
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 09 January 2023
Sec. Pediatric Immunology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1055091
This article is part of the Research Topic Case Reports in Pediatric Immunology 2022 View all 11 articles
 Mayla Sgrulletti1,2
Mayla Sgrulletti1,2 Cristina Cifaldi3
Cristina Cifaldi3 Silvia Di Cesare4
Silvia Di Cesare4 Barbara Kroegler5Elisabetta Del Duca1
Barbara Kroegler5Elisabetta Del Duca1 Valentina Ferradini6
Valentina Ferradini6 Simona Graziani1
Simona Graziani1 Mario Bengala7
Mario Bengala7 Gigliola Di Matteo4
Gigliola Di Matteo4 Viviana Moschese1*
Viviana Moschese1*
Over the last decades, Inborn Errors of Immunity (IEI) characterized by an immune dysregulatory picture, isolated or combined with infections, have been increasingly identified and referred as Primary Immune Regulatory Disorders (PIRD). PIRD diagnosis may be difficult due to heterogeneity of time onset, sequence of clinical manifestations and laboratory abnormalities. Moreover, the dissection of a PIRD vs. a secondary immunodeficiency (SID) might be a real challenge since the same indications for immunosuppressant treatments might represent per se a PIRD clinical expression. Here we report a female patient with a history of recurrent respiratory and urinary tract infections since early infancy and a diagnosis of Rheumatoid Arthritis in adulthood. After poor response to several biologicals she was treated with Rituximab and sent to immunology referral for a severe hypogammaglobulinemia. Clinical and immunological features matched a diagnosis of common variable immunodeficiency and when IgG replacement therapy and antibiotic prophylaxis were added a good infectious control was obtained. Next generation sequencing analysis has revealed a novel heterozygous VUS in the IKBKB gene (c.1465A > G; p.Ser489Gly). Functional analysis has shown a reduced capacity of B lymphocytes and CD4 positive T cells in inducing IκBα degradation, with negative impact on NF-kB pathway. Due to recurrent infections attributed to a common condition in childhood and to an exclusive autoimmunity-centered approach in adulthood, both diagnosis and suitable treatment strategies have suffered a significant delay. To reduce the diagnostic delay, pediatricians, general practitioners and specialists should be aware of IEI and the challenges to differentiate them from SID. Furthermore, genetic characterization and functional analysis may contribute to a personalized approach, in a perspective of targeted or semi-targeted therapy.
Primary Immunodeficiencies, most recently termed as “Inborn errors of immunity” (IEI), refer to a wide group of inherited defects of one or more component of the innate and/or adaptive immune system (1). Susceptibility to recurrent and/or severe infectious diseases that take longer to resolve or need for hospitalization, use of intravenous antibiotics, atypical infections for localization or causing pathogen, historically represent warning signs for IEI (2). On the other hand, infections are common in childhood, so that accurate family and personal history together with anatomical and environmental factors should be considered before the suspicion of an IEI. Over the last decade, it has been increasingly acknowledged that immune-dysregulation, expressed with autoimmunity, hyper/auto-inflammation, granulomas, lymphoproliferative disorders and malignancies, might occur in patients with IEI (3). The term Primary Immune Regulatory Disorders (PIRD) coins a subset of IEI with non-infectious immune-mediated pathology stemming from cellular and molecular mechanisms leading to immune tolerance failure (4, 5). The recognition of molecular mechanisms underlying PIRDs is paving the way for targeted or semi-targeted therapeutic approaches (3). Approximately 20% of IEI patients might suffer from isolated immune dysregulation or combined with infectious recurrence (6). Diagnosis of a PIRD may be cumbersome due to their heterogeneity in terms of time onset, sequence of clinical manifestations and laboratory abnormalities. Not every infectious history or dysregulation is promptly associated with an IEI causing a significant diagnostic delay and a poor outcome. To make the history even more puzzling, some immunological treatments used for the same clinical indications that might be expressed in a IEI/PIRD condition may cause a iatrogenic secondary immunodeficiency (SID) (7). In fact, iatrogenic SIDs have been exponentially reported (8), in tandem with the progressive increase in the incidence of autoimmune diseases and the wider use of standard immunosuppressant drugs (i.e., corticosteroids and DMARDs) as well as biological therapies (bDMARDs) in several specialty settings (i.e., hematological, rheumatological, neurological, etc). These conditions mainly derive from B-cell targeting biologicals, with the anti-CD20 monoclonal antibody rituximab (RTX) as the cornerstone (9). Delayed manifestations of genetic immunological disorders and the increasing recognition of patients with previously undiagnosed IEI receiving biologics, renders the dissection of a IEI vs. a SID extremely challenging. Increasing awareness among the different subspecialties and appropriate investigation will favor early diagnosis as well as optimal treatment for a better outcome and quality of life.
