 Chikako Senju1,2,3,4,5Yuka Nakazawa3,4,5Mayuko Shimada3,4,5Dai Iwata3,4
Chikako Senju1,2,3,4,5Yuka Nakazawa3,4,5Mayuko Shimada3,4,5Dai Iwata3,4 Michiko Matsuse6Katsumi Tanaka2Yasushi Miyazaki1Shinichi Moriwaki7
Michiko Matsuse6Katsumi Tanaka2Yasushi Miyazaki1Shinichi Moriwaki7 Norisato Mitsutake6
Norisato Mitsutake6 Tomoo Ogi3,4,5*
Tomoo Ogi3,4,5*
- 1Department of Hematology, Atomic Bomb Disease Institute, Nagasaki University Graduate School of Biomedical Sciences, Nagasaki, Japan
- 2Department of Plastic and Reconstructive Surgery, Nagasaki University Graduate School of Biomedical Sciences, Nagasaki, Japan
- 3Department of Genetics, Research Institute of Environmental Medicine, Nagoya University, Nagoya, Japan
- 4Department of Human Genetics and Molecular Biology, Graduate School of Medicine, Nagoya University, Nagoya, Japan
- 5Department of Genome Repair, Atomic Bomb Disease Institute, Nagasaki University, Nagasaki, Japan
- 6Department of Radiation Medical Sciences, Atomic Bomb Disease Institute, Nagasaki University, Nagasaki, Japan
- 7Department of Dermatology, Osaka Medical and Pharmaceutical University, Takatsuki, Japan
Aicardi–Goutières syndrome (AGS) is a rare genetic disorder characterised by progressive encephalopathy, involving microcephaly, intracranial calcification, and cerebrospinal fluid lymphocytosis with increased interferon-α concentrations. The clinical features of AGS overlap with fetal cerebral anomalies caused by congenital infections, such as TORCH (toxoplasmosis, other, rubella, cytomegalovirus, and herpes), or with those of other genetic disorders showing neonatal microcephaly, including Cockayne syndrome (CS) with transcription-coupled DNA repair deficiency, and Seckel syndrome (SS) showing aberrant cell-cycle checkpoint signaling. Therefore, a differential diagnosis to confirm the genetic cause or a proof of infection should be considered. In this report, we describe an individual who showed primordial dwarfism and encephalopathy, and whose initial diagnosis was CS. First, we conducted conventional DNA repair proficiency tests for the patient derived fibroblast cells. Transcription-coupled nucleotide excision repair (TC-NER) activity, which is mostly compromised in CS cases, was slightly reduced in the patient's cells. However, unscheduled DNA synthesis (UDS) was significantly diminished. These cellular traits were inconsistent with the diagnosis of CS. We further performed whole exome sequencing for the case and identified a compound heterozygous loss-of-function variants in the SAMHD1 gene, mutations in which are known to cause AGS. As SAMHD1 encodes deoxyribonucleoside triphosphate triphosphohydrolase, we reasoned that the deoxyribonucleoside triphosphate (dNTP) pool size in the patient's cells was elevated, and the labeling efficiency of UDS-test was hindered due to the reduced concentration of phosphorylated ethynyl deoxyuridine (EdU), a nucleoside analogue used for the assay. In conclusion, UDS assay may be a useful diagnostic tool to distinguish between AGS with SAMHD1 mutations and other related diseases.
Introduction
Aicardi–Goutières syndrome (AGS) is a rare neurodegenerative autoimmune disorder, which was first described by Aicardi and Goutieres in 1984 (1, 2). Initially, AGS was thought to be caused by an in utero infection on the basis of clinical similarities to known congenital infectious diseases, such as TORCH (toxoplasmosis, other, rubella, cytomegalovirus, and herpes) infection (3). However, AGS turned out to be a hereditary disease due to a malfunction of the antiviral interferon response system. To date, pathogenic variants associated with typical AGS have been reported in nine genes (TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, IFIH1, LSM11 and RNU7–1) (4–11). The common symptoms of AGS include psychomotor impairment, progressive microcephaly, basal ganglia calcification, spasticity, and dystonia. Patients with AGS also develop systemic lupus erythematosus (SLE)-associated autoantibodies (12, 13). They also show erythema with photosensitivity (14), which are common characteristic of infantile SLE, and their symptoms resemble those of Cockayne syndrome (CS). CS is an autosomal recessive neurodegenerative disorder characterised by photosensitivity, microcephaly, progressive growth failure, intellectual disability, and neuropsychiatric symptoms (15). CS is associated with a dysfunction in transcription-coupled nucleotide excision repair (TC-NER), which is a sub-pathway of nucleotide excision repair (NER) (16–18). TC-NER removes ultraviolet (UV)-induced photolesions and bulky DNA adducts preferentially from actively transcribed regions.
Because of an overlap of clinical manifestations between AGS and CS, their differential diagnosis is often challenging. In this report, we describe a patient with an initial diagnosis of CS, which eventually turned out to be AGS with mutations in the SAMHD1 gene. From our regular DNA repair proficiency tests performed for typical-CS cases, the patient had been initially thought to be an NER-deficient disease (19), but no obvious aberration was detected in known NER genes (20, 21). Whole exome sequencing (WES) identified biallelic mutations in the SAMHD1 gene, which encodes an antiviral protein that acts as a deoxyribonucleoside triphosphate triphosphohydrolase (dNTPase) (6, 22). The dNTPase activity of SAMHD1 decreases the deoxyribonucleoside triphosphate (dNTP) pool and inhibits reverse transcription of the viral genome (23). Because of the increased dNTP pool size in the patient's cells, the labeling efficiency of unscheduled DNA synthesis (UDS) test, which is usually used for the purpose of measuring NER capacity (19, 24, 25), was hindered. We examined the NER function of SAMHD1 and further discuss the differences between AGS and CS symptoms. The present findings suggest that UDS test is a useful tool for the differential diagnosis of AGS with mutations in SAMHD1 and other related diseases such as CS.
