 Aleksandra Krstic1,2
Aleksandra Krstic1,2 Fatemah Rezayee1,2
Fatemah Rezayee1,2 Leonie Saft3,4
Leonie Saft3,4 Anna Hammarsjö1,2Petter Svenberg5,6
Anna Hammarsjö1,2Petter Svenberg5,6 Gisela Barbany1,2*
Gisela Barbany1,2*
- 1Clinical Genetics, Karolinska University Hospital, Stockholm, Sweden
- 2Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden
- 3Clinical Pathology and Cancer Diagnostics, Karolinska University Hospital, Stockholm, Sweden
- 4Department of Clinical Pathology and Oncology, Karolinska Institute, Stockholm, Sweden
- 5Pediatric Oncology, Karolinska University Hospital, Stockholm, Sweden
- 6Department of Women’s and Children’s Health, Karolinska Institutet, Stockholm, Sweden
In the present report, we applied whole genome sequencing (WGS) to genetically characterize a case of pediatric T-cell acute lymphoblastic leukemia (ALL) refractory to standard therapy. WGS identified a novel JAK2 fusion, with CCDC88C as a partner. CCDC88C encodes a protein part of the Wnt signaling pathway and has previously been described in hematological malignancies as fusion partner to FLT3 and PDGFRB. The novel CCDC88C::JAK2 fusion gene results in a fusion transcript, predicted to produce a hybrid protein, which retains the kinase domain of JAK2 and is expected to respond to JAK2 inhibitors. This report illustrates the potential of WGS in the diagnostic setting of ALL.
Introduction
Current treatment protocols for pediatric acute lymphoblastic leukemia (ALL) use information regarding genomic aberrations in the leukemic blasts together with clinical features at presentation, and initial therapy response is used to assign patients to different risk categories and treatment intensities (1). Survival of pediatric acute ALL in risk-adapted chemotherapy trials exceeds 90% (2); however, further improvements in outcome are not likely to be achieved with conventional chemotherapy but will require alternative treatment approaches such as immunotherapy or targeted therapy.
High throughput sequencing technologies have shown enormous potential in the genetic characterization of hematological malignancies, particularly in pediatric ALL (3), and are paving the way to personalized treatment strategies, although implementation in clinical practice is still meager (4). Technologies that interrogate the whole genome with high resolution are increasingly being adopted in the diagnostic workout of pediatric ALL (5). Here, we report a novel JAK2 fusion in a pediatric case of therapy-refractory T-ALL that was detected using paired-end whole genome sequencing (WGS) in the diagnostic setting.
Case presentation
A 7-year-old boy presenting with a leukocyte count 800 × 109/L and peripheral lymphadenopathy was diagnosed with precursor T-ALL with extensive involvement of the bone marrow (BM) and peripheral blood. Multiparameter flow cytometry showed an immature T-lymphoblast population (95%) of cortical phenotype (EGIL III; CD45dim/CD7+/cytCD3+/Tdt+//CD4/CD8dim+/CD10−/CD1ahetero/CD56/16−/CD99++/CD5dim+/ CD2+/CD48+/CD38+//TCRa/b−/TCRg/d−/HLA−DR− {Bene, 1995 #41}). The blasts were negative for myeloid markers (cytMPO and CD33) and for B-cell markers (CD19 and cytCD79a). According to the standard of care (SoC) genomic characterization, the patient had a normal karyotype and none of the targeted fluorescence in situ hybridization (FISH) analysis was positive, i.e., no KMT2A- or BCR::ABL rearrangements and no ABL-class rearrangement were detected. A biallelic CDKN2A/B deletion was the only clonal aberration detected. The patient responded poorly to standard four-drug induction (dexamethasone, pegylated-asparaginase, daunorubicin, and vincristine) and was switched to consolidation at day 19 (6-mercaptopurine, cytarabine, and cyclophosphamide). Two weeks later, he still had 80% blasts in the bone marrow and was considered refractory and treatment with nelarabine initiated. Following two rounds of nelarabine and one additional high-risk chemotherapy block, remission was achieved, and he subsequently underwent hematopoietic stem cell transplantation (HSCT) from his haploidentical mother. The conditioning regimen consisted of ATG (grafalon), fludarabine, and thiotepa together with total body irradiation (TBI) (12 Gy in four fractions) and rituximab after which he received an alpha/beta T-cell-depleted peripheral stem cell graft. Initially, mycophenolate mofetil was switched to cyclosporin together with prednisolone after 45 days due to acute gut and skin graft versus host disease (GVHD). He is currently 8 months post HSCT, with no sign of disease but still on a low dose prednisolone.
At the time of initial failure, the genetic laboratory was engaged to screen for potential targets for experimental therapy. DNA isolated from the bone marrow sample taken at diagnosis was sequenced using a PCR-free, paired-end WGS protocol with a 30× coverage on an Illumina HiSeq X platform at Clinical Genomics (SciLifeLab, Stockholm) and annotated to the Human GRCh37 (hg19) build. Variants were identified using the MIP (6) validated for routine diagnostics and visualized in the SCOUT interface (7). MIP performs structural variant (SV) detection using Delly (8), TIDDIT V2.0 (9), as well as Manta (10) and single nucleotide variant (SNV) detection using GATK HaplotypeCaller (11). Subsequently, filtering was performed using the list of targetable genes described in the INFORM study (12). This analysis resulted in 260 SNVs and 18 SVs. Further filtering of the SNVs based on gnomAD or local observations as well as annotations in ClinVar narrowed the list to 11 SNVs that were manually inspected and dismissed, as no targetable SNV was identified. Similarly, the 11 SVs were manually curated based on recurrency in the local database and inspection in Integrated Genomic Viewer (IGV) (13) leaving only three SVs that interestingly affected regions recurrently involved in ALL.
Two of these SVs affected the JAK2 locus on 9p24.1 and inspection in IGV revealed a shared breakpoint suggesting that both SVs had occurred in the same genomic rearrangement (Supplementary Figure 1). The first SV consisted in a 122 kb-long intrachromosomal inversion with breakpoints that mapped to intron 15 (NM_001322194.2) and telomeric to the JAK2 locus. The second SV consisted in an interchromosomal event and shared the breakpoint in intron 15 of JAK2, while the second breakpoint was located at 14q32.11 and mapped to intron 25 in the CCDC88C locus (NM_001080414). Both events are visualized in the Circos plot in Figure 1A. The rearrangement of JAK2 could be verified with metaphase FISH that confirmed that the 5′ signal at the JAK2 locus was translocated to chromosome 14 while the 3′ signal was retained on derivative chromosome 9 (Figure 1B). As the rearrangement involves the most distal regions of chromosomal arms 9p and 14q, it is beyond the resolution of chromosome banding analysis and cannot be detected by karyotype analysis. In addition, SV analysis also detected a deletion on 9p21.3 encompassing 39 kb and supported by 95% of reads that resulted in a biallelic loss of CDKN2A/B that had been previously found in SoC testing.
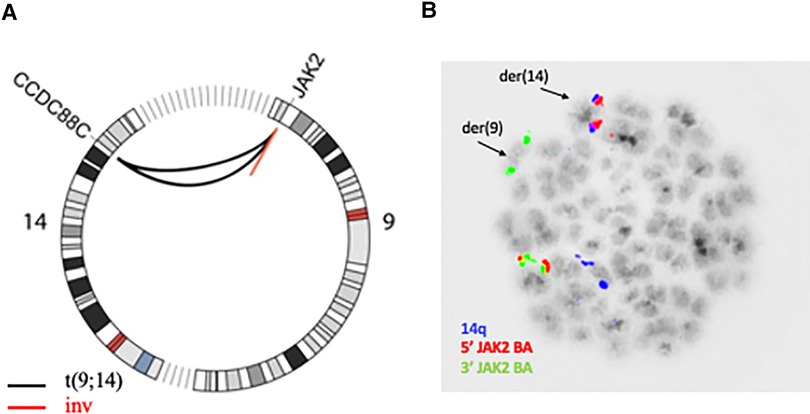
Figure 1. (A) Circos plot showing chromosomes 9 and 14; the black lines represent the reciprocal translocation together with the concomitant inversion of the small fragment from 9p23.1 (red line). (B) Aberrant metaphase hybridized with JAK2 break apart probe (red/green signal, CytoTest) and 14 centromere probe (blue signal) showing the distal red signal (5′ end) of JAK2 translocated to 14q.
Taken together the data indicated a reciprocal translocation t(9;14)(p24.1;q32.11) with a concomitant inversion of a 122-kb-long fragment from 9p that resulted in the creation of a fusion gene joining intron 25 of CCDC88C to intron 15 of JAK2 on derivative chromosome 9. As a result of the inversion from the 9p fragment, the genes are arranged in opposite orientations and thus no reciprocal fusion is created at derivative 14 (Figure 2).
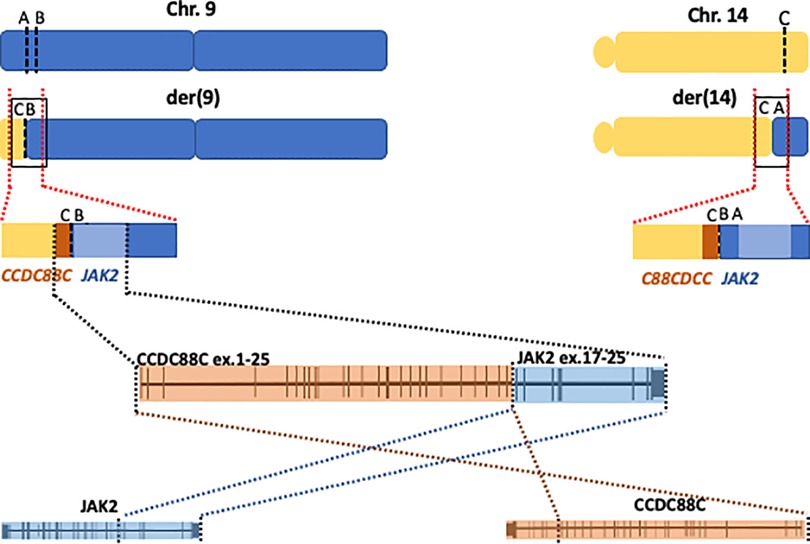
Figure 2. Cartoon showing a schematic representation of chromosomes 9 (blue) and 14 (yellow) at the derivative chromosomes 9 and 14 (upper panel), the black squares mark the junctions. Middle cartoon represents a blow up of the junctions illustrating the creation of the fusion gene on derivative chromosome 9 (der9). The lower panel represents a magnification of the fusion gene showing exons 1–25 in CCDC88C joined to exons 16–25 in JAK2.
According to the WGS findings, the rearrangement juxtaposes exons 1 to 25 of CCDC88C to exons 16 through 25 of JAK2, creating a novel fusion gene (Figure 2). The expression of the fusion transcript joining exon 25 of CCDC88C (3′ partner) to exon 16 in JAK2 gene (5′ partner) was confirmed with RT-PCR followed by Sanger sequencing. The fusion gene will thus result in an in-frame hybrid protein with the kinase domain of JAK2 at the carboxy-terminal.
Discussion
JAK2 encodes a nonreceptor tyrosine kinase involved in cell proliferation and differentiation through the JAK/STAT signaling pathway. Aberrant JAK2 is found in several cancers including both myeloid and lymphoid malignancies (14). Fusions involving JAK2 were found in Philadelphia-like ALL (15), a subset of high-risk B-ALL characterized by a distinctive gene expression profile resembling Philadelphia-positive ALL, but lacking the canonical BCR::ABL fusion (16, 17). A fraction of Philadelphia-like ALL harbors JAK2 fusions where JAK2 is the 5′ partner and all the fusions described so far include exons 19–25 and retain the kinase domain that is constitutively activated in the fusions (18). To this date, over 30 genes have been found as potential 3′ partners to JAK2, but this is the first report of a CCDC88C::JAK2 fusion. JAK2 inhibitors have shown antileukemic effect in ex vivo experiments in cells carrying JAK2 fusions (19, 20), that include the kinase domain of JAK2; however, the experience in vivo is limited (21). JAK2 fusions have only been described in isolated cases of T-ALL (18), although activating point mutations in members of the JAK/STAT pathway has been reported in up to 25% of patients with T-ALL (22). Patients carrying genomic alterations that activate the JAK/STAT pathway either through gain of function mutations or gene fusion that include the JAK2 kinase domain are potential candidates for targeted therapy (23) (24). Pediatric patients with ALL and genomic features consistent with JAK/STAT pathway activation are currently recruited in ongoing trials (clinicaltrials.gov NCT03117751 and NCT02723994) for targeted treatment with ruxolitinib (selective JAK1/2 inhibitor) in addition to chemotherapy. Since our patient responded to nelarabine, he was never considered for therapy with JAK inhibitors nor were cells available to perform in vitro sensitivity tests.
CCDC88C is a ubiquitous protein, involved in the Wnt signaling pathway, with multiple functions that include determination of cell polarity, development of the nervous system, and tumor suppressor activity (25). CCDC88C protein is highly expressed in B- and T-cell pediatric leukemias (https://pecan.stjude.cloud/proteinpaint/CCDC88C). CCDC88C has been reported a handful occasions in hematological malignancies as fusion partner with FLT3 (26) as well as PDGFRB (27) including a case report of pediatric patient with a myeloproliferative disorder (28). It has also been reported as the 5′ fusion partner to PDGFRB in a young adult with Philadelphia-like ALL that responded to imatinib (29).
The genetic landscape of T-ALL is widely heterogeneous, and despite several recurrent aberrations, to date no single lesion is used to risk-stratify T-ALL patients. However, certain aberrations such as BCR-ABL and other ABL-class fusions or KMT2A rearrangements, although rare, contribute important information to individualized patient management.
Since our patient responded to nelarabine, he could be bridged to transplantation and experimental therapy with JAK2 inhibitors was not considered. However, this report illustrates the power of WGS in the diagnostic setting of ALL and describes a novel CCDC88C::JAK2 fusion in T-ALL, adding CCDC88C to the list of potential JAK2 partners. While the validation of WGS in the diagnostic setting of acute leukemias is still ongoing in our laboratory, the present report serves as a real-life example of the power of WGS in the diagnostic setting of acute leukemia, as it enables timely recognition of potential therapeutic targets in high-risk pediatric ALL.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The study was approved by the Ethical Review Board at Stockholm County. Written informed consent was obtained from the patient’s legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article. The study was conducted in accordance with the World Medical Association's Declaration of Helsinki.
Author contributions
AK, LS and AH performed data acquisition, AK, FR, and GB interpreted the data and wrote the manuscript. PS provided patient care and contributed to writing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
We acknowledge the support from the Swedish Childhood Cancer Fund (Barncancerfonden) through grants TJ 2015-0047 and PR 2019-0072.
Acknowledgments
The authors are grateful to the patient and his parents, the staff at the Pediatric Oncology Unit, the Clinical Genetics Department at Karolinska University Hospital as well as at the Clinical Genomics Platform at Science for Life Laboratory (SciLife Lab), Stockholm.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.1082986/full#supplementary-material.
References
1. Schwab C, Harrison CJ. Advances in B-cell precursor acute lymphoblastic leukemia genomics. Hemasphere. (2018) 2(4):e53. doi: 10.1097/HS9.0000000000000053
2. Toft N, Birgens H, Abrahamsson J, Griskevicius L, Hallbook H, Heyman M, et al. Results of NOPHO ALL2008 treatment for patients aged 1-45 years with acute lymphoblastic leukemia. Leukemia. (2018) 32(3):606–15. doi: 10.1038/leu.2017.265
3. Brady SW, Roberts KG, Gu Z, Shi L, Pounds S, Pei D, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet. (2022) 54(9):1376–89. doi: 10.1038/s41588-022-01159-z
4. Pavlovic S, Kotur N, Stankovic B, Zukic B, Gasic V, Dokmanovic L. Pharmacogenomic and pharmacotranscriptomic profiling of childhood acute lymphoblastic leukemia: paving the way to personalized treatment. Genes (Basel). (2019) 10(3):1–31. doi: 10.3390/genes10030191
5. Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. (2020) 105(11):2524–39. doi: 10.3324/haematol.2020.247031
6. MIP. Mutation Identification Pipeline. Available from: https://mip-api.readthedocs.io/en/latest/
7. SCOUT. Scout. Available from: https://github.com/Clinical-Genomics/scout
8. Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. (2012) 28(18):i333–9. doi: 10.1093/bioinformatics/bts378
9. Eisfeldt J, Vezzi F, Olason P, Nilsson D, Lindstrand A. TIDDIT, an efficient and comprehensive structural variant caller for massive parallel sequencing data. F1000Res. (2017) 6:664. doi: 10.12688/f1000research.11168.1
10. Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. (2016) 32(8):1220–2. doi: 10.1093/bioinformatics/btv710
11. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20(9):1297–303. doi: 10.1101/gr.107524.110
12. Worst BC, van Tilburg CM, Balasubramanian GP, Fiesel P, Witt R, Freitag A, et al. Next-generation personalised medicine for high-risk paediatric cancer patients—the INFORM pilot study. Eur J Cancer. (2016) 65:91–101. doi: 10.1016/j.ejca.2016.06.009
13. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. (2013) 14(2):178–92. doi: 10.1093/bib/bbs017