Here we report the case of a currently 47-year-old female patient who suffered with recurrent and severe infections since early infancy and developed in adulthood a Rheumatoid Arthritis with poor response to several biological DMARDs. Immunology referral allowed a clinical diagnosis of Common Variable Immunodeficiency (CVID) and targeted Next Generation Sequencing (NGS) analysis identified a novel heterozygous variant in IKBKB gene. This study aims to reinforce the notion that pediatricians, general pratictioners (GPs) and different subspecialties should be aware of the diverse temporality and spectrum of IEI/PIRD disorders and enhance a multidisciplinary approach for continual improvement in the field.
The work was conducted in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The patient gave her informed consent to perform immunological and genetic analysis. According to best general practice, immunological work-up included the evaluation of serum Ig level by nephelometry, serum IgG subclasses by radial immunodiffusion, T and B cell immunophenotype, specific IgG antibody response to pneumococcus vaccine, autoantibodies, complement C3 and C4 and specific serum IgE. Targeted Next-Generation sequencing.
DNA was extracted by QIAamp DNA Blood Mini Kit (Qiagen) and prepared, sequenced and analyzed according to manufacturer's protocol as previously reported (10). Our CVID custom Ion Torrent panel, including 62 known genes (Supplementary Table S1), was designed with Ampliseq Designer software using GRCh38 as references. Sanger sequencing on gDNA isolated from total PBMCs was performed to confirm the presence of mutations in IKBKB gene (ABI PRISM 3130, Applied Biosystems, Foster City, CA).
All flow cytometric analyses were performed on EDTA blood samples within 24 h of venipuncture. After red blood cell lysis with ammonium chloride, lymphocytes were washed, resuspended in PBS, and stained with the following mouse anti-human antibodies to identify T and B cell subsets: CD45RA (clone T6D11; Miltenyi Biotec), CD3 (clone BW264/56; Miltenyi Biotec), CCR7 (clone 3D12; eBioscience), CD4 (clone OKT4; Becton Dickinson), CD8 PE- (clone RPA-T8; Becton Dickinson), CD19 (clone SJ25C1; Becton Dickinson), CD16 (clone 3G8), CD56, CD27 (clone M-T271, Becton Dickinson), TCR α-beta (clone T10B9; Becton Dickinson), TCR gamma-delta (11F3; Miltenyi Biotec), CD21 (clone B-ly4; Becton Dickinson), CD24 (clone ML5; Becton Dickinson), IgD (clone IA6-2; Becton Dickinson), Goat F(ab)2 anti-Human IgM (µ) (Jackson ImmunoResearch), and CD38 (clone HIT2; Becton Dickinson). Cells were incubated with the appropriate antibody cocktail for 30 min at 4 °C and then washed with PBS and resuspended in PBS for flow cytometric acquisition. At least 50,000 events were acquired within the lymphogate. Data were acquired on a FACSCanto II (Becton Dickinson) and analyzed with FlowJo software (Tree Star Inc, version 9.3.2).
PBMCs from patient and healthy controls were isolated by density gradient centrifugation on Ficoll-Paque PLUS (GE Healthcare), washed twice in PBS, and maintained in complete RPMI (Sigma-Aldrich) containing 10% FBS, 2 mM l-glutamine (Sigma-Aldrich), and 100 U/ml penicillin and streptomycin (Sigma-Aldrich).
p65 phosphorylation: Total PBMC (300.000 per tube) were stimulated with phorbol 12-myristate 13-acetate (PMA; Sigma, Cat# P1585) /Ionomycin (Ionomycin; Sigma Cat #I0634) at 37 °C for 0, 5, 10, 15 min. Stimulation was blocked and cells were fixed using Fix Buffer I (BD Biosciences, Cat #557870,) for 10 min at 37 °C then centrifuged, washed twice with FACS buffer (PBS supplemented with 2% FBS and 1 mM EDTA) and permeabilized for 10 min on ice with 1 ml Phosflow Perm Buffer III (BD Biosciences, Cat #558050). After two washes, the cells were stained for anti-CD4+, anti-CD19 + antibodies and with anti-phospho-p65 (BD Biosciences, Cat #558422) for 30 min at RT. Samples were washed three times and data were collected on FACSCanto II (Becton Dickinson).
IκBα degradation: Total PBMC (300.000 per tube) were stimulated with phorbol 12-myristate 13-acetate (PMA)/Ionomycin (PMA 100 ng/ml, Ionomycin 1 μg/ml) at 37 °C for 0, 15, 30, 60 min.
Cells were washed with FACS buffer. Fc receptors (BD Biosciences, Cat#553141) were blocked for 5 min at RT followed by a 20 min of staining on ice with anti- CD4+, CD19+ antibodies and incubated for 30 min at 4 °C. After two washes, cells were permeabilized for 30 min at RT, according to manufacturer's protocol, centrifuged and washed twice with FACS buffer. Cells were stained with anti-monoclonal IкBα (H-4) (Santa Cruz Cat #sc-1643) for 30 min at RT then washed and incubated with anti-Mouse IgG (eBiosciences, Cat# 11-4011-85). After two washes, data were collected on FACSCanto II (Becton Dickinson). All Data were analyzed with FlowJo software (Tree Star Inc, version 9.3.2). Data were analyzed with Graph-Pad Prism, version 6.2 (Graph Pad Software, la Jolla, CA).