Materials and methods
Cell culture
The following primary fibroblasts were used. 48BR was from a normal human subject (26). CS10LO was derived from a patient with CS complementation group B (CS-B). XP21BR was derived from a patient with xeroderma pigmentosum complementation group C (XP-C) (24). XP15BR was derived from a patient with xeroderma pigmentosum complementation group A (XP-A) (27). CS213NG was derived from the present case. All fibroblasts were cultured in Dulbecco's modified Eagle's medium (Wako; Cat. No. 043–30085) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific; Cat. No. 10437–028) and 1% penicillin-streptomycin (Wako; Cat. No. 168–23191).
NER activity
Unscheduled DNA synthesis (UDS) and recovery of RNA synthesis (RRS) assays
NER activity was measured using unscheduled DNA synthesis (UDS) and recovery of RNA synthesis (RRS) assays. Details of the experimental procedures were described previously (23, 24). Normal 48BR, CS patient-derived CS10LO (CS-B), XP patient-derived XP21BR (XP-C) and XP15BR (XP-A), as well as CS213NG patient fibroblasts were plated into 96-well plastic dishes. In UDS assay, cells were irradiated with 20 J/m2UV (254 nm) and immediately incubated with 5 μM 5-ethynyl-2′-deoxyuridine (EdU, Thermo Fisher Scientific; Cat. No. A10044) for 4 h to measure non-S phase DNA synthesis activity associated with global genome repair (GGR), which is compromised in typical XP-patient cells. In RRS assay, cells were irradiated with UV (13 J/m2), followed by 18 h of recovery and subsequent 2 h incubation with a medium containing 5-ethynyluridine (EU, Thermo Fisher Scientific; Cat. No. E10345). This measures transcription recovery, activity of which is diminished in CS patient cells. After EdU- or EU-incorporation, the cells were fixed, conjugated with Alexa Fluor 488 azide (Thermo Fisher Scientific, A10266), and stained with DAPI (Dojindo; Cat. No. D523). Incorporated EdU or EU in each cell nucleus were measured using an automated image acquisition and processing system (Cellomics ArrayScan VTI; Thermo Scientific). As UDS assay measures EdU incorporation levels of non-S phase cells, we select an appropriate intensity cutoff level of EdU nuclear fluorescence to exclude high-intensity cells under S-phase DNA replication.
Lentivirus complementation assay
Experimental details of the production of lentivirus particles expressing the human XPA-XPG and ERCC1 cDNAs have been described previously (19, 28, 29). Virus complementation experiments for UDS assay were performed using CS213NG cells. These cells were plated on 96-well plastic dishes and infected with the lentiviruses. The cells were subjected to UDS assay at 66–72 h after viral infection, followed by EdU-incorporation and detection.
Immunoblotting
Cells were lysed in 2.5% sodium dodecyl sulfate sample buffer, followed by 5 min incubation at 96 °C. Equivalent amounts of protein samples were resolved with 5%–20% gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels, and blotted onto polyvinylidene difluoride membranes. The membranes were blocked with 10% skim-milk in TBS-T (0.05% Tween-20), followed by overnight incubation at 4 °C with anti-SAMHD1 antibodies (Cell Signaling Technology; Cat. No. 12,361S; source: rabbit) at a 1:250 dilution, or anti-β-actin antibodies (Santa Cruz Biotechnology; Clone C-4; Cat. No. sc-47,778; source: mouse) at a 1:1,000 dilution. After extensive washing with TBS-T (0.05% Tween-20), the membranes were incubated with secondary antibodies against anti-rabbit immunoglobulin G (Cell Signaling Technology; Cat. No. 7074) at a 1:500 dilution, or anti-mouse immunoglobulin G (Cell Signaling Technology; Cat. No. 7076) at a 1:1,000 dilution. The proteins were detected using an enhanced chemiluminescence system (Luminescent Image Ana-Lyzer LAS-3000; Fujifilm).
Measurement of cyclobutane pyrimidine dimers and 6–4 photoproducts by enzyme-linked immunosorbent assay
DNA repair activity was measured by residual UV-photolesions. Cells were plated in 10 cm dishes and cultured to reach confluence. The cells were then 15 J/m2 irradiated by UV, followed by incubation for different times (cyclobutane pyrimidine dimers [CPDs]: 24, 48, 72, and 96 h; 6–4 photoproducts [6–4PPs]: 1, 3, 6, and 24 h). After harvesting the cells, genomic DNA was extracted using a QIAamp DNA Mini Kit (Qiagen; Cat. No. 51306). Genomic DNA samples were prepared respectively at 0.2 μg/ml for CPD measurements and at 4.0 μg/ml for 6–4PP measurements. DNA samples were denatured at 100 °C for 10 min and placed on ice for 15 min. A 50 μl aliquot of the sample DNA solution was added to each well of a protamine sulfate-coated enzyme-linked immunosorbent assay plate (Cosmo Bio; Cat. No. NM-MA-P001) and completely dried by overnight incubation at 37 °C. Non-specific antibody-binding sites of the DNA samples were depleted by blocking with 2% fetal bovine serum in phosphate-buffered saline (PBS) at 150 μl/well. The DNA samples were then incubated with anti-CPD antibodies (TDM-2, Cosmo Bio; Cat. No. NMDND001) or with anti-6–4PPs antibodies (64M-2, Cosmo Bio; Cat. No. NMDND002), and both samples were diluted in PBS (1:1000) at 100 μl/well for 30 min at 37 °C. After extensive washing with PBS-T (0.05% Tween-20), the plates were incubated with horseradish peroxidase-conjugated rat-anti-mouse secondary antibodies (Cell Signaling Technology; Cat. No. 7076) diluted in PBS (1:1000) at 100 μl/well for 30 min at 37 °C. After five times wash with PBS-T (0.05% Tween-20) at 150 μl/well, 100 μl of tetramethylbenzidine peroxidase substrate solution (Sigma; Cat. No. T0440) was added to each well and incubated for 30 min at room temperature in the dark. The reaction was stopped with 100 μl of 0.5 M H2SO4. The fluorescence intensity was measured at 450 nm with an enzyme-linked immunosorbent assay plate reader (Multiskan FC; Thermo Scientific).