14. Tang G, Sydney Sir Philip JK, Weinberg O, Tam W, Sadigh S, Lake JI, et al. Hematopoietic neoplasms with 9p24/JAK2 rearrangement: a multicenter study. Mod Pathol. (2019) 32(4):490–8. doi: 10.1038/s41379-018-0165-9
15. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. (2014) 371(11):1005–15. doi: 10.1056/NEJMoa1403088
16. Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. (2008) 453(7191):110–4. doi: 10.1038/nature06866
17. Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. (2009) 10(2):125–34. doi: 10.1016/S1470-2045(08)70339-5
18. Downes CE, McClure BJ, McDougal DP, Heatley SL, Bruning JB, Thomas D, et al. JAK2 alterations in acute lymphoblastic leukemia: molecular insights for superior precision medicine strategies. Front Cell Dev Biol. (2022) 10:942053. doi: 10.3389/fcell.2022.942053
19. Roberts KG, Yang YL, Payne-Turner D, Lin W, Files JK, Dickerson K, et al. Oncogenic role and therapeutic targeting of ABL-class and JAK-STAT activating kinase alterations in Ph-like ALL. Blood Adv. (2017) 1(20):1657–71. doi: 10.1182/bloodadvances.2017011296
20. Steeghs EMP, Jerchel IS, de Goffau-Nobel W, Hoogkamer AQ, Boer JM, Boeree A, et al. JAK2 aberrations in childhood B-cell precursor acute lymphoblastic leukemia. Oncotarget. (2017) 8(52):89923–38. doi: 10.18632/oncotarget.21027
21. Ding YY, Stern JW, Jubelirer TF, Wertheim GB, Lin F, Chang F, et al. Clinical efficacy of ruxolitinib and chemotherapy in a child with Philadelphia chromosome-like acute lymphoblastic leukemia with GOLGA5-JAK2 fusion and induction failure. Haematologica. (2018) 103(9):e427–31. doi: 10.3324/haematol.2018.192088
22. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. (2017) 49(8):1211–8. doi: 10.1038/ng.3909
23. Inaba H, Azzato EM, Mullighan CG. Integration of next-generation sequencing to treat acute lymphoblastic leukemia with targetable lesions: the St. Jude children’s research hospital approach. Front Pediatr. (2017) 5:258. doi: 10.3389/fped.2017.00258
24. Brivio E, Baruchel A, Beishuizen A, Bourquin JP, Brown PA, Cooper T, et al. Targeted inhibitors and antibody immunotherapies: novel therapies for paediatric leukaemia and lymphoma. Eur J Cancer. (2022) 164:1–17. doi: 10.1016/j.ejca.2021.12.029
25. Ear J, Dunkel Y, Mittal Y, Lim BBC, Liu L, Holda MK, et al. Two isoforms of the guanine nucleotide exchange factor, daple/CCDC88C cooperate as tumor suppressors. Sci Rep. (2019) 9(1):12124. doi: 10.1038/s41598-019-48420-w
26. Kurihara Y, Mizuno H, Honda A, Shimura A, Fujioka Y, Maki H, et al. CCDC88C-FLT3 gene fusion in CD34-positive haematopoietic stem and multilineage cells in myeloid/lymphoid neoplasm with eosinophilia. J Cell Mol Med. (2022) 26(3):950–2. doi: 10.1111/jcmm.17143
27. Gosenca D, Kellert B, Metzgeroth G, Haferlach C, Fabarius A, Schwaab J, et al. Identification and functional characterization of imatinib-sensitive DTD1-PDGFRB and CCDC88C-PDGFRB fusion genes in eosinophilia-associated myeloid/lymphoid neoplasms. Genes Chromosomes Cancer. (2014) 53(5):411–21. doi: 10.1002/gcc.22153
28. Bielorai B, Leitner M, Goldstein G, Mehrian-Shai R, Trakhtenbrot L, Fisher T, et al. Sustained response to imatinib in a pediatric patient with concurrent myeloproliferative disease and lymphoblastic lymphoma associated with a CCDC88C-PDGFRB fusion gene. Acta Haematol. (2019) 141(2):119–27. doi: 10.1159/000495687
Keywords: pediatric T-ALL, whole genome sequencing, JAK2 fusions, targeted therapy, precision medicine
Citation: Krstic A, Rezayee F, Saft L, Hammarsjö A, Svenberg P and Barbany G (2023) Case Report: Whole genome sequencing identifies CCDC88C as a novel JAK2 fusion partner in pediatric T-cell acute lymphoblastic leukemia. Front. Pediatr. 10:1082986. doi: 10.3389/fped.2022.1082986
Received: 28 October 2022; Accepted: 12 December 2022;
Published: 10 January 2023.
Edited by:
Hasan Hashem, King Hussein Cancer Center, JordanReviewed by:
Takeshi Isoda, Tokyo Medical and Dental University, JapanTomasz Szczepanski, Medical University of Silesia, Poland
© 2023 Krstic, Rezayee, Saft, Hammarsjö, Svenberg and Barbany. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gisela Barbany R2lzZWxhLmJhcmJhbnlAa2kuc2U=
Specialty: This article was submitted to Pediatric Hematology and Hematological Malignancies, a section of the journal Frontiers in Pediatrics