A 47-year-old woman affected with Rheumatoid Arthritis (RA) was referred from the Rheumatology outpatient clinic to the Regional Center for Immunodeficiency Diseases at Tor Vergata University Hospital, Rome, Italy for hypogammaglobulinemia and recurrent infections.
Her family history was positive for Hashimoto thyroiditis, psoriasis, hepatocarcinoma and lymphoma. Past medical history revealed an intensive care unit admission at birth for respiratory distress, with non-invasive ventilation until the first month of life. Since early infancy, she suffered from one or two episodes per month of upper respiratory tract infections (URTI) that repeatedly required oral antibiotic treatment and the use of parenteral antibiotic therapy 2–3 times/year. In adolescenthood, in addition to recurrent URTI, she suffered from recurrent lower respiratory tract infections (LRTI) and recurrent urinary tract infections (UTI). At the age of seventeen, after a routine dental procedure, a mandibular osteitis occurred that required surgical curettage. This was complicated by Staphylococcus aureus infection with the need of parenteral antibiotic treatment for 3 months. Also, at the age of twenty-two, tonsillectomy offered no improvement of infections (Figure 1A).
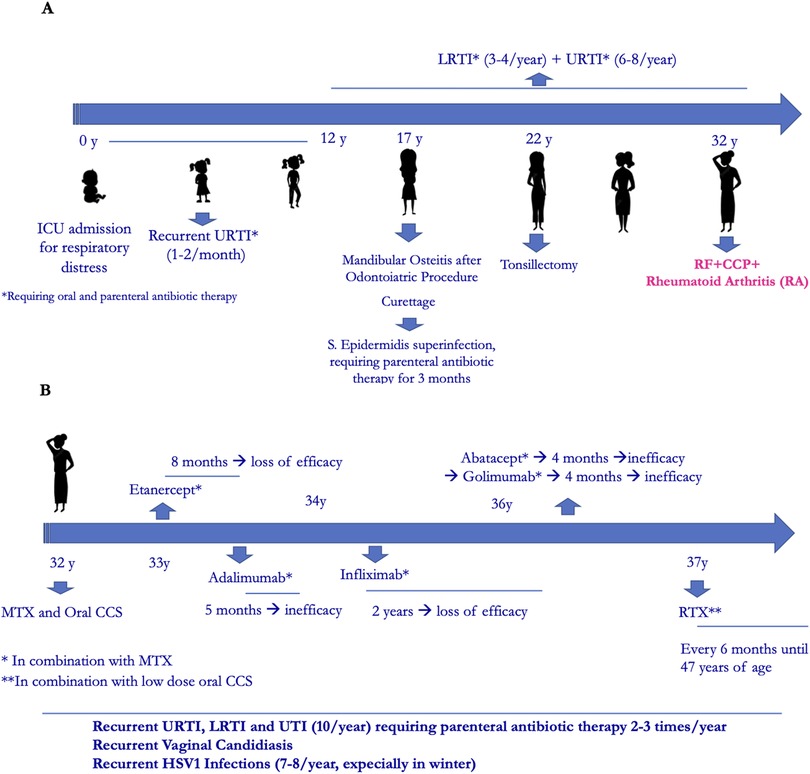
Figure 1. (A) Clinical history timeline. (B) Rheumatological treatment timeline.
The patient was diagnosed with RA at the age of 32, with main involvement of hands and feet small joints. After transient and poor response to oral corticosteroids and conventional Disease Modifying Antirheumatic Drugs (DMARDs) such as methotrexate (MTX), leflunomide and cyclosporine, she started anti-TNFα therapy with Etanercept, which was discontinued after 8 months for secondary inefficacy. Other biological- DMARDs were later administered, always in combination with MTX, with poor efficacy. During this time recurrent infections continued, including vaginal candidiasis and HSV1 infections (7–8 times/year). At the age of 37 she started RTX (2 cycles/year), with good rheumatological response (Figure 1B). No immunological investigations had been performed except for a pre-RTX immunoglobulin dosage which showed low IgM (41 mg/dl) with normal IgG and IgA (793 and 170 mg/dl, respectively). Due to further worsening of infections and the observation of severe hypogammaglobulinemia (IgG 383 mg/dl, IgA 50 mg/dl, IgM 12 mg/dl mg/dl), she was referred to our Immunology Unit.