Exome sequencing
To identify pathogenetic variants in the patient's genome, WES was conducted as described previously (28). DNA fragments were enriched by using the Agilent Sure Select Human All Exon Kit v.6 (Agilent, Santa Clara, CA, United States). The captured exonic genomic DNA fragments were sequenced on the Illumina Hiseq 2,500 platform (Illumina, San Diego CA, United States), with paired-end (PE) flow cells to obtain 100–150 base pair reads of 100–200 × exon coverage. The next-generation sequencing data were analysed using our standard exome pipeline. All of the variants were annotated with ANNOVAR (30) on the basis of GENCODE release 19 (GRCh37).
Results
Case report
The patient (CS213NG) was a boy born uneventfully at 40 weeks of gestation with a birth weight of 2452 g. At 3 years of age, his body weight was 8.6 kg (−2 standard deviations [SDs]), his height was 82 cm (−3SDs), and his head circumference was 45.7 cm (−2.4SDs). The patient showed psychomotor impairment and spastic paralysis of both lower extremities. A rash due to photosensitivity was observed on both cheeks. Calcification was noted in the bilateral basal ganglia on a head computed tomography scan. His symptoms resembled those of CS.
NER activity in the patient's cells
Because the patient displayed CS-like clinical features, we initially measured NER activity in the patient's CS213NG fibroblasts. Global genome NER (GG-NER) removes damages throughout the genome, while TC-NER removes damages from actively transcribed genes. GG-NER and TC-NER activities can be measured by UDS (31) and RRS (32) assays, respectively.
In UDS assay, normal 48BR and CS-B patient-derived CS10LO fibroblasts showed normal levels of EdU incorporation after UV irradiation and were proficient in GG-NER, while XPC-deficient XP21BR and XPA-deficient XP15BR fibroblasts showed no EdU incorporation due to the lack of GG-NER activity (Figure 1A). Unexpectedly, UV-induced EdU incorporation in CS213NG cells was significantly reduced compared with that in 48BR cells, which indicated that the patient's CS213NG cells had substantial UDS deficiency (Figure 1A). In RRS assay, CS10LO and XP15BR cells showed impaired TC-NER function, with decreased EU incorporation after UV-irradiation, while 48BR and XP21BR cells showed normal RRS levels (Figure 1B). Contrary to our expectation, RRS activity was only mildly decreased in the patient's CS213NG cells. These findings suggested that the patient did not have typical CS (Figure 1B).
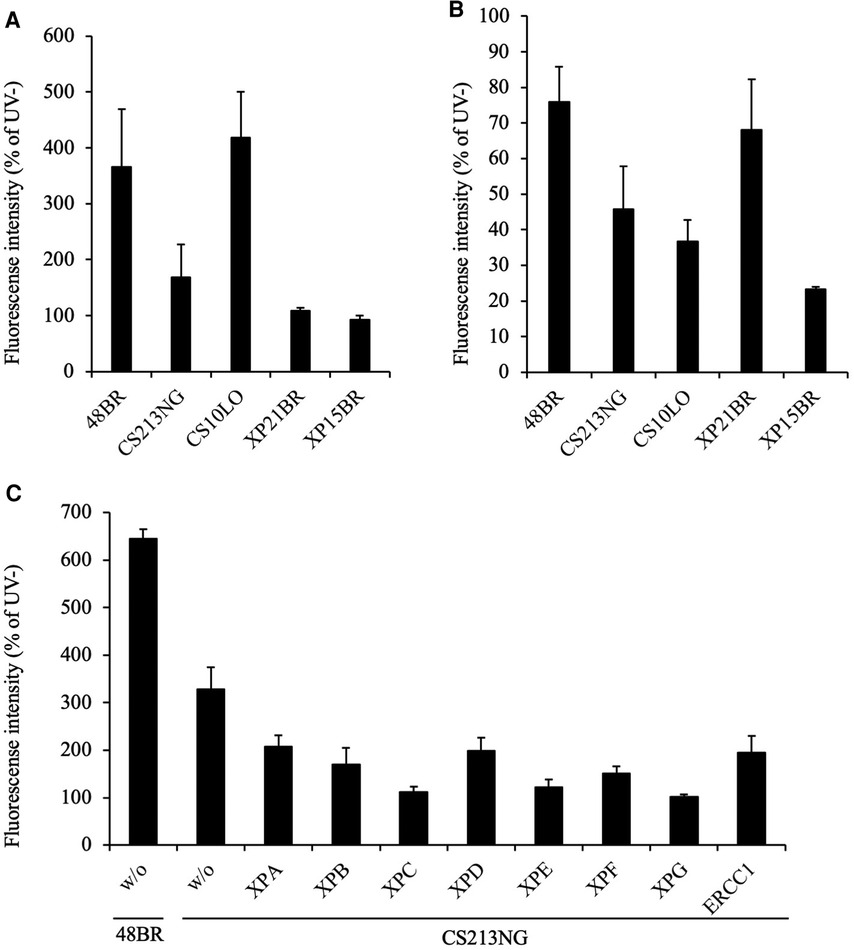
Figure 1. Examination of NER activity by UDS and RRS assays. (A) UDS activity after UV irradiation was impaired in CS213NG cells. 48BR (Normal), CS10LO (CS-B), XP21BR (XP-C), XP15BR (XP-A), and the patient-derived CS213NG cells were irradiated with 20 J/m2 UVC followed by UDS measurements (*p < 0.05, two-tailed homoscedastic t-test, 48BR vs. CS213NG). (B) Normal but slightly reduced RRS activity in CS213NG cells (*p < 0.05, 48BR vs. CS213NG; 13 J/m2 UVC irradiation). UDS and RRS were normalised to activity in non-irradiated cells. Error bars represent the standard deviation (SD) of the mean nuclear fluorescent intensity in triplicate samples (A, B). (C) UDS virus complementation assay (representative data). CS213NG cells were infected with lentiviruses expressing either of the XPA-XPG as well as ERCC1 cDNAs. Error bars represent the standard deviation of four wells.
