayzel PJ Jr, Naureckas ET, Robinson KA, Mueller G, Hadjiliadis D, Hoag JB, et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med. (2013) 187(7):680–9. doi: 10.1164/rccm.201207-1160oe
31. Que C, Cullinan P, Geddes D. Improving rate of decline of FEV1 in young adults with cystic fibrosis. Thorax. (2006) 61(2):155–7. doi: 10.1136/thx.2005.043372
32. Waters V, Stanojevic S, Atenafu EG, Lu A, Yau Y, Tullis E, et al. Effect of pulmonary exacerbations on long-term lung function decline in cystic fibrosis. Eur Respir J. (2012) 40(1):61–6. doi: 10.1183/09031936.00159111
33. Horsley A, Siddiqui S. Putting lung function and physiology into perspective: cystic fibrosis in adults. Respirology. (2015) 20(1):33–45. doi: 10.1111/resp.12382
34. Aquino CSB, Rodrigues JC, Silva-Filho LVRFD. Routine spirometry in cystic fibrosis patients: impact on pulmonary exacerbation diagnosis and FEV1 decline. J Bras Pneumol. (2022) 48(3):e20210237. doi: 10.36416/1806-3756/e20210237
35. Morgan WJ, VanDevanter DR, Pasta DJ, Foreman AJ, Wagener JS, Konstan MW. Forced expiratory volume in 1 s variability helps identify patients with cystic fibrosis at risk of greater loss of lung function [published correction appears in J Pediatr. (2018) 197:322]. J Pediatr. (2016) 169:116–21.e2. doi: 10.1016/j.jpeds.2015.08.042
36. Redding GJ, Restuccia R, Cotton EK, Brooks JG. Serial changes in pulmonary functions in children hospitalized with cystic fibrosis. Am Rev Respir Dis. (1982) 126(1):31–6. doi: 10.1164/arrd.1982.126.1.31
37. Rosenberg SM, Schramm CM. Predictive value of pulmonary function testing during pulmonary exacerbations in cystic fibrosis. Pediatr Pulmonol. (1993) 16(4):227–35. doi: 10.1002/ppul.1950160404
38. Collaco JM, Green DM, Cutting GR, Naughton KM, Mogayzel PJ Jr. Location and duration of treatment of cystic fibrosis respiratory exacerbations do not affect outcomes. Am J Respir Crit Care Med. (2010) 182(9):1137–43. doi: 10.1164/rccm.201001-0057OC
39. Sanders DB, Bittner RC, Rosenfeld M, Redding GJ, Goss CH. Pulmonary exacerbations are associated with subsequent FEV1 decline in both adults and children with cystic fibrosis. Pediatr Pulmonol. (2011) 46(4):393–400. doi: 10.1002/ppul.21374
40. Hoppe JE, Wagner BD, Sagel SD, Accurso FJ, Zemanick ET. Pulmonary exacerbations and clinical outcomes in a longitudinal cohort of infants and preschool children with cystic fibrosis. BMC Pulm Med. (2017) 17(1):188. doi: 10.1186/s12890-017-0546-8
41. de Jong PA, Nakano Y, Lequin MH, Mayo JR, Woods R, Paré PD, et al. Progressive damage on high resolution computed tomography despite stable lung function in cystic fibrosis. Eur Respir J. (2004) 23(1):93–7. doi: 10.1183/09031936.03.00006603
42. Gustafsson PM, De Jong PA, Tiddens HA, Lindblad A. Multiple-breath inert gas washout and spirometry versus structural lung disease in cystic fibrosis. Thorax. (2008) 63(2):129–34. doi: 10.1136/thx.2007.077784
43. Singer F, Kieninger E, Abbas C, Yammine S, Fuchs O, Proietti E, et al. Practicability of nitrogen multiple-breath washout measurements in a pediatric cystic fibrosis outpatient setting. Pediatr Pulmonol. (2013) 48(8):739–46. doi: 10.1002/ppul.22651
44. Parisi GF, Pignatone E, Papale M, Mulé E, Manti S, Leonardi S. Lung clearance index: a new measure of ventilation inhomogeneity in childhood respiratory diseases. Curr Respir Med Rev. (2021) 17(4):209–25. doi: 10.2174/1573398X17666211201092525