As reported in Table 1, an extended immunological work-up confirmed the hypogammaglobulinemia (IgG 488 mg/dl, IgA 50 mg/dl, IgM 25 mg/dl) and detected low serum free light chains (kappa sFLC 6.58 mg/L, lambda sFLC 7.45 mg/L). The immunophenotype showed a moderate increase of central memory CD4+ T cells (CD3 + CD4 + CD27 + CD45RA + 60.5%), with a remarkable reduction of effector memory and EMRA CD4 T cells (CD3 + CD4 + CD27-CD45RA- 4% and CD3 + CD4 + CD27-CD45RA + 1.3%, respectively), low naïve CD8 T cells (CD3 + CD8 + CCR7 + CD45RA + 16.7%) with expansion of the effector and terminal memory compartment (CD3 + CD8 + CCR7-CD45RA- 49,8%, CD3 + CD8 + CCR7-CD45RA + 30%). Despite normal frequencies of B cells, dramatically low switched (CD27 + IgD-IgM- 0.08%) and low IgM memory B cells (CD27 + IgD + IgM + 3.5%) were identified. Based on clinical and immunological results a presumptive diagnosis of CVID was made. Specific antibody response could not be determined since a clinical worsening during an acute infection promptly required immunoglobulin replacement treatment. After the use of RTX, the patient started a treatment with Sulfasalazine and low dose oral corticosteroids in addition to regular IgG replacement therapy, with good control of infections and autoimmunity. She is now doing well with 500 mg/kg/4 weeks of intravenous immunoglobulin (IVIG) replacement therapy (obtaining an IgG level of approximately 10 g/L) and azithromycin prophylaxis.
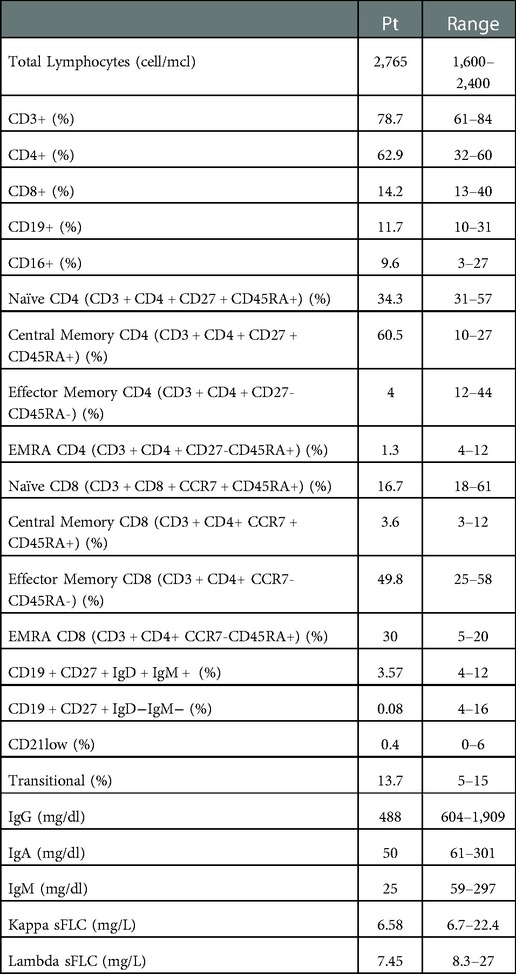
Table 1. Immunological data.
Targeting NGS analysis including 62 genes (Supplementary Table S1) causing primary antibody defects has been performed. We identified a novel heterozygous variant of uncertain significance (VUS) in IKBKB gene (c.1465A > G; p.Ser489Gly) leading to the substitution of the conserved serine in position 489 with glycine (SCV002758753 - https://submit.ncbi.nlm.nih.gov/clinvar/). This variant was not found in gnomAD exomes and genomes.
To evaluate its pathogenic role, we investigated the NF-kB pathway. The p65 phosphorylation was investigated in CD4+ T cells and CD19+ B cells at different time points after PMA/Ionomycin stimulation. We observed a delayed and reduced capacity to phosphorylate p65 overtime in CD4+ T cells (Figure 2A) and an almost absent response in CD19+ B cells (Figure 2A). Further, IκBα degradation was normally regulated in CD4+ T cells and impaired in CD19+ B cells where protein degradation was particularly reduced (Figure 2B).