On the basis of the reduced UDS activity, we anticipated that the patient carried pathogenic mutations either in the GG-NER related genes. We tested complementation of the UDS defects by infecting the patient's CS213NG cells with lentiviruses expressing one of the genes (cDNAs) associated with the entire NER pathway. Unexpectedly, neither gene complemented the UDS activity compared with 48BR cells. Therefore, NER-associated genes were not responsible for the disease (Figure 1C).
Exome sequencing identifies loss of function variants in SAMHD1
To identify disease causative variants carried in the patient, we then performed exome sequencing (WES). Under a recessive model of inheritance, we identified potentially pathogenic mutations in the SAMHD1 gene, which is known to be associated with AGS. The identified variants were c.G724T in exon 7, resulting a truncation mutation, p. Glu242Ter (pathogenic - PVS1/PM2/PP3, based on the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guideline, [ACMG/AMP] 2015; no variant allele was found in gnomad v.2.1.1), and c.A464G in exon 4 leading to an amino acid substitution, p.Tyr155Cys (likely pathogenic - PS3 (33, 34)/PM1/PM2/PP3, based on the ACMG/AMP 2015; rs1306109528; 1 in ∼250 k alleles in gnomad v.2.1.1) (Supplementary Table S1); these mutations were confirmed by Sanger sequencing (Figure 2A).
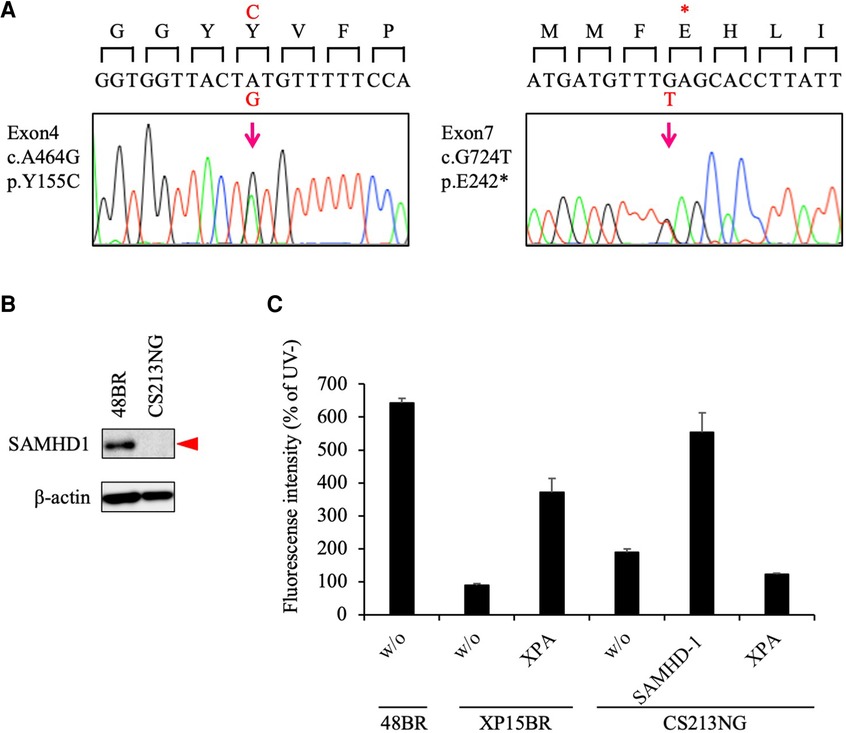
Figure 2. Pathogenic variants identified in the SAMHD1 gene. (A) Sanger sequencing confirmed that CS213NG is compound heterozygous for the SAMHD1 c.A464G and c.G724T. (B) Lack of the SAMHD1 protein expression in CS213NG cells. Bata-actin was a loading control. (C) Ectopic expression of the SAMHD1 cDNA complemented the UDS defect of CS213NG cells. CS213NG and XP15BR cells were infected with lentiviruses expressing either of the XPA or SAMHD1 cDNAs (representative data). Error bars represent the standard deviation of four wells.