45. Horsley AR, Gustafsson PM, Macleod KA, Sanders C, Greening AP, Porteous DJ, et al. Lung clearance index is a sensitive, repeatable and practical measure of airways disease in adults with cystic fibrosis. Thorax. (2008) 63(2):135–40. doi: 10.1136/thx.2007.082628
46. Stanojevic S, Davis SD, Perrem L, Shaw M, Retsch-Bogart G, Davis M, et al. Determinants of lung disease progression measured by lung clearance index in children with cystic fibrosis. Eur Respir J. (2021) 58(1):2003380. doi: 10.1183/13993003.03380-2020
47. Perrem L, Stanojevic S, Shaw M, Jensen R, McDonald N, Isaac SM, et al. Lung clearance index to track acute respiratory events in school-age children with cystic fibrosis. Am J Respir Crit Care Med. (2021) 203(8):977–86. doi: 10.1164/rccm.202006-2433OC
48. Walicka-Serzysko K, Postek M, Milczewska J, Sands D. Lung clearance index in children with cystic fibrosis during pulmonary exacerbation. J Clin Med. (2021) 10(21):4884. doi: 10.3390/jcm10214884
49. Rayment JH, Stanojevic S, Davis SD, Retsch-Bogart G, Ratjen F. Lung clearance index to monitor treatment response in pulmonary exacerbations in preschool children with cystic fibrosis. Thorax. (2018) 73(5):451–8. doi: 10.1136/thoraxjnl-2017-210979
50. Eyns H, Piérard D, De Wachter E, Eeckhout L, Vaes P, Malfroot A. Respiratory bacterial culture sampling in expectorating and non-expectorating patients with cystic fibrosis. Front Pediatr. (2018) 6:403. doi: 10.3389/fped.2018.00403
51. D’Sylva P, Caudri D, Shaw N, Turkovic L, Douglas T, Bew J, et al. Induced sputum to detect lung pathogens in young children with cystic fibrosis. Pediatr Pulmonol. (2017) 52(2):182–9. doi: 10.1002/ppul.23636
52. Kos R, Brinkman P, Neerincx AH, Paff T, Gerritsen MG, Lammerse A, et al. Targeted exhaled breath analysis for detection of Pseudomonas aeruginosa in cystic fibrosis patients. J Cyst Fibros. (2022) 21(1):e28–e34. doi: 10.1016/j.jcf.2021.04.015
53. Sly PD, Gangell CL, Chen L, Ware RS, Ranganathan S, Mott LS, et al. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med. (2013) 368(21):1963–70. doi: 10.1056/NEJMoa1301725
54. Caudri D, Turkovic L, de Klerk NH, Rosenow T, Murray CP, Steyerberg EW, et al. A screening tool to identify risk for bronchiectasis progression in children with cystic fibrosis. Pediatr Pulmonol. (2022) 57(1):122–31. doi: 10.1002/ppul.25712
55. Parisi GF, Papale M, Tardino L, Nenna R, Midulla F, Leonardi S. Biomarkers in pediatric lung diseases including cystic fibrosis. Curr Respir Med Rev. (2019) 15(3):163–73. doi: 10.2174/1573398X15666190521112824
56. Ordoñez CL, Henig NR, Mayer-Hamblett N, Accurso FJ, Burns JL, Chmiel JF, et al. Inflammatory and microbiologic markers in induced sputum after intravenous antibiotics in cystic fibrosis. Am J Respir Crit Care Med. (2003) 168(12):1471–5. doi: 10.1164/rccm.200306-731OC
57. Colombo C, Costantini D, Rocchi A, Cariani L, Garlaschi ML, Tirelli S, et al. Cytokine levels in sputum of cystic fibrosis patients before and after antibiotic therapy. Pediatr Pulmonol. (2005) 40(1):15–21. doi: 10.1002/ppul.20237
58. Mayer-Hamblett N, Aitken ML, Accurso FJ, Kronmal RA, Konstan MW, Burns JL, et al. Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am J Respir Crit Care Med. (2007) 175(8):822–8. doi: 10.1164/rccm.200609-1354OC
59. Lepissier A, Addy C, Hayes K, Noel S, Bui S, Burgel PR, et al. Inflammation biomarkers in sputum for clinical trials in cystic fibrosis: current understanding and gaps in knowledge. J Cyst Fibros. (2022) 21(4):691–706. doi: 10.1016/j.jcf.2021.10.009
60. Montuschi P, Kharitonov SA, Ciabattoni G, Corradi M, van Rensen L, Geddes DM, et al. Exhaled 8-isoprostane as a new non-invasive biomarker of oxidative stress in cystic fibrosis. Thorax. (2000) 55(3):205–9. doi: 10.1136/thorax.55.3.205
61. Lucidi V, Ciabattoni G, Bella S, Barnes PJ, Montuschi P. Exhaled 8-isoprostane and prostaglandin E2 in patients with stable and unstable cystic fibrosis. Free Radic Biol Med. (2008) 45(6):913–9. doi: 10.1016/j.freeradbiomed.2008.06.026
62. Robroeks CM, Rosias PP, van Vliet D, Jöbsis Q, Yntema JL, Brackel HJL, et al. Biomarkers in exhaled breath condensate indicate presence and severity of cystic fibrosis in children. Pediatr Allergy Immunol. (2008) 19(7):652–9. doi: 10.1111/j.1399-3038.2007.00693.x
63. Toprak Kanık E, Yilmaz O, Ozdogru E, Alper H, Ulman C, Kanik A, et al. Relevance between clinical status and exhaled molecules related to neutrophilic inflammation in pediatric cystic fibrosis. J Breath Res. (2020) 14(4):046007. doi: 10.1088/1752-7163/ab670d
64. Nguyen AV, Haas D, Bouchard M, Quon BS. Metabolomic biomarkers to predict and diagnose cystic fibrosis pulmonary exacerbations: a systematic review. Front Pediatr. (2022) 10:896439. doi: 10.3389/fped.2022.896439
65. Shoki AH, Mayer-Hamblett N, Wilcox PG, Sin DD, Quon BS. Systematic review of blood biomarkers in cystic fibrosis pulmonary exacerbations. Chest. (2013) 144(5):1659–70. doi: 10.1378/chest.13-0693
66. Girón-Moreno RM, Justicia JL, Yamamoto S, Valenzuela C, Cisneros C, Gómez-Punter RM, et al. Role of C-reactive protein as a biomarker for prediction of the severity of pulmonary exacerbations in patients with cystic fibrosis. BMC Pulm Med. (2014) 14:150. doi: 10.1186/1471-2466-14-150
67. Gray RD, Imrie M, Boyd AC, Porteous D, Innes JA, Greening AP. Sputum and serum calprotectin are useful biomarkers during CF exacerbation. J Cyst Fibros. (2010) 9(3):193–8. doi: 10.1016/j.jcf.2010.01.005
68. Reid PA, McAllister DA, Boyd AC, Innes JA, Porteous D, Greening AP, et al. Measurement of serum calprotectin in stable patients predicts exacerbation and lung function decline in cystic fibrosis. Am J Respir Crit Care Med. (2015) 191(2):233–6. doi: 10.1164/rccm.201407-1365LE
69. Sagel SD, Thompson V, Chmiel JF, Montgomery GS, Nasr SZ, Perkett E, et al. Effect of treatment of cystic fibrosis pulmonary exacerbations on systemic inflammation. Ann Am Thorac Soc. (2015) 12(5):708–17. doi: 10.1513/AnnalsATS.201410-493OC
70. Twigg MS, Brockbank S, Lowry P, FitzGerald SP, Taggart C, Weldon S. The role of serine proteases and antiproteases in the cystic fibrosis lung. Mediators Inflamm. (2015) 2015:293053. doi: 10.1155/2015/293053
71. Wu CC, Khorashadi L, Abbott GF, Gilman MD. Common blind spots on chest CT: where are they all hiding? Part 1–airways, lungs, and pleura. Am J Roentgenol. (2013) 201(4):W533–8. doi: 10.2214/AJR.12.9354
72. FitzMaurice TS, McCann C, Nazareth DS, McNamara PS, Walshaw MJ. Use of dynamic chest radiography to assess treatment of pulmonary exacerbations in cystic fibrosis. Radiology. (2022) 303(3):675–81. doi: 10.1148/radiol.212641
73. Kuo W, Soffers T, Andrinopoulou ER, Rosenow T, Ranganathan S, Turkovic L, et al. Quantitative assessment of airway dimensions in young children with cystic fibrosis lung disease using chest computed tomography. Pediatr Pulmonol. (2017) 52(11):1414–23. doi: 10.1002/ppul.23787
74. Kuo W, de Bruijne M, Petersen J, Nasserinejad K, Ozturk H, Chen Y, et al. Diagnosis of bronchiectasis and airway wall thickening in children with cystic fibrosis: objective airway-artery quantification. Eur Radiol. (2017) 27(11):4680–9. doi: 10.1007/s00330-017-4819-7
75. Davis SD, Fordham LA, Brody AS, Noah TL, Retsch-Bogart GZ, Qaqish BF, et al. Computed tomography reflects lower airway inflammation and tracks changes in early cystic fibrosis. Am J Respir Crit Care Med. (2007) 175(9):943–50. doi: 10.1164/rccm.200603-343OC
76. Byrnes CA, Vidmar S, Cheney JL, Carlin JB, Armstrong DS, Cooper PJ, et al. Prospective evaluation of respiratory exacerbations in children with cystic fibrosis from newborn screening to 5 years of age. Thorax. (2013) 68(7):643–51. doi: 10.1136/thoraxjnl-2012-202342
77. Diab-Cáceres L, Girón-Moreno RM, García-Castillo E, Pastor Sanz MT, Olveira C, García-Clemente MM, et al. Predictive value of the modified Bhalla score for assessment of pulmonary exacerbations in adults with cystic fibrosis. Eur Radiol. (2021) 31(1):112–20. doi: 10.1007/s00330-020-07095-y
78. Shah RM, Sexauer W, Ostrum BJ, Fiel SB, Friedman AC. High-resolution CT in the acute exacerbation of cystic fibrosis: evaluation of acute findings, reversibility of those findings, and clinical correlation. Am J Roentgenol. (1997) 169(2):375–80. doi: 10.2214/ajr.169.2.9242738
79. Kuo W, Ciet P, Tiddens HA, Zhang W, Guillerman RP, van Straten M. Monitoring cystic fibrosis lung disease by computed tomography. Radiation risk in perspective. Am J Respir Crit Care Med. (2014) 189(11):1328–36. doi: 10.1164/rccm.201311-2099CI
80. Sheahan KP, Glynn D, Joyce S, Maher MM, Boland F, O’Connor OJ. Best practices: imaging strategies for reduced-dose chest CT in the management of cystic fibrosis-related lung disease. Am J Roentgenol. (2021) 217(2):304–13. doi: 10.2214/AJR.19.22694
81. Rapp JB, Ho-Fung VM, Ramirez KI, White AM, Otero HJ, Biko DM. Dual-source computed tomography protocols for the pediatric chest—scan optimization techniques [published online ahead of print, 2022 Aug 11]. Pediatr Radiol. (2022):1–12. doi: 10.1007/s00247-022-05468-7
82. Rosenow T, Oudraad MC, Murray CP, Turkovic L, Kuo W, de Bruijne M, et al. PRAGMA-CF. A quantitative structural lung disease computed tomography outcome in young children with cystic fibrosis. Am J Respir Crit Care Med. (2015) 191(10):1158–65. doi: 10.1164/rccm.201501-0061OC
83. Dournes G, Hall CS, Willmering MM, Brody AS, Macey J, Bui S, et al. Artificial intelligence in computed tomography for quantifying lung changes in the era of CFTR modulators. Eur Respir J. (2022) 59(3):2100844. doi: 10.1183/13993003.00844-2021
84. Dournes G, Menut F, Macey J, Fayon M, Chateil JF, Salel M, et al. Lung morphology assessment of cystic fibrosis using MRI with ultra-short echo time at submillimeter spatial resolution. Eur Radiol. (2016) 26(11):3811–20. doi: 10.1007/s00330-016-4218-5
85. Ciet P, Serra G, Bertolo S, Spronk S, Ros M, Fraioli F, et al. Assessment of CF lung disease using motion corrected PROPELLER MRI: a comparison with CT. Eur Radiol. (2016) 26(3):780–7. doi: 10.1007/s00330-015-3850-9
86. Landini N, Orlandi M, Occhipinti M, Nardi C, Tofani L, Bellando-Randone S, et al. Ultrashort echo-time magnetic resonance imaging sequence in the assessment of systemic sclerosis-interstitial lung disease. [published online ahead of print, 2022 Feb 04]. J Thorac Imaging. (2022) :10.1097/RTI.0000000000000637. doi: 10.1097/RTI.0000000000000637
87. Dournes G, Yazbek J, Benhassen W, Benlala I, Blanchard E, Truchetet ME, et al. 3D Ultrashort echo time MRI of the lung using stack-of-spirals and spherical k -space coverages: evaluation in healthy volunteers and parenchymal diseases. J Magn Reson Imaging. (2018) 48(6):1489–97. doi: 10.1002/jmri.26212
88. Dournes G, Grodzki D, Macey J, Girodet PO, Fayon M, Chateil JF, et al. Quiet submillimeter MR imaging of the lung is feasible with a PETRA sequence at 1.5 T. Radiology. (2015) 276(1):258–65. doi: 10.1148/radiol.15141655
89. Campbell-Washburn AE, Malayeri AA, Jones EC, Moss J, Fennelly KP, Olivier KN, et al. T2-weighted lung imaging using a 0.55-T MRI system. Radiol Cardiothorac Imaging. (2021) 3(3):e200611. doi: 10.1148/ryct.2021200611
90. Azour L, Condos R, Keerthivasan MB, Bruno M, Pandit Sood T, Landini N, et al. Low-field 0.55 T MRI for assessment of pulmonary groundglass and fibrosis-like opacities: inter-reader and inter-modality concordance [published online ahead of print, 2022 Sep 8]. Eur J Radiol. (2022) 156:110515. doi: 10.1016/j.ejrad.2022.110515
91. Ciet P, Bertolo S, Ros M, Andrinopoulou ER, Tavano V, Lucca F, et al. Detection and monitoring of lung inflammation in cystic fibrosis during respiratory tract exacerbation using diffusion-weighted magnetic resonance imaging. Eur Respir J. (2017) 50(1):1601437. doi: 10.1183/13993003.01437-2016
92. Ciet P, Serra G, Andrinopoulou ER, Bertolo S, Ros M, Catalano C, et al. Diffusion weighted imaging in cystic fibrosis disease: beyond morphological imaging. Eur Radiol. (2016) 26(11):3830–9. doi: 10.1007/s00330-016-4248-z
93. Benlala I, Hocke F, Macey J, Bui S, Berger P, Laurent F, et al. Quantification of MRI T2-weighted high signal volume in cystic fibrosis: a pilot study. Radiology. (2020) 294(1):186–96. doi: 10.1148/radiol.2019190797
94. Wielpütz MO, Puderbach M, Kopp-Schneider A, Stahl M, Fritzsching E, Sommerburg O, et al. Magnetic resonance imaging detects changes in structure and perfusion, and response to therapy in early cystic fibrosis lung disease. Am J Respir Crit Care Med. (2014) 189(8):956–65. doi: 10.1164/rccm.201309-1659OC
95. Ciet P, Tiddens HA, Wielopolski PA, Wild JM, Lee EY, Morana G, et al. Magnetic resonance imaging in children: common problems and possible solutions for lung and airways imaging. Pediatr Radiol. (2015) 45(13):1901–15. doi: 10.1007/s00247-015-3420-y
96. Munidasa S, Couch MJ, Rayment JH, Voskrebenzev A, Seethamraju R, Vogel-Claussen J, et al. Free-breathing MRI for monitoring ventilation changes following antibiotic treatment of pulmonary exacerbations in paediatric cystic fibrosis. Eur Respir J. (2021) 57(4):2003104. doi: 10.1183/13993003.03104-2020
97. Cheeney SHE, Maloney E, Iyer RS. Safety considerations related to intravenous contrast agents in pediatric imaging [published online ahead of print, 2022 Aug 9]. Pediatr Radiol. (2022) :10.1007/s00247-022-05470-z. doi: 10.1007/s00247-022-05470-z
98. Tiddens HAWM, Stick SM, Wild JM, Ciet P, Parker GJM, Koch A, et al. Respiratory tract exacerbations revisited: ventilation, inflammation, perfusion, and structure (VIPS) monitoring to redefine treatment: respiratory tract exacerbations revisited. Pediatr Pulmonol. (2015) 50(S40):S57–S65. doi: 10.1002/ppul.23266
99. Harrington SG, Jaimes C, Weagle KM, Greer MC, Gee MS. Strategies to perform magnetic resonance imaging in infants and young children without sedation. Pediatr Radiol. (2022) 52(2):374–81. doi: 10.1007/s00247-021-05062-3
100. Barton K, Nickerson JP, Higgins T, Williams RK. Pediatric anesthesia and neurotoxicity: what the radiologist needs to know. Pediatr Radiol. (2018) 48(1):31–6. doi: 10.1007/s00247-017-3871-4
101. Stefanidis K, Moser J, Vlahos I. Imaging of diffuse lung disease in the intensive care unit patient. Radiol Clin North Am. (2020) 58(1):119–31. doi: 10.1016/j.rcl.2019.08.005
102. Soldati G, Demi M. What is COVID 19 teaching us about pulmonary ultrasound? Diagnostics (Basel). (2022) 12(4):838. doi: 10.3390/diagnostics12040838
103. Landini N, Orlandi M, Fusaro M, Ciet P, Nardi C, Bertolo S, et al. The role of imaging in COVID-19 pneumonia diagnosis and management: main positions of the experts, key imaging features and open answers. J Cardiovasc Echogr. (2020) 30(Suppl 2):S25–S30. doi: 10.4103/jcecho.jcecho_59_2
104. Hughes M, Bruni C, Cuomo G, Delle Sedie A, Gargani L, Gutierrez M, et al. The role of ultrasound in systemic sclerosis: on the cutting edge to foster clinical and research advancement. J Scleroderma Relat Disord. (2021) 6(2):123–32. doi: 10.1177/2397198320970394
105. Bruni C, Mattolini L, Tofani L, Gargani L, Landini N, Roma N, et al. Lung ultrasound B-lines in the evaluation of the extent of interstitial lung disease in systemic sclerosis. Diagnostics (Basel). (2022) 12(7):1696. doi: 10.3390/diagnostics12071696
106. Peixoto AO, Al Marson F, Dertkigil SS, Dertkigil RP, Souza TH, Fraga AMA, et al. The use of ultrasound as a tool to evaluate pulmonary disease in cystic fibrosis. Respir Care. (2020) 65:293–303. doi: 10.4187/respcare.07038
Keywords: cystic fibrosis, respiratory tract exacerbations, inflammation, imaging, lung function, computed tomography, magnetic resonance imaging, chest radiography
Citation: Landini N, Ciet P, Janssens HM, Bertolo S, Ros M, Mattone M, Catalano C, Majo F, Costa S, Gramegna A, Lucca F, Parisi GF, Saba L, Tiddens HAWM and Morana G (2023) Management of respiratory tract exacerbations in people with cystic fibrosis: Focus on imaging. Front. Pediatr. 10:1084313. doi: 10.3389/fped.2022.1084313
Received: 30 October 2022; Accepted: 28 December 2022;
Published: 6 February 2023.
Edited by:
Laurence Delhaes, Université de Bordeaux, FranceReviewed by:
Arturo Solis-Moya, Dr. Carlos Sáenz Herrera National Children’s Hospital, Costa RicaOliver Weinheimer, Heidelberg University Hospital, Germany
© 2023 Landini, Ciet, Janssen, Bertolo, Ros, Mattone, Catalano, Majo, Costa, Gramegna, Lucca, Parisi, Saba, Tiddens and Morana. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pierluigi Ciet cC5jaWV0QGVyYXNtdXNtYy5ubA==
Specialty Section: This article was submitted to Pediatric Pulmonology, a section of the journal Frontiers in Pediatrics