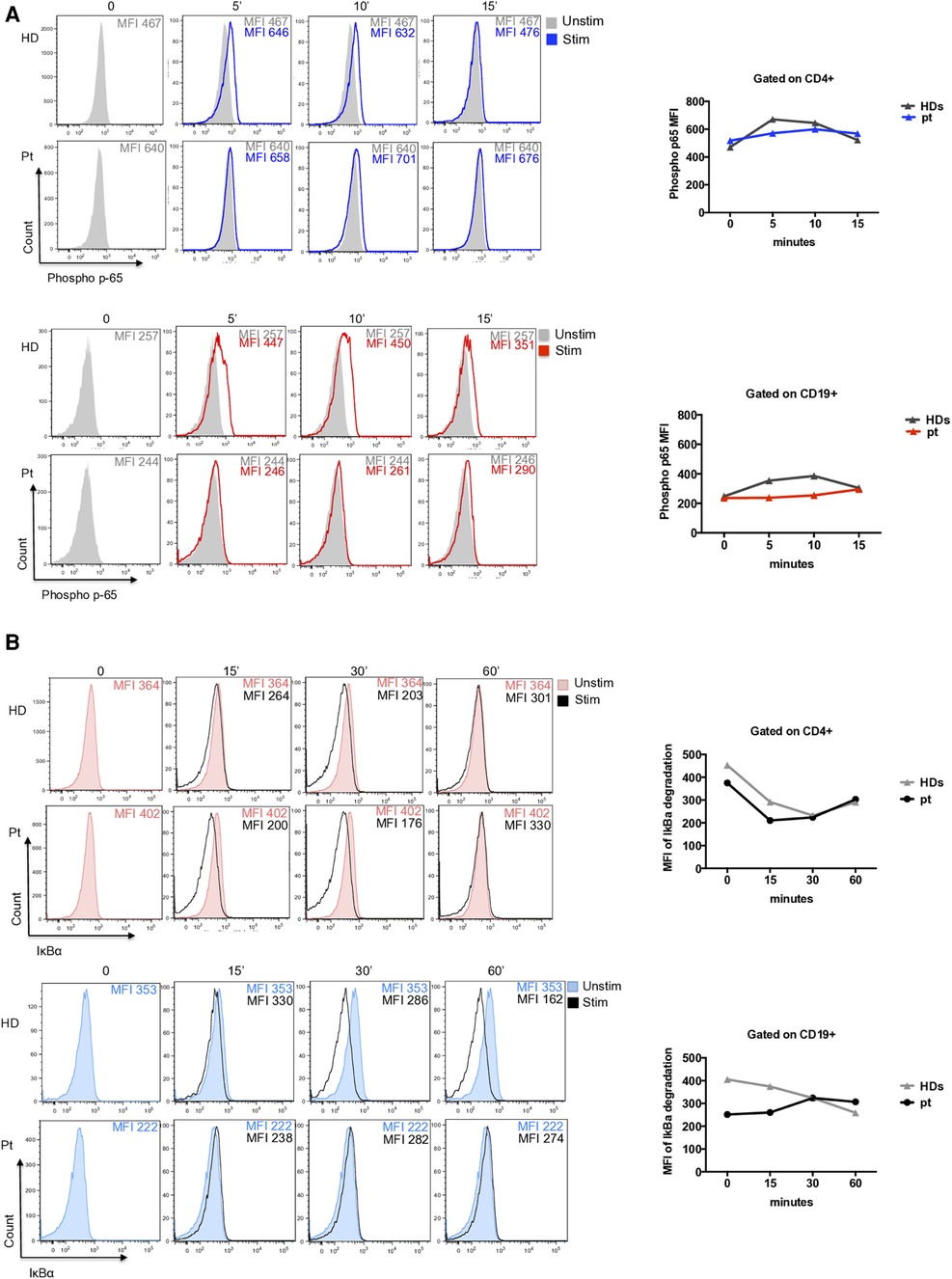
Figure 2. (A) Representative histograms of phospho-p65 expression in CD4+ T cells (blue histograms) and CD19+ B cells (red histograms) from patient and unrelated control. Graphs show the mean of two independent experiments. (B) Representative histograms of IκBα degradation (a reduction of IkBa expression is showed due to degradation) in CD4+ T cells (red filled) and CD19+ B cells (blue filled) from patient and an unrelated control. Graphs show the mean of two independent experiments.
Here we present the case of a 47-year-old female patient who suffered with recurrent infections since early infancy, who received a diagnosis of Rheumatoid Arthritis at age 32 but refractory to several biological DMARDs before she was treated with Rituximab. In the setting of a worsening recurrence of infections, hypogammaglobulinemia was observed, and an extensive immunological work-up allowed a clinical diagnosis of Common Variable Immunodeficiency (CVID) and genetic analysis identified a novel heterozygous variant in IKBKB gene.
Infectious recurrence, mainly involving the respiratory tract, represent an extremely common feature during pediatric age (11) with approximately 10%–20% of children <10 years of age suffering from recurrent respiratory tract infections, requiring antibiotic treatment and, in some cases, hospitalization (12). Many children with IEI conditions may remain undiagnosed or misdiagnosed for a longtime since infections might be underestimated and each infection treated as an isolated episode while the underlying cause and appropriate investigations are missed. IEI may present at any age and with a variable clinical presentation. Approximately 25%–30% of patients with IEI may suffer with manifestations of immune dysregulation, isolated or with coexisting infections (6). Autoimmune cytopenias, rheumatologic diseases and inflammatory bowel disease as more common. In some cases, the evolution of additional medical problems complicate matters and unveiling a PIRD condition may be a daunting task. PIRD patients require a multispecialistic approach but, since the autoimmune manifestation might appear as predominant (6), these patients are often at first evaluated by non-IEI addicted specialists who might tend to manage the immune dysregulation with no vigilance on a potential underlying PIRD condition. Therefore, it's not unusual that IEI diagnosis is delayed. Also, the infectious recurrence as a potential warning sign for an IEI, may be misinterpreted as a common complication of the extended use of a wide range of biologicals/immunosuppressant treatments. Additionally, prolonged B cell deficiency have been documented in 30%–56% of patients receiving B-cell targeting agents such as RTX for the treatment of B cell lymphoproliferative diseases and autoimmune disorders (8, 13–19). Although the precise role of RTX is not easily discernible due to the heterogeneity of previous treatments, a significant increase in the percentage of patients developing isolated or combined immunoglobulin deficiency over RTX treatment has been observed for all three isotypes. Some of these patients present symptomatic hypogammaglobulinemia and require immunoglobulin replacement to prevent infectious complications (20). In our patient, at pre-RTX use IgM deficiency was detected while some years post-RTX a severe hypogammaglobulinemia occurred. In a previous study we have reported that persistent hypogammaglobulinemia after RTX may occur in a subset of children with autoimmune cytopenia, but this should not always be interpreted as iatrogenic secondary hypogammaglobulinemia since it may unveil an IEI disorder (13). Several studies have evaluated the presence of post-RTX hypogammaglobulinemia in patients with autoimmune disorders, including rheumatologic disease (14, 21–23). An imbalance between naïve and memory B cells with low switched memory B cells may mimic a CVID condition and establishing the pathogenetic role of B cell perturbation beyond a pure iatrogenic effect may be challenging, however this is not only necessary but dutiful (23).
Rheumatologic diseases are more frequently observed in patients with IEI, especially in female adults with CVID (24). However, it is not unusual to make a diagnosis of rheumatologic disease before IEI diagnosis. In our patient, either during infancy and in adulthood, a comprehensive care of the patient missed evaluation and eventually reevaluation of the patient's immune system over time for a proper and timely management.
Among PIRD-related genetic variants found in cohorts of rheumatologic patients with hypogammaglobulinemia, both monoallelic variants in NF-kB subunits deficiency and post-translational modification of NF-kB pathway proteins have been detected. These could be responsible for either immunodeficiency, autoimmune diseases (RA, SLE and inflammatory bowel diseases), or both. IKBKB gene is implicated in NF-κB transcription signaling and particularly in the activation of the canonical NF-κB pathway, which is relevant for lymphocyte activation, homeostasis and control of self-tolerance. Upon receptor ligation, signals induce activation of the IKK complex, which includes the kinases IKKalpha, IKKbeta and NEMO that are encoded by IKBKA, IKBKB and IKBKG genes, respectively. Then, phosphorylation of the inhibitory protein IκBα allows release of NF-κB molecules p65 (RelA), c-Rel, and p50 to the nucleus that act as transcription factors with induction of a multitude of pro-inflammatory cytokines (i.e., IL-1, IL-2, IL6, IL8, IL12 and TNFα), chemokines (i.e., CXCL1 and CXCL10) and other inflammatory mediators (i.e., adhesion molecules as ICAM-1, VCAM-1, ECAM-1, anti-apoptotic factors as Fas, BCL-2, Caspase, BFL-1 and cell cycle regulator as PAI2 and Cyclin) (25, 26). Thus, it is not surprising that IKK/NFKB aberrations can cause a variety of immune-related disorders.
In our patient, we identified a novel heterozygous variant of uncertain significance (VUS) in IKBKB gene (c.1465A > G; p.Ser489Gly), in a strong conserved serine in position 489. Functional analysis revealed a strongly reduced capacity to phosphorylate the p65 overtime in CD4+ T cells and an almost absent response to stimulation in CD19+ B cells. Further, IκBα degradation was normally regulated in CD4+ T cells and impaired in CD19+ B cells. Whether this patient's functional deficiency is due to the underlying genetic defect or, alternatively, directly induced by RTX and other immunosuppressive treatments is a matter of concern. The use of immunosuppressive drugs as CCS and RTX overtime, could affect NF-kB signaling and alter T and B cell homeostasis. Only in vitro experiments using transducing vectors might confirm the pathogenicity of this variant.
In this scenario, it goes without saying that increasing awareness of pediatricians and other specialists who take care of these patients is warranted for an early immunology referral to prompt diagnosis and timely treatment with significant prognostic and socio-economic implications for the patient, family and community. The wider use of new laboratory-based genetic technologies and functional immune studies are leading to a better understanding of IEI/PIRDs pathophysiology. In IEI-addicted immunology centers, molecular and functional characterization of patients are started simultaneously with PIRD suspicion and with the initiation of first-line treatment for immune dysregulation. Molecular characterization of each distinct PIRD patient may expedite the therapeutic approach, by the use of immune-modifying biologicals directly targeting the altered immune pathway. This is crucial to optimize treatment efficacy and minimize possible adverse events (21). Conversely, in non-IEI addicted setting, molecular and functional characterization are explored after first and second lines of treatment (27). Thus, patients receiving RTX and/or other immunosuppressants should not exclusively regarded as affected with a iatrogenic immunodeficiency since a higher degree of suspicion for a previously undiagnosed IEI is warranted when atypical clinical and immunological features occur. In these cases, a prompt immunological referral and work-up, i.e., extensive B cell immunophenotype, Ig isotypes, vaccine responses, free serum light chains as well as evaluation and function of candidate genes by targeted NGS or whole exome/genome sequencing is recommended (10, 20). Growing knowledge on the molecular mechanisms sustaining immune dysregulation will be extremely beneficial for applying precision medicine and for continual translational progress in the field (28).
In conclusion, this case highlights the importance of an early suspicion for IEI disorders since childhood and of clinical and immunological reevaluations over time for a correct interpretation of more complicated courses. Collaborations among pediatricians, expert clinicians, geneticists, are required to expand the spectrum of diagnosed IEI disorders and benefit those patients who present manifestations spanning multiple disease domains. Targeted or semi-targeted treatment options might control or balance altered immune signaling pathways, however further studies are needed to highlight the off-target effects of biologics.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/ - SCV002758753.
The studies involving human participants were reviewed and approved by Policlinico Tor Vergata Ethics Committee, Viale Oxford 81, 00133 Rome, Italy. The patients/participants provided their written informed consent to participate in this study.
VM and GDM conceived and designed the study and revised the work critically for intellectual content. VM, MS, EDD, SG, BK provided care of the patient, clinical samples and clinical data. MS wrote the initial draft. VF and GDM performed tNGS experiments and contributed to data interpretation. MB provided genetic counselling. GDM, CC, SDC performed experiments and analyzed the data. MS, CC, SDC, SG contributed to the study design and data interpretation. VM, MS, GDM, CC, SDC contributed to writing the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported in part by 2021 University of Tor Vergata Research Project grant (PID-SID) to VM and by Bambino Gesù Children Hospital with a 5X1000 grant (202205_INFETT_CIFALDI) to CC.
This work was carried out in the tutorial framework of Master in Advanced Pediatric Allergy and Immunology at University of Rome Tor Vergata as well as of PhD program in Immunology, Molecular Medicine and Applied Biotechnology at University of Rome Tor Vergata.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.1055091/full#supplementary-material.
1. Eddens T, Mack M, McCormick M, Chong H, Kalpatthi R. Trends in pediatric primary immunodeficiency: incidence, utilization, transplantation, and mortality. J Allergy Clin Immunol Pract. (2022) 10(1):286–296.e3. doi: 10.1016/j.jaip.2021.10.033
2. Jeffrey Modell Foundation (JMF). (2022). (http://www.info4pi.org/library/ educational-materials/10-warning-signs).
3. Costagliola G, Peroni DG, Consolini R. Beyond infections: new warning signs for inborn errors of immunity in children. Front Pediatr. (2022) 10:855445. doi: 10.3389/fped.2022.855445
4. Chan AY, Torgerson TR. Primary immune regulatory disorders: a growing universe of immune dysregulation. Curr Opin Allergy Clin Immunol. (2020) 20(6):582–90. doi: 10.1097/ACI.0000000000000689
5. Sogkas G, Atschekzei F, Adriawan IR, Dubrowinskaja N, Witte T, Schmidt RE. Cellular and molecular mechanisms breaking immune tolerance in inborn errors of immunity. Cell Mol Immunol. (2021) 18(5):1122–40. doi: 10.1038/s41423-020-00626-z
6. Thalhammer J, Kindle G, Nieters A, Rusch S, Seppänen MRJ, Fischer A, et al. Initial presenting manifestations in 16,486 patients with inborn errors of immunity include infections and noninfectious manifestations. J Allergy Clin Immunol. (2021) 148(5):1332–1341.e5. doi: 10.1016/j.jaci.2021.04.015
7. Tuano KS, Seth N, Chinen J. Secondary immunodeficiencies: an overview. Ann Allergy Asthma Immunol. (2021) 127(6):617–26. doi: 10.1016/j.anai.2021.08.413
8. Kaplan B, Bonagura VR. Secondary hypogammaglobulinemia: an increasingly recognized complication of treatment with immunomodulators and after solid organ transplantation. Immunol Allergy Clin North Am. (2019) 39(1):31–47. doi: 10.1016/j.iac.2018.08.005
9. Leandro MJ. Infections related to biologics: agents targeting B cells. Infect Dis Clin North Am. (2020) 34(2):161–78. doi: 10.1016/j.idc.2020.02.013
10. Cifaldi C, Brigida I, Barzaghi F, Zoccolillo M, Ferradini V, Petricone D, et al. Targeted NGS platforms for genetic screening and gene discovery in primary immunodeficiencies. Front Immunol. (2019) 10:316. doi: 10.3389/fimmu.2019.00316
11. Yousefzadegan S, Tavakol M, Abolhassani H, Nadjafi A, Mansouri S, Yazdani R, et al. Systematic investigation for underlying causes of recurrent infections in children: surveillance of primary immunodeficiency. Eur Ann Allergy Clin Immunol. (2018) 50(2):72–80. doi: 10.23822/EurAnnACI.1764-1489.39
12. Peeters D, Verhulst P, Vaessen-Verberne AA, van den Tweel XW, Noordzij JG, Driessen GJA. Low prevalence of severe underlying pathology in children with recurrent respiratory tract infections. Pediatr Infect Dis J. (2021) 40(11):e424–6. doi: 10.1097/INF.0000000000003256
13. Ottaviano G, Marinoni M, Graziani S, Sibson K, Barzaghi F, Bertolini P, et al. Rituximab unveils hypogammaglobulinemia and immunodeficiency in children with autoimmune cytopenia. J Allergy Clin Immunol Pract. (2020) 8:273–82. doi: 10.1016/j.jaip.2019.07.032
14. Labrosse R, Barmettler S, Derfalvi B, Blincoe A, Cros G, Lacombe-Barrios J, et al. Rituximab-induced hypogammaglobulinemia and infection risk in pediatric patients. J Allergy Clin Immunol. (2021) 148:523–532.e8. doi: 10.1016/j.jaci.2021.03.041
15. Kaplan B, Kopyltsova Y, Khokhar A, Lam F, Bonagura V. Rituximab and immune deficiency: case series and review of the literature. J Allergy Clin Immunol Pract. (2014) 2(5):594–600. doi: 10.1016/j.jaip.2014.06.003
16. Bonagura VR. Successful rituximab treatment for lymphoma, secondary immunodeficiency causing debilitating sinusitis: underlying primary immunodeficiency disease, and alternative treatments to improve the quality of life? J Clin Immunol. (2019) 39(3):229–30. doi: 10.1007/s10875-019-00636-1
17. Viallard JF, Parrens M, Rieux-Laucat F. Fatal hypogammaglobulinemia 3 years after rituximab in a patient with immune thrombocytopenia: an underlying genetic predisposition? Case Rep Immunol. (2019) 28(2019):2543038. doi: 10.1155/2019/2543038
18. Bussel JB, Lee CS, Seery C, Imahiyerobo AA, Thompson MV, Catellier D, et al. Rituximab and three dexamethasone cycles provide responses similar to splenectomy in women and those with immune thrombocytopenia of less than two years duration. Haematologica. (2014) 99(7):1264–71. doi: 10.3324/haematol.2013.103291
19. Levy R, Mahévas M, Galicier L, Boutboul D, Moroch J, Loustau V, et al. Profound symptomatic hypogammaglobulinemia: a rare late complication after rituximab treatment for immune thrombocytopenia. Report of 3 cases and systematic review of the literature. Autoimmun Rev. (2014) 13(10):1055–63. doi: 10.1016/j.autrev.2014.08.036
20. Ottaviano G, Sgrulletti M, Moschese V. Secondary rituximab-associated versus primary immunodeficiencies: the enigmatic border. Eur J Immunol. (2022) 52(10):1572–80. doi: 10.1002/eji.202149667
21. Walter JE, Ayala IA, Milojevic D. Autoimmunity as a continuum in primary immunodeficiency. Curr Opin Pediatr. (2019) 31(6):851–62. doi: 10.1097/MOP.0000000000000833
22. Roberts DM, Jones RB, Smith RM, Alberici F, Kumaratne DS, Burns S, et al. Rituximab-associated hypogammaglobulinemia: incidence, predictors and outcomes in patients with multi-system autoimmune disease. J. Autoimmun. (2015) 57:60–5. doi: 10.1016/j.jaut.2014.11.009
23. Tieu J, Smith RM, Gopaluni S, Kumararatne DS, McClure M, Manson A, et al. Rituximab associated hypogammaglobulinemia in autoimmune disease. Front. Immunol. (2021) 12:671503. doi: 10.3389/fimmu.2021.671503
24. Resnick ES, Cunningham-Rundles C. The many faces of the clinical picture of common variable immune deficiency. Curr Opin Allergy Clin Immunol. (2012) 12(6):595–601. doi: 10.1097/ACI.0b013e32835914b9
25. Cardinez C, Miraghazadeh B, Tanita K, da Silva E, Hoshino A, Okada S, et al. Gain-of-function IKBKB mutation causes human combined immune deficiency. J Exp Med. (2018) 215(11):2715–24. doi: 10.1084/jem.20180639
26. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:17023–. doi: 10.1038/sigtrans.2017.23
27. Seidel MG. Treatment of immune-mediated cytopenias in patients with primary immunodeficiencies and immune regulatory disorders (PIRDs). Hematology Am Soc Hematol Educ Program. (2020) 2020(1):673–9. doi: 10.1182/hematology.2020000153
Keywords: inborn errors of immunity, primary immunodeficiency, secondary hypogammaglobulinemia, rheumatic disease, immune dysregulation
Citation: Sgrulletti M, Cifaldi C, Di Cesare S, Kroegler B, Del Duca E, Ferradini V, Graziani S, Bengala M, Di Matteo G and Moschese V (2023) Case Report: Crossing a rugged road in a primary immune regulatory disorder. Front. Pediatr. 10:1055091. doi: 10.3389/fped.2022.1055091
Received: 27 September 2022; Accepted: 19 December 2022;
Published: 9 January 2023.
Edited by:
Jutte Van Der Werff Ten Bosch, ZNA Queen Paola Children's Hospital, BelgiumReviewed by:
Ramsay Fuleihan, Columbia University Irving Medical Center, United States© 2023 Sgrulletti, Cifaldi, Di Cesare, Kroegler, Del Duca, Ferradini, Graziani, Bengala, Di Matteo and Moschese. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viviana Moschese bW9zY2hlc2VAbWVkLnVuaXJvbWEyLml0
Specialty Section: This article was submitted to Pediatric Immunology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.