As the identified variants have not yet been reported previously as any congenital disease-causing mutations, we next studied the consequences of the variants. While the SAMHD1 protein was detected at 72 kDa by immunoblotting in normal 48BR cells, this band was not detectable in the patient's CS213NG cells (Figure 2B). This result is consistent with previous reports describing the instability nature of the missense variant, p. Y155C, in vitro (34), or in leukemia patients' cells with this mutation (33).
We further performed a lentivirus complementation test. Overexpression of the wild-type SAMHD1 cDNA in the patient's cells restored normal UDS (Figure 2C). These data indicated that the identified SAMHD1 variants were causal for the patient's cellular phenotype and new pathogenic mutations. Therefore, we concluded that the patient did not have CS, but had AGS instead.
Reduced EdU incorporation capacity of the patient's cells due to an increase in the dNTP pool size
The SAMHD1 gene encodes a major dNTPase, which depletes cellular dNTPs. Therefore, we deduced that the compromised UDS test of the SAMHD1-deficient CS213NG cells was due to an increase in the cellular dNTP pool size and an eventual reduction in the EdU labeling efficiency, rather than a lack of repair DNA synthesis associated with NER activity. To test this possibility, we performed further UDS tests of the patient's cells under various concentrations of EdU (Figure 3A). We have previously shown that the EdU incorporation efficiency is dependent on its concentration relative to endogenously synthesized thymine nucleotides in the nucleotide pool (24). In the normal 48BR cells, UDS activity was increased with high EdU concentrations and reached a plateau at 5 μM. While in the SAMHD1-deficient CS213NG cells, the UDS activity remained lowish at 0.5–50 μM of EdU. These data strongly suggest that the dNTP concentration in the SAMHD1-deficient AGS patient cells is much higher than that of normal cells. Collectively, the abnormal UDS result of CS213NG is a consequence of an increase of dNTP pool size rather than an NER defect.
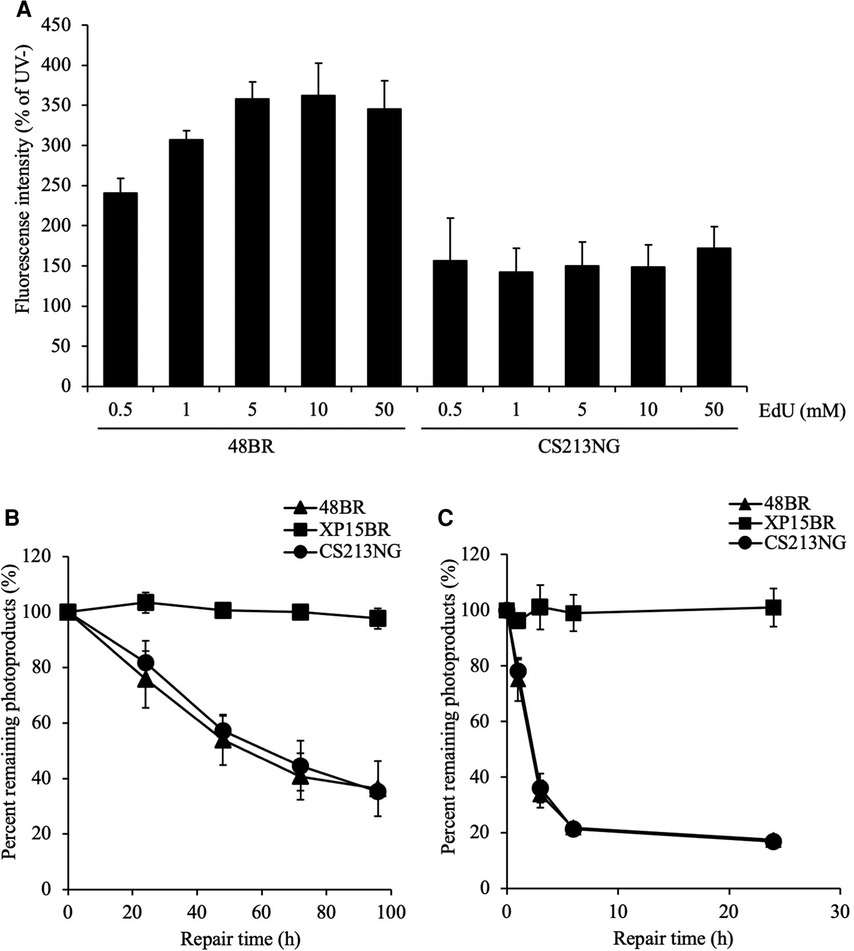
Figure 3. Reduced UDS in CS213NG cells is associated with an increased dNTP pool size. (A) A UDS assay was performed with various concentrations of EdU. 48BR and CS213NG cells were incubated with increasing EdU concentrations from 0.5 to 50 μM. Error bars represent the SD of means of nuclear fluorescent intensity in triplicate samples. (B, C) Normal DNA repair kinetics in CS213NG cells. Detection of CPD (B) and 6-4PP (C) damages. Cells were irradiated by UV at 15 J/m2 and incubated for various times to allow repair. The error bars represent the SD of triplicate experiments.
Normal DNA repair activity confirmed by CPD and 6–4pp repair assays
Considering that DNA repair activity is proficient in SAMHD1-deficient cells, we further assessed the NER function and repair kinetics of major photolesions, cyclobutane pyrimidine dimers (CPDs) and pyrimidine-pyrimidone 6–4 photoproducts (6–4PPs), in CS213NG cells (Figures 3B,C). While NER-deficient XP15BR cells exhibited no repair of CPDs and 6–4PPs, CS213NG cells displayed similar repair kinetics to those of normal 48BR cells, which indicates that CS213NG cells are proficient in NER. This result is consistent with a previous report describing a proficiency (maybe rather efficient) in DNA repair of major photolesions in SAMHD-deficient fibroblasts from AGS patients (35). Collectively, the disease-responsible gene in the patient CS213NG was SAMHD1. The lack of SAMHD1 protein apparently reduces UDS activity, but its NER function is proficient.
Discussion
Prompt establishment of a definite diagnosis in patients with rare disorders is important from the point of view of the patient's quality of life. As summarised in Table 1, AGS and CS have various overlapping phenotypes, including basal ganglia calcification, intellectual disability, developmental delay, muscle stiffness, microsomia, microcephaly, and hepatosplenomegaly. While CS shows characteristic erythema after UV irradiation, AGS may have a clinical overlap with SLE resulting in a lupus-like rash. Because of these similar clinical manifestations, the differential diagnosis of CS and AGS, as well as other related disorders, is often difficult.
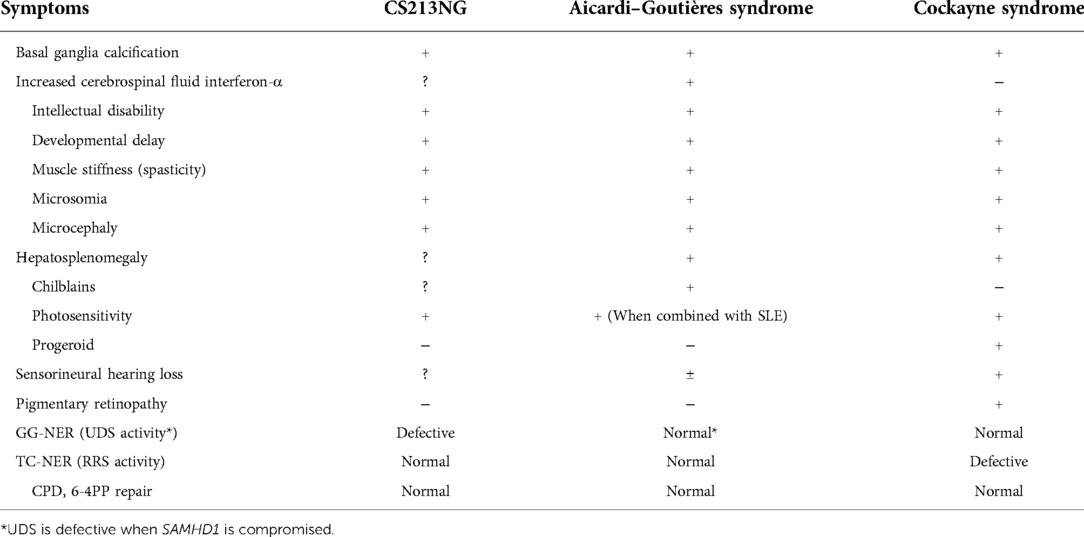
Table 1. Common clinical manifestations in Aicardi–Goutières syndrome and cockayne syndrome, and symptoms observed in the present patient (CS213NG).
In the present case, CS213NG, the initial diagnosis based on the clinical appearance was CS. Most typical CS cases are caused by a deficiency of the CSA or CSB gene. Because the CSA/CSB protein complex roles in the initiation process of TC-NER, by ubiquitinating DNA damage stalled RNA polymerase IIo molecules, CSA/CSB-compromised patients cells display TC-NER deficiency (36). Therefore, laboratory-based CS clinical diagnoses rely on UDS and RRS tests to determine the cellular NER capacity. RRS-negative is a key endpoint of CS-patient cells compromised in the CSA/CSB protein complex. RRS-proficiency can eliminate the possibility of non-CS conditions, such as TORCH infections showing congenital intracranial calcification, and hereditary microcephalic disorders including Seckel syndrome. In addition, UDS-deficiency is observed in patients with specific minor mutations in the NER responsible XPB, XPD, and XPG genes; they display the combined features of CS and another genodermatosis, xeroderma pigmentosum (XP), termed XPCS. According to the circumstances, even typical CS cases with mutations in the CSA or CSB gene might display an intermediate RRS deficiency. In the present case, the RRS reduction was not significant enough to determine a TC-NER defect; however, the decrease of UDS was prominent. Therefore, we initially presumed that the patient falls into XPCS with mutations either in the GG-NER gene. Unexpectedly, the lentivirus complementation test revealed that neither of the XPA-XPG as well as ERCC1 complemented the UDS defect of the patient's CS213NG cells. At this point, we anticipated a possibility that the patient may carry pathogenic variants in a gene for which CS-causing mutations have not yet been identified.
The use of next generation sequencing (NGS) has become popular for clinical genetics. We routinely perform whole exome sequencing (WES) for the diagnosis of uncharacterised genetic disorders. Although WES provides a rapid diagnosis compared to conventional cellular functional analyses, the overall diagnostic rate of rare diseases by WES is still insufficient in general clinical practice. This is particularly because of the low detection power of non-coding variants as well as the necessity of extra evaluations of identified variants of unknown significance (VUSs). For these reasons, in case of CS genetic tests, we ordinary rely on the target Sanger sequencing of the CSA/CSB genes. Because the cellular NER property of CS213NG was inconsistent with the diagnosis of CS, we further performed WES on the case to search for potentially pathogenic variants. The WES identified 18 candidate genes with homozygous or compound heterozygous mutations under a recessive model of inheritance (Supplementary Table S1). We focused on the SAMHD1 gene as mutations in this gene are known to cause AGS, a disease with similar pathology to CS. According to the ACMG guideline, the variants identified in the SAMHD1 gene were evaluated as a pathogenic stopgain mutation, p. E242X, and a likely-pathogenic amino acid substitution, p. Y155C. These WES evaluations were sufficient to suspect that the patient has AGS, but are still insufficient to provide a definite diagnosis; therefore, further experimental evaluation of variants was needed.
The identified SAMHD1 variants were both proved to be loss-of-function pathogenic mutations because the SAMHD1 protein expression was absolutely absent and the UDS defect was complemented by the ectopic expression of the SAMHD1 cDNA in the patient's cells. As SAMHD1 encodes a major dNTPase, the reason for the UDS deficiency in the patient's cells was due to an increase of the dNTP pool size and, as a consequence, a decrease of the EdU labeling efficiency of DNA synthesis associated with NER. This deduction is intelligible for the explanation of ΔSAMHD1 cellular phenotype; however, in terms of the use of UDS and RRS tests for the diagnosis of NER-deficient disorders such as CS and XP, the UDS-deficiency was unexpected. The current experience indicates that UDS test is also effective for a cytological examination of SAMHD1-deficient AGS cases. In conclusion, UDS and RRS assays are useful diagnostic tools to distinguish between AGS with SAMHD1 mutations and other similar disorders.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Affected individuals and normal control samples were obtained with local ethical approvals (the Ethics Committee for Human Genome Studies in Research Institute of Environmental Medicine, Nagoya University, the Nagasaki University Ethical, Legal and Social Implications (ELSI) Committee, medical ethic committees at Osaka Medical and Pharmaceutical University). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author contributions
CS and TO designed the study and the experiments. CS, YN, MS, NM, and TO analyzed the genetic data. CS, YN, DI, MM, and TO performed molecular and cell biological experiments. CS, SM, and TO analyzed clinical manifestations of the affected individuals. SM provided patients’ samples. CS and TO wrote the manuscript. All authors commented on the manuscript and approved the final version for submission. All authors contributed to the article and approved the submitted version.
Acknowledgments
This work was supported by AMED under Grant Number JP20ek0109488 and JP21ek0109549, JSPS KAKENHI Grant Number JP20H00629, The Uehara Memorial foundation and Takeda Science Foundation to TO. This work was supported by AMED under Grant Number JP20ak0101146, JSPS KAKENHI Grant Number JP21K19844 and JP21H02399, Takeda Science Foundation, The Naito Foundation and Daiichi Sankyo Foundation of Life Science to YN.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.1048002/full#supplementary-material.
References
1. Aicardi J, Goutieres F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. (1984) 15:49–54. doi: 10.1002/ana.410150109
2. Goutieres F. Aicardi-Goutieres syndrome. Brain Dev. (2005) 27:201–6. doi: 10.1016/j.braindev.2003.12.011
3. Megli CJ, Coyne CB. Infections at the maternal-fetal interface: an overview of pathogenesis and defence. Nat Rev Microbiol. (2022) 20:67–82. doi: 10.1038/s41579-021-00610-y
4. Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutiéres syndrome and mimic congenital viral brain infection. Nat Genet. (2006) 38:910–6. doi: 10.1038/ng1842
5. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the gene encoding the 3 ‘-5 ‘ DNA exonuclease TREX1 cause Aicardi-Goutiéres syndrome at the AGS1 locus. Nat Genet. (2006) 38:917–20. doi: 10.1038/ng1845
6. Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, et al. Mutations involved in Aicardi-Goutiéres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. (2009) 41:829–32. doi: 10.1038/ng.373
7. Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, et al. Mutations in ADAR1 cause Aicardi-Goutiéres syndrome associated with a type I interferon signature. Nat Genet. (2012) 44:1243–8. doi: 10.1038/ng.2414
8. Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, et al. Aicardi-Goutieres syndrome is caused by IFIH1 mutations. Am J Hum Genet. (2014) 95:121–5. doi: 10.1016/j.ajhg.2014.06.007
9. Rice GI, Duany Y DT, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. (2014) 46:503–9. doi: 10.1038/ng.2933
10. Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. (2007) 81:713–25. doi: 10.1086/521373
11. Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher MT, et al. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet. (2020) 52:1364–72. doi: 10.1038/s41588-020-00737-3
12. Gunther C, Kind B, Reijns MA, Berndt N, Martinez-Bueno M, Wolf C, et al. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J Clin Invest. (2015) 125:413–24. doi: 10.1172/JCI78001
13. Ramantani G, Kohlhase J, Hertzberg C, Innes AM, Engel K, Hunger S, et al. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutiéres syndrome. Arth Rheum. (2010) 62:1469–77. doi: 10.1002/art.27367
14. De Laet C, Goyens P, Christophe C, Ferster A, Mascart F, Dan B. Phenotypic overlap between infantile systemic lupus erythematosus and Aicardi-Goutiéres syndrome. Neuropediatrics. (2005) 36:399–402. doi: 10.1055/s-2005-873058
15. Laugel V. Cockayne syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington (1993). https://www.ncbi.nlm.nih.gov/books/NBK1342/ [Updated Aug 29, 2019]
16. Jia N, Guo C, Nakazawa Y, van den Heuvel D, Luijsterburg MS, Ogi T. Dealing with transcription-blocking DNA damage: repair mechanisms, RNA polymerase II processing and human disorders. DNA Repair (Amst). (2021) 106:103192. doi: 10.1016/j.dnarep.2021.103192
17. van den Heuvel D, van der Weegen Y, Boer DEC, Ogi T, Luijsterburg MS. Transcription-Coupled DNA repair: from mechanism to human disorder. Trends Cell Biol. (2021) 31:359–71. doi: 10.1016/j.tcb.2021.02.007
18. Lans H, Hoeijmakers JHJ, Vermeulen W, Marteijn JA. The DNA damage response to transcription stress. Nat Rev Mol Cell Biol. (2019) 20:766–84. doi: 10.1038/s41580-019-0169-4
19. Jia N, Nakazawa Y, Guo C, Shimada M, Sethi M, Takahashi Y, et al. A rapid, comprehensive system for assaying DNA repair activity and cytotoxic effects of DNA-damaging reagents. Nat Protoc. (2015) 10:12–24. doi: 10.1038/nprot.2014.194
20. Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. (2014) 15:465–81. doi: 10.1038/nrm3822
21. de Boer J, Hoeijmakers JH. Nucleotide excision repair and human syndromes. Carcinog. (2000) 21:453–60. doi: 10.1093/carcin/21.3.453
22. Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. (2011) 480:379–82. doi: 10.1038/nature10623
23. Kim B, Nguyen LA, Daddacha W, Hollenbaugh JA. Tight interplay among SAMHD1 protein level, cellular dNTP levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J Biol Chem. (2012) 287:21570–4. doi: 10.1074/jbc.C112.374843
24. Limsirichaikul S, Niimi A, Fawcett H, Lehmann A, Yamashita S, Ogi T. A rapid non-radioactive technique for measurement of repair synthesis in primary human fibroblasts by incorporation of ethynyl deoxyuridine (EdU). Nucleic Acids Res. (2009) 37:e31. doi: 10.1093/nar/gkp023
25. Nakazawa Y, Yamashita S, Lehmann AR, Ogi T. A semi-automated non-radioactive system for measuring recovery of RNA synthesis and unscheduled DNA synthesis using ethynyluracil derivatives. DNA Repair (Amst). (2010) 9:506–16. doi: 10.1016/j.dnarep.2010.01.015
26. Arlett CF, Green MH, Priestley A, Harcourt SA, Mayne LV. Comparative human cellular radiosensitivity: i. The effect of SV40 transformation and immortalisation on the gamma-irradiation survival of skin derived fibroblasts from Normal individuals and from ataxia-telangiectasia patients and heterozygotes. Int J Radiat Biol. (1988) 54(6):911–28. doi: 10.1080/09553008814552321
27. Arlett CF, Plowman PN, Rogers PB, Parris CN, Abbaszadeh F, Green MH, et al. Clinical and cellular ionizing radiation sensitivity in a patient with xeroderma pigmentosum. Br J Radiol. (2006) 79(942):510–7. doi: 10.1259/bjr/83726649
28. Nakazawa Y, Sasaki K, Mitsutake N, Matsuse M, Shimada M, Nardo T, et al. Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat Genet. (2012) 44:586–92. doi: 10.1038/ng.2229
29. Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes cockayne syndrome, xeroderma pigmentosum, and fanconi anemia. Am J Hum Genet. (2013) 92:807–19. doi: 10.1016/j.ajhg.2013.04.007
30. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38(16):e164. doi: 10.1093/nar/gkq603
31. Lehmann AR, Stevens S. A rapid procedure for measurement of DNA repair in human fibroblasts and for complementation analysis of xeroderma pigmentosum cells. Mutat Res. (1980) 69(1):177–90. doi: 10.1016/0027-5107(80)90187-6
32. Lehmann AR, Thompson AF, Harcourt SA, Stefanini M, Norris PG. Cockayne's syndrome: correlation of clinical features with cellular sensitivity of RNA synthesis to UV irradiation. J Med Genet. (1993) 30:679–82. doi: 10.1136/jmg.30.8.679
33. Clifford R, Louis T, Robbe P, Ackroyd S, Burns A, Timbs AT, et al. SAMHD1 Is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood. (2014) 123:1021–31. doi: 10.1182/blood-2013-04-490847
34. Bowen NE, Temple J, Shepard C, Oo A, Arizaga F, Kapoor-Vazirani P, et al. Structural and functional characterization explains loss of dNTPase activity of the cancer-specific R366C/H mutant SAMHD1 proteins. J Biol Chem. (2021) 297:101170. doi: 10.1016/j.jbc.2021.101170
35. Franzolin E, Coletta S, Ferraro P, Pontarin G, D'Aronco G, Stevanoni M, et al. SAMHD1-deficient Fibroblasts from Aicardi-Goutiéres syndrome patients can escape senescence and accumulate mutations. FASEB J. (2020) 34:631–47. doi: 10.1096/fj.201902508R
Keywords: Aicardi-Goutières syndrome (AGS), SAMHD1, unscheduled DNA synthesis (UDS), cockayne syndrome (CS), transcription coupled repair (TCR)
Citation: Senju C, Nakazawa Y, Shimada M, Iwata D, Matsuse M, Tanaka K, Miyazaki Y, Moriwaki S, Mitsutake N and Ogi T (2022) Aicardi–Goutières syndrome with SAMHD1 deficiency can be diagnosed by unscheduled DNA synthesis test. Front. Pediatr. 10:1048002. doi: 10.3389/fped.2022.1048002
Received: 19 September 2022; Accepted: 14 October 2022;
Published: 4 November 2022.
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Chiara Rampazzo, University of Padua, ItalyGillian Inara Rice, The University of Manchester, United Kingdom
© 2022 Senju, Nakazawa, Shimada, Iwata, Matsuse, Tanaka, Miyazaki, Moriwaki, Mitsutake and Ogi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomoo Ogi dG9naUByaWVtLm5hZ295YS11LmFjLmpw
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics