
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 04 November 2022
Sec. Neonatology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1041919
This article is part of the Research TopicEndotyping and Phenotyping Prematurity and its ComplicationsView all 5 articles
 Giacomo Simeone Amelio1
Giacomo Simeone Amelio1 Livia Provitera1
Livia Provitera1 Genny Raffaeli1,2*Matteo Tripodi1
Genny Raffaeli1,2*Matteo Tripodi1 Ilaria Amodeo1
Ilaria Amodeo1 Silvia Gulden1
Silvia Gulden1 Valeria Cortesi1,2Francesca Manzoni1,2Gaia Cervellini1,2Andrea Tomaselli1,2Valentina Pravatà1
Valeria Cortesi1,2Francesca Manzoni1,2Gaia Cervellini1,2Andrea Tomaselli1,2Valentina Pravatà1 Felipe Garrido3
Felipe Garrido3 Eduardo Villamor4
Eduardo Villamor4 Fabio Mosca1,2
Fabio Mosca1,2 Giacomo Cavallaro1*
Giacomo Cavallaro1*
Millions of infants are born prematurely every year worldwide. Prematurity, particularly at lower gestational ages, is associated with high mortality and morbidity and is a significant global health burden. Pregnancy complications and preterm birth syndrome strongly impact neonatal clinical phenotypes and outcomes. The vascular endothelium is a pivotal regulator of fetal growth and development. In recent years, the key role of uteroplacental pathologies impairing endothelial homeostasis is emerging. Conditions leading to very and extremely preterm birth can be classified into two main pathophysiological patterns or endotypes: infection/inflammation and dysfunctional placentation. The first is frequently related to chorioamnionitis, whereas the second is commonly associated with hypertensive disorders of pregnancy and fetal growth restriction. The nature, timing, and extent of prenatal noxa may alter fetal and neonatal endothelial phenotype and functions. Changes in the luminal surface, oxidative stress, growth factors imbalance, and dysregulation of permeability and vascular tone are the leading causes of endothelial dysfunction in preterm infants. However, the available evidence regarding endothelial physiology and damage is limited in neonates compared to adults. Herein, we discuss the current knowledge on endothelial dysfunction in the infectious/inflammatory and dysfunctional placentation endotypes of prematurity, summarizing their molecular features, available biomarkers, and clinical impact. Furthermore, knowledge gaps, shadows, and future research perspectives are highlighted.
Preterm birth is defined as birth prior to 37 weeks of gestational age (GA). Prematurity, particularly in the lowest gestational age ranges, is the leading cause of infant mortality and morbidity, accounting for up to 35% of all deaths among newborns, and also may lead to severe complications as well as prolonged stays in neonatal intensive care units (NICUs) (1–3). Although considerable progress has been made in identifying factors that contribute to preterm birth and its complications, several gaps remain in our understanding of the pathogenesis, pathology, and clinical management of the condition. Preterm delivery is induced by the interaction of various medical and genetic causes but also by environmental and socioeconomic determinants (4, 5). In recent years, two broad pathophysiological patterns have been identified as responsible for the majority of very and extremely preterm births (i.e., GA less than 32 weeks): intrauterine infection/inflammation (Table 1) and dysfunctional placentation (Table 2) (10). These patterns involve specific cellular and biochemical pathways, representing two distinct endotypes of prematurity (11, 12).

Table 1. Infectious/inflammatory endotype.

Table 2. Dysfunctional placentation endotype.
The term endotype refers to “a subtype of a condition, which is defined by a distinct functional or pathophysiological mechanism” (13). Endotypes are thus a different form of classification from clinical phenotypes and describe distinct disease entities with a defining etiology and/or a consistent pathophysiological mechanism (13, 14). Although the term endotype has been applied mainly to studies of respiratory and allergic diseases, its use is increasingly extended to other areas of medicine (14–19). In line with the approach of personalized medicine, endotyping will facilitate the design of more targeted therapeutic and prognostic approaches (14–17, 19). Various epidemiological studies have suggested a correlation between endotypes of prematurity and neonatal phenotypes, although much remains to be explained about how antenatal events determine the outcomes (8, 9, 11, 20).
The vascular endothelium (VE) is a monolayer of cells covering the luminal surface of blood vessels (21). The VE plays an essential role in vascular homeostasis and regulates several pathways that could reveal information about the neonatal endotype-phenotype correlation (22). In particular, endothelial cells and pericytes interact with soluble mediators of inflammation, modulate vascular permeability and coagulation balance, drive angiogenesis, and regulate vascular tone through continuous endocrine-paracrine activity (23, 24). Conversely, VE undergoes changes in its properties when exposed to injuries, shifting towards a proinflammatory, procoagulative, and vasoconstrictive phenotype, resulting in the condition known as endothelial dysfunction (25).
Over the past two decades, endothelial pathways have proved to be an effective diagnostic and therapeutic target in several adult inflammatory diseases, increasing interest in the role played by VE in many medical areas, including prematurity-related complications. To date, examples of this research are already available, such as antiangiogenetic therapy with anti-Vascular Endothelial Growth Factor (VEGF) drugs for retinopathy of prematurity (ROP) (26). Moreover, several studies documented endothelial dysfunction in former premature adults, with increased incidence of high blood pressure, type 2 diabetes, and stroke, suggesting that prematurity may be the origin of an unfavorable endothelial phenotype (20, 27–29). Interestingly, the placental and pregnancy studies revealed that this endothelial dysfunction may have started very early and be related to the adverse intrauterine environment induced by the pregnancy complications that lead to preterm birth (30, 31).
Gaps in knowledge about endothelial dysfunction in preterm infants limit progress on prevention, early diagnosis, and treatment of the condition. Furthermore, characterizing endothelial profiles in fetuses and preterm infants is highly challenging due to the “endothelial developmental physiology” process (32–34). Several factors, such as GA, partial arterial oxygen pressure (PaO2), and placental vascular growth factors, expose the immature VE to continuous dynamic events during the antenatal period. Additionally, simultaneous maturation of the hemostatic and immune systems, closely connected to the VE, adds additional complexity to understanding the process (35, 36). Thus, we should not consider fetal endothelial physiology as static but consider it a developmental process that continues in postnatal life and progressively approaches the adult phenotype.
Classifying prematurity into endotypes can provide a systematic model for approaching the physiopathology of “endothelial phenotypes” commonly encountered in NICU, such as systemic hypotension, pulmonary hypertension, generalized tissue edema, and thrombotic or hemorrhagic events.
Rather than surveying all known mechanisms of endothelial damage in prematurity, this review focuses on the main endothelial features of the infectious/inflammatory and dysfunctional placentation endotypes, highlighting their peculiar pathways and clinical implications in the neonatal setting.
The infectious/inflammatory endotype is associated with preterm labor, premature rupture of membranes (PROM) out of labor, cervical insufficiency, and placental abruption (10, 37, 38) (Table 1). These conditions, even though distinct, are frequently associated with chorioamnionitis, defined as an acute inflammation of the membranes and chorion of the placenta, typically caused by an ascending microbial infection (39, 40). Of note, placental abruption can also be associated with placental vascular abnormalities and severe hypertensive disorders, requiring a heedful anamnestic evaluation to classify the endotype properly (6, 7).
The incidence of chorioamnionitis becomes higher as GA decreases and can be as high as 90% in infants at 24 weeks of GA and then progressively decreases to 10% in late preterm infants (GA > 34 weeks) (41). In addition, a growing number of individual studies and meta-analyses show an association between chorioamnionitis and increased risk of developing complications of prematurity such as necrotizing enterocolitis (NEC), early- and late-onset sepsis, periventricular leukomalacia, bronchopulmonary dysplasia (BPD), ROP, intraventricular hemorrhage, or patent ductus arteriosus (8, 42–51). However, a common conundrum in perinatal medicine is the extent to which chorioamnionitis, as well as other pregnancy complications, harms preterm infants through triggering prematurity or through disturbances in fetal homeostasis and development.
During chorioamnionitis, the infection invades the decidua and the amniotic cavity leading to neutrophilic infiltration of the chorioamnion and causing an increase in proinflammatory cytokine concentrations in the amniotic fluid (52, 53). As inflammation progresses, immune cells can penetrate blood vessels and infiltrate the umbilical cord, resulting in funisitis, which reflects a systemic inflammatory response syndrome called Fetal Inflammatory Response Syndrome (FIRS) (54).
The term FIRS was initially coined in 1998 to identify an elevated concentration of fetal plasma interleukin (IL)-6 in response to an intra-amniotic infection in spontaneous preterm labor (54). However, the type, kinetics, and effects of its numerous proinflammatory markers were characterized only subsequently and still remain incomplete. Moreover, due to practical and ethical limitations, most of the information available derives from preclinical studies (30).
The incidence of FIRS, likewise for histological chorioamnionitis, is inversely related to GA, and its extent is proportional to the maturity of the immune system (55). In this context, premature infants exposed to an infectious/inflammatory endotype may manifest a broad spectrum of endothelial dysfunction phenotypes. However, the FIRS model is the condition that best characterizes the endothelial response of the fetus and preterm infant to this group of adverse antenatal conditions. Therefore, in the following paragraphs, we will summarize the endothelial pathways with an increased risk of being affected by a systemic inflammatory response syndrome in the setting of an infectious/inflammatory endotype of prematurity (Figure 1, Table 1).
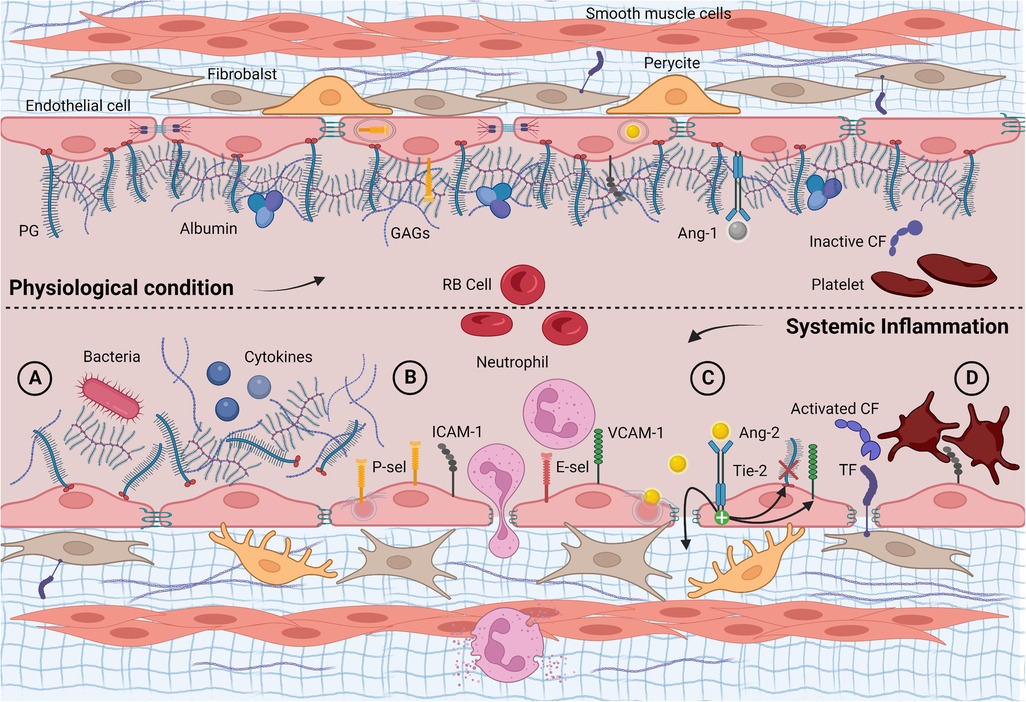
Figure 1. Longitudinal section of a blood vessel: endothelial phenotype in physiological conditions and infectious/inflammatory endotype. (A) Acute glycocalyx shedding following endogenous and/or exogenous proinflammatory stimuli; (B) unveiling and overexpression of membrane glycoproteins, adhesion, and diapedesis of neutrophils; (C) Ang-2 exocytosis with increased vascular leakage, further glycocalyx shedding via heparanase and up-regulation of adhesion molecules; (D) subendothelial TF exposure, activation, and consumption of clotting factors and platelets. Ang-1, angiopoietin 1; Ang-2, angiopoietin 2; CF, clotting factor; E-sel, E-selectin; GAGs, glycosaminoglycans; PG, proteoglycan; P-sel, P-selectin; RB cell, red blood cell; TF, tissue factor; Tie-2, tyrosine kinase receptor. Created with BioRender.com.
Communication between the bloodstream and endothelial surface plays a crucial role during systemic inflammation. Under physiological conditions, this interaction is mediated by the endothelial glycocalyx, an antiadhesive and anticoagulant layer that covers the luminal endothelial surface (56). The endothelial glycocalyx is mainly composed of proteoglycans, macromolecules with a protein core anchored to the endothelial surface or released soluble in the extracellular compartment (57). A large amount of glycosaminoglycan, mostly heparan sulfate and hyaluronan, gives the endothelial glycocalyx great hydrophilicity, which allows for trapping a fixed noncirculating plasma volume of approximately one liter in adults (58). The result is a soluble gel-like network, whose geometry changes depending on the vascular bed, volume load, and blood flow rates, and that acts as a mechanotransducer, translating shear stress into cellular signaling processes (59). Moreover, the negative charge of glycosaminoglycans repels endothelial glycocalyx interaction with negatively charged proteins as well as white and red blood cells and platelets, maintaining an oncotic gradient that limits vascular leakage (60). Conversely, entrapped plasma- and endothelium-derived proteins help in reducing hydraulic conductivity, maintaining an anticoagulant phenotype, and guaranteeing cell signaling (61).
In addition, glycoproteins are essential for leukocyte recruitment, platelet activation, and hemostatic function (59). The endothelial glycocalyx coating provides a physical barrier that prevents the indiscriminate activation of all these pathways. However, it undergoes significant changes in acute inflammation, especially when exposed to lipopolysaccharides (LPS) (62). Following neutrophilic activation, the release of reactive oxygen species (ROS), reactive nitrogen species (RNS), and tumor necrosis factor-α (TNF-α) lead to glycocalyx degradation via heparanase (62, 63). This process promotes endothelial glycocalyx shedding and severely affects its functions (64).
Experimental models showed that endothelial glycocalyx is expressed early in ontogenesis for normal vasculogenesis and angiogenesis (65, 66). Many proteins constituting the glycosaminoglycans are expressed even before the onset of vascular circulation (67). Furthermore, during embryological development, endothelial glycocalyx appears from the very first moment blood flow emerges, and the glycosaminoglycan structure shows a regular composition similar to the adult in a quail embryo model (68). A human in vivo study investigated endothelial glycocalyx thickness by testing the perfused boundary region, an inverse measure of glycocalyx thickness, in a population of full-term and preterm infants (69). Interestingly, premature neonates in the lowest GA group had the largest endothelial glycocalyx dimensions at birth, which highlights the relevance of endothelial glycocalyx in the development of the circulatory system (69).
All inflammation conditions, such as sepsis, autoinflammatory diseases, surgery, and trauma, can cause endothelial glycocalyx damage (60, 70). Although the evidence is not robust, this process may also occur in preterm infants. At follow-up, the cohort of neonates in the lowest GA group showed a progressive thinning of the glycocalyx, expressed as an increase of the perfused boundary region, reaching lower glycocalyx dimensions compared to infants of higher GA. This suggests that peri- and postnatal inflammatory insults may damage glycocalyx integrity (69). Moreover, degradation of the endothelial glycocalyx layer (shedding) has also been demonstrated in a cohort of neonates undergoing cardiac surgery (71). Interestingly, high preoperative doses of corticosteroids seem to confer protection to the endothelial glycocalyx through unresolved mechanisms, opening the hypothesis of benefits on endothelial glycocalyx induced by antenatal steroids (72, 73).
Syndecan-1 (an anchored protein core), heparan sulfate, and hyaluronan are the three well-established biomarkers of endothelial glycocalyx disruption (60, 70). The products of glycosaminoglycan chains increased in the plasma and urine of patients during and after the inflammatory event, generating a further amplification of the inflammatory cascade. Vascular loss of syndecan-1 resulted in reduced expression of shear stress compensating pathways, such as nitric oxide (NO) production (74). Moreover, hyaluronan fragments activate nuclear factor-κβ (NF-κβ) signaling, which produces proinflammatory cytokines and chemokines. At the same time, serum heparan sulfate induces myocardial dysfunction, which plays a crucial role in systemic hypotension and organ hypoperfusion during neonatal septic shock (64, 75, 76).
Clinical studies targeting the perinatal role of the endothelial glycocalyx in the infectious/inflammatory endotype may have great diagnostic and therapeutic perspectives for patient-tailored care. The endothelial glycocalyx breakdown could be crucial in septic preterms and infants with non-infectious hemodynamic imbalances (76). Unstable hemodynamic conditions are expected in very preterm infants in the first days of life and are usually treated with high intravenous volume administration and inotropes (77). Nevertheless, fluid overload may promote adverse outcomes through endothelial glycocalyx disruption, worsening of myocardial dysfunction, vascular leak, systemic inflammatory cascade, and organ damage (60). Likewise, excessive use of vasoactive amines has a potentially harmful effect on endothelial glycocalyx (78).
Interestingly, preclinical data showed that colloids favor endothelial glycocalyx restoration (79–83). Therefore, besides volume, the choice of fluid therapy could also affect the endothelial phenotype (79). Indeed, albumin is a physiological component of endothelial glycocalyx and possesses pleiotropic physiological activities, including antioxidant effects and protective effects on vessel wall integrity (84). However, so far, there are no supporting trials for albumin first-line use in this clinical setting (85). In conclusion, targeted therapies that preserve endothelial glycocalyx shedding may potentially have protective cardiovascular effects. However, these clinical applications still need a long way to go.
As a result of damage to the endothelial glycocalyx, adhesion molecules are exposed to the denuded VE, which induces the recruitment of leukocytes and platelets, promoting endothelial dysfunction (86, 87). The endothelial glycoproteins responsible for leukocyte adhesion mainly belong to the selectins family (P- and E-selectin) and the endothelial immunoglobulin superfamily, such as the intercellular adhesion molecule 1 (ICAM-1) and the vascular cell adhesion molecule 1 (VCAM- 1). Their constitutive expression varies significantly between tissues, and their kinetic upregulation is diversified (88). Especially, ICAM-1 has a broad physiological expression in most vascular beds, P-selectin is stored and rapidly mobilized by Weibel-Palade bodies, while E-selectin and VCAM-1 have a low constitutive expression, rising mainly during inflammatory stimulus via nuclear transcription (89–91).
The complex cascade mechanisms triggered by adhesion molecules are finely regulated (89, 91, 92). Briefly, initial adhesive interactions between leukocytes and venular endothelium are carried out by P- and E-selectins via low-affinity interactions that lead to leukocyte tethering and rolling on the endothelial surface. Subsequently, leukocyte activation reinforces this interaction, exposing leukocyte integrins and binding the endothelial immunoglobulin superfamily (89). Then, having reached a stable adhesion, platelet endothelial cell adhesion molecule-1 (PECAM-1) and other endothelial immunoglobulins permit leukocyte diapedesis through the intercellular spaces (93).
The systemic inflammatory response, including FIRS, intensifies this process by overexpressing adhesion molecules. The mechanism involves several molecular pathways: cytokines, endotoxins, shear stress, ROS, RNS, and imbalance of glucose and cholesterol are the main biological regulators (90).
Despite some differences due to a reduced baseline exposure of glycoproteins and delayed recruitment of neutrophils in tissue, evidence suggests that the adhesion molecules of preterm infants are not anergic to proinflammatory stimuli (76). Ex vivo studies demonstrated that human umbilical vein endothelial cells (HUVECs) of extremely low birth weight infants (ELBW) could upregulate E-selectin, VCAM-1, and ICAM-1 in response to TNF-α and IL-1β (94). This setting is reliable as the fetal sheep undergoes cytokine changes as early as 5 h after the intraamniotic injection of LPS and up to 15 days after the onset of inflammation (95). Furthermore, the comparative study of neonatal and adult selectins showed effective functionality in vitro, with no differences in rolling speed or number of adherent cells (96). However, in vivo characterization of adhesion molecules on premature neonates has been scarcely investigated.
The cord blood sample of ELBW infants showed enhanced exposure and shedding of soluble E-selectin and ICAM-1 in patients with funisitis, demonstrating that chorioamnionitis with funisitis induces a significant glycoprotein overexpression with soluble forms increased in the fetal circulation (97).
The transient vascular shedding of glycoprotein suggested using soluble adhesion molecules as a surrogate marker to assess the endothelial inflammatory response severity. Several studies on term infants with sepsis exploited this process, finding greater sensitivity and predictivity than C-reactive protein alone (98–100). However, the clinical use of these markers in preterm infants is limited by physiological fetal immaturity and a lack of normality curves adapted for GA (96, 101, 102). This mainly concerns selectins, whose transcription factor, NF-κβ has a GA-dependent capacity (94). Amelio et al. supported these findings by showing lower plasma E-selectin levels in preterm infants compared to a control group of healthy full-term infants, despite having a predominantly inflammatory endotype with higher VCAM-1 level (103). Therefore, the clinical application of these markers requires further research.
Emerging evidence points toward a “double barrier concept” in which both the endothelial cell layer with its multiple intercellular junctions and endothelial glycocalyx play a role in maintaining the vascular barrier (104). Although both layers must be compromised to obtain a major increase in vascular permeability, a slight increase in cell permeability in a damaged endothelial glycocalyx may be sufficient to generate vascular leakage (105). In addition, paracellular and transcellular pathways are linked to water and macromolecule leakage from the endothelial cell layer (106). The first is regulated by endothelial cleft, with ROS and TNF-α stimulating the opening of tight, adherent, and gap junctions (107, 108). The second is controlled by proinflammatory mediators known to accelerate caveolae formation, leading to trans-endothelial hyperpermeability (109). Several growth factors and receptor systems are involved in these processes. However, the angiopoietin (Ang)/tyrosine kinase receptor with immunoglobulin and epidermal growth factor homology domain 2 (Tie-2) signaling pathway seems particularly relevant.
The angiopoietin family (Ang-1, Ang-2, Ang-3, Ang-4) was initially discovered as a regulator of angiogenesis and subsequently demonstrated a key role in several inflammatory diseases through modulation of the Tie-2 receptor (22). Ang-1 is a potent stabilizer of endothelial cell contacts, modulating the VEGF/NO pathway (110). It is constitutively expressed by pericytes, smooth muscle cells, and fibroblasts and promotes endothelial quiescence by enhancing cell survival and downregulating proinflammatory and pro-coagulating pathways (111). In addition, Ang-1 has cardiovascular protective effects, showing a therapeutic impact in the post-ischemic myocardium by improving cardiac function, scar thickness, and scar area fraction (112).
Ang-2 is a competitive inhibitor of Ang-1. It is undetectable in quiescent VE but increases dramatically via rapid exosomal secretion during hypoxic or inflammatory injury. Assisted by VEGF, Ang-2 promotes destabilization of both layers of the vascular barrier: it reduces endothelial glycocalyx thickness by activating heparanase and increasing soluble heparan sulfate. Moreover, Ang-2 subverts intercellular junctions inducing paracellular gap formation and increasing the surface expression of cell adhesion molecules, worsening edema and tissue neutrophil recruitment (105, 113). All these properties indicate Ang-2 as an important mediator of LPS-induced endothelial glycocalyx damage (105, 114). Ang-3 in mice and Ang-4 in humans are interspecies orthologs (115). They are both Tie-2 agonists, although they are still poorly characterized (116).
Plasma imbalance of the Ang-2/Ang-1 ratio has been demonstrated in acute lung injury, sepsis, trauma, and multi-organ dysfunction in full-term neonates, children, and adults, also exhibiting a predictive value on mortality (117–120).
Ang/Tie-2 pathway is directly involved in the infectious/inflammatory endotype of prematurity. A placental immunohistochemical study demonstrated a significant increase in Tie-2 angiopoietin receptors in chorioamnionitis and funisitis (121). Furthermore, a higher concentration of Ang-2 was found in the amniotic fluid of women with preterm labor and intra-amniotic inflammation compared to women with preterm birth without inflammation (122). A plasma imbalance towards Ang-2 was shown from the first hours of life in a cohort of very preterm infants, suggesting an antenatal origin of this endothelial phenotype (103). Moreover, Ang-2 concentrations were inversely related to GA, suggesting a co-dependence between prematurity and endothelial permeability (103, 122).
Finally, the Ang/Tie-2 system may play a role in the close correlation between the infectious/inflammatory endotype and adverse neurological outcomes. During FIRS, the impaired integrity of the blood-brain barrier (BBB) increases the permeability to proinflammatory cells and cytokines, which causes a direct injury to neurons and oligodendrocytes of the developing brain (123). Preclinical models show that Ang-2 increases BBB permeability via paracellular and transcellular routes (124). Furthermore, Ang-2 levels were upregulated in patients affected by cerebrovascular disorders associated with BBB alterations (124). However, to our knowledge, there are still no studies on the correlation between Ang-2 and neurological outcomes in preterms.
Hemostatic changes in infants with an infectious/inflammatory endotype also deserve attention. Besides leukocyte diapedesis and vascular leakage, vascular surface mutation activates platelets and coagulation. Moreover, inflammation downregulates anticoagulant proteins and inhibits the fibrinolytic system, according to the immunothrombosis process (125, 126). On the other hand, in advanced septic states, an indiscriminate activation leads to the consumption of hemostatic factors, resulting in disseminated intravascular coagulation (DIC) and subsequent hypocoagulability (127). However, the evidence for a hyper- or hypocoagulant endothelial phenotype during FIRS is currently scarce.
Despite expressing different concentrations of pro- and anticoagulant factors, premature infants are hemostatically competent, and viscoelastic tests of infants with systemic inflammation have shown significant but not univocal alterations of the hemostatic balance (128–136). In addition, recent studies have observed a lengthening of coagulation times during neonatal sepsis and post-surgery, so the bleeding risk in these newborns must be considered (130). However, as in adults, an opposite prothrombotic switch cannot be excluded in the initial phase of the infectious/inflammatory endotype (137).
In this regard, the high levels of IL-1β observed in FIRS showed procoagulant and permeabilizing effects on HUVECs through a rapid and dose-dependent increase in tissue factor activity (138). Similarly, HPA increased coagulation activity via the stimulation of tissue factor expression in endothelial cells (139). Therefore, exploring the tissue factor pathway could bridge coagulation, angiopoietins, and endothelial glycocalyx shedding in the perinatal period (103). Further viscoelastic studies will help to better characterize the perinatal hemostatic peculiarities related to this endotype.
The placenta is the maternal-fetal interface, and its vascular development is crucial for maintaining proper fetal growth through efficiently delivering nutrients and oxygen (140). The unique characteristics of the placental and umbilical cord vascular endothelium provide information on the events occurring on both sides. Therefore, several studies have investigated the physiology and diseases of placentogenesis, providing helpful information on the aberrant pathways leading to several disorders of pregnancy (141, 142).
The dysfunctional placentation endotype mainly encloses hypertensive disorders of pregnancy, including gestational hypertension, preeclampsia, and eclampsia, as well as fetal growth restriction (FGR), defined as the failure of a fetus to achieve its genetic growth potential (10, 141).
Severe dysfunctional placentation endotype may require the earlier birth of the infant, balancing the neonatal risks of prematurity with the fetal risk of carrying on a pregnancy burdened by pathology.
These conditions are among the most significant contributing factor to perinatal morbidity and mortality, affecting up to 12%–16% of pregnant patients (143, 144). Furthermore, FGR is the leading risk factor for stillbirth and may increase the risk of developing several complications of prematurity, including BPD and ROP (9, 145–148).
Preeclampsia and FGR are often associated and share similar pathogenesis, such as reduced trophoblast invasion and impaired perfusion in the uteroplacental compartment, leading to harmful effects to the fetus (149, 150). Usually, the expression of integrins, cadherins, and metalloproteases allows the cytotrophoblasts to penetrate the uterine wall between the 6th and 8th week of gestation, differentiating into an endothelial phenotype and transforming the spiral arteries into large vessels with low vascular resistance (140). These steps increase the blood flow directed to the intervillous space and the fetus during early development. Conversely, failure of this process leads to narrow, high-strength, high-pressure spiral arteries, resulting in chronic placental hypoperfusion (151). The hallmarks of this impaired uteroplacental blood flow are maternal vascular underperfusion lesions on placental histology (152).
The mismatch between fetoplacental blood demand and adequate uteroplacental supply promotes blood flow turbulence, hyperinflammatory state, and placental endothelial dysfunction (151, 153, 154). An increase in maternal inflammatory cytokines, acute phase proteins, and systemic endothelitis is well documented in severe preeclampsia, mainly when associated with FGR (155, 156). On the fetal side, the consequence is a preferential blood flow redistribution to the vital organs (brain, myocardium, and adrenal glands) at the expense of other organs (157). Furthermore, the high placental resistance associated with FGR may lead to increased cardiac afterload, resulting in myocardial remodeling and hypertrophy, increased vascular tone, vascular stiffness, and risk of perinatal hypotension and persistent pulmonary hypertension (157).
Despite overlaps with previously described inflammatory pathways, the different triggers and chronic exposure of this endotype lead to typical molecular and phenotypic features that are reviewed below and summarized in Figure 2 and Table 2.
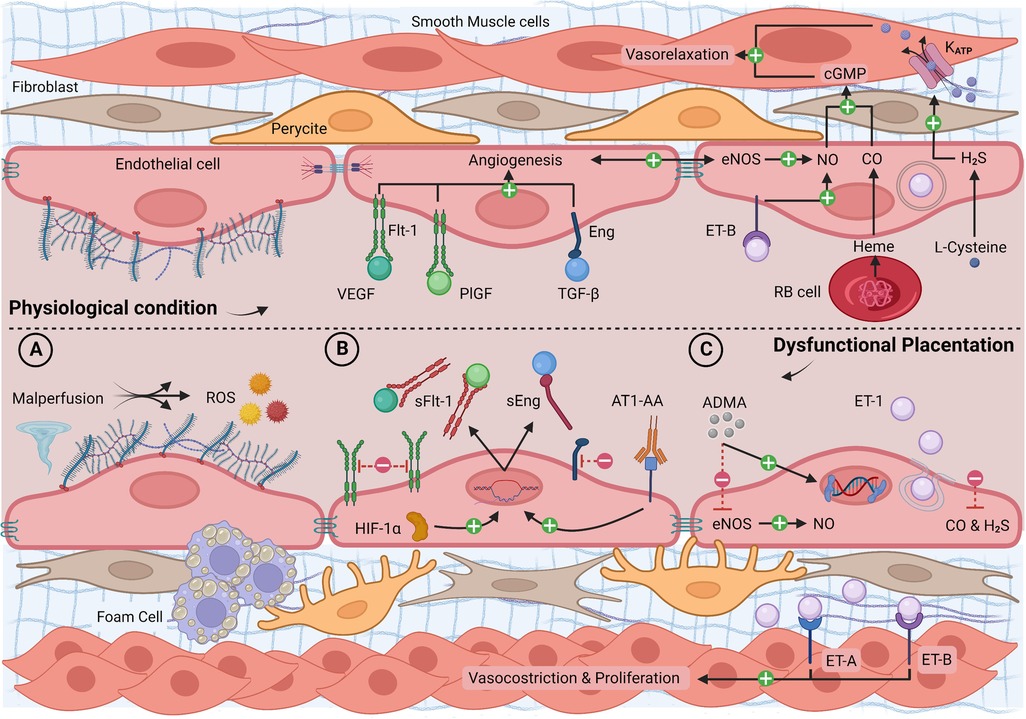
Figure 2. Longitudinal section of a blood vessel: endothelial phenotype in physiological conditions and dysfunctional placentation endotype. (A) Malperfusion phenomena with shear stress, hypoxia, reperfusion injury, atherosclerosis, and ROS production; (B) high levels of HIF-1α and AT1-AA with increased synthesis of sFlt-1 and sEng, reduced free circulating VEGF, PlGF, TGF-β, and reduced angiogenesis; (C) high levels of ADMA and ET-1 and low levels of gasotransmitters with increased vasoconstriction, smooth muscle layer proliferation, and epigenetic mutations. ADMA, asymmetric dimethylarginine; AT1-AA, angiotensin II type 1 agonistic receptor autoantibody; cGMP, cyclic guanosine monophosphate; CO, carbon monoxide; eNOS, endothelial nitric oxide synthase; ET-1, endothelin-1; ET-A/B, endothelin-1 receptor A/B; H2S, hydrogen sulfide; HIF-1α, hypoxia-inducible factor 1α; Katp, potassium channel; PlGF, placental growth factor; RB cell, red blood cell; ROS, reactive oxygen species; sEng, soluble endoglin; sFlt-1, soluble fms-like tyrosine kinase-1; TGF-β, transforming growth factor-β; NO, nitric oxide; VEGF, vascular endothelial growth factor. Created with BioRender.com.
Deficient spiral arterial remodeling leads to high velocity and pulsatile inflow in the intervillous space, greater intermittent perfusion of the placenta, atherotic changes with vascular wall deposition of foam cells, platelet aggregation, and fibrinoid necrosis (150). In addition, impaired perfusion induces hypoxia/reperfusion injury with increased oxidative stress (158).
Highly reactive free radicals can negatively affect lipids, proteins, or nucleic acids (158). ROS and RNS play an important physiological role at normal levels, acting as second messengers in many signaling pathways (159, 160). However, cell damage and cell death are triggered when ROS and RNS production overwhelms antioxidant capacity (158, 161–168).
Pregnancy represents, per se, a state of oxidative stress resulting from the increased metabolic activity in the placental mitochondria and the reduced scavenging power of antioxidants but this imbalance increases as a consequence of placental dysfunction (169–171). Indeed, free radical species inevitably rise with cascade effects on inflammation, angiogenesis, and vascular contractility. The overexpression of free radicals and acute phase proteins is an established process in pregnancies complicated by preeclampsia or FGR, involving even severe cardiovascular effects (172, 173).
Several studies investigated, through cord blood samples analysis, the oxidant and antioxidant state of newborns of preeclamptic women, with or without FGR, compared to newborns from physiological pregnancies (156, 174–183). Different analytical methods and some biomarkers have been used to measure oxidative stress, such as direct detection of ROS (e.g., hydrogen peroxide, H2O2), indirect research of the peroxidation products of lipids and proteins (e.g., malondialdehyde and protein carbonyl levels), and activity evaluation of antioxidant enzymes (e.g., glutathione peroxidase). Many studies found a cord blood profile comparable to the maternal one, with a higher level of ROS and lower antioxidant activity in the offspring of preeclamptic women, suggesting that the mother and fetus are subjected to a pro-oxidant environment (156, 174–176). Moreover, preclinical data from ewe's model support that chronic hypoxia exposure significantly increases fetal plasma urate concentration from oxidative stress (184).
Neonates of preeclamptic women with associated hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome showed higher oxidative stress up to seven days of life compared to a group with preeclampsia but without HELLP (185). Hence, fetal oxidative stress appears to be proportional to the extent of maternal endothelial dysfunction and remains even after the interruption of placental circulation. In addition, preterm infants have a lower antioxidant reserve, being more susceptible to oxidative stress injury (186). Moreover, for the same GA, lower-weight infants have less tolerance to oxidative stress, especially when comparing small for gestation age (SGA) and adequate for gestational age (AGA) (187–189).
The direct correlation between oxidative stress and early neonatal outcomes has been the subject of a few studies with inconclusive results (179, 180, 190). Conversely, late consequences of oxidative stress from dysfunctional placentation have been more extensively investigated. Lipid peroxidation levels remain higher until adolescence in infants exposed to perinatal complications, including an adverse intrauterine environment (191). Moreover, FGR rat models proved that impairment of hepatic and muscular mitochondrial oxidative processes predisposes to improper glucose metabolism, insulin resistance, and diabetes typical of these infants (192–194). Finally, preliminary data show that the maternal total antioxidant status may predict infant motor development at one year of corrected age in preeclampsia (178).
All these findings report a persistent cellular inheritance in subjects exposed to placental dysfunction, suggesting developmental programming influenced by oxidative stress (195). In this direction, studies indicate that using a ROS-targeted therapy with antioxidants may be a promising approach in perinatal medicine, especially for fetal neuroprotection (196). However, many proposed drugs are still under investigation for routine clinical use, such as vitamins, melatonin, lycopene, selenium, acetylsalicylic acid, L-ergothioneine, and mitochondria-specific drugs (197–210).
The main effect of malperfusion and oxidative stress is the decreasing activity of three pivotal factors, VEGF, placental growth factor (PlGF), and transforming growth factor-β (TGF-β), which play a crucial role in placental and fetal angiogenesis (31, 211). Indeed, soluble fms-like tyrosine kinase-1 (sFlt-1), an inhibitory receptor for VEGF and PlGF, and soluble endoglin (sEng), a co-receptor for TGF-β, are over-expressed during malperfusion contributing to reducing the activity of VEGF, PlGF, and TGF-β (31). Their antiangiogenic role is exerted by binding their circulating ligands and preventing proangiogenic effects on native endothelial cell-surface receptors (31, 154). In turn, the persistent increase in sFlt-1 and the decrease in VEGF can worsen oxidative stress and impair vascular reactivity, contributing to endothelial dysfunction (212).
There are mainly two pathways that regulate angiogenic factors imbalance. First, hypoxia-inducible factors (HIFs) are oxygen-sensitive transcriptional factors that regulate the expression of the multiple vascular growth factors involved in angiogenesis (213). Specifically, HIF-1α is a subunit of HIF-1 that is rapidly inactivated and degraded in normoxia, stimulating the sFlt-1 and sEng transcription (214). In a transgenic mouse model, embryonic overexpression of HIF-1α leads to placental malformations (215). HIF-1α increases in women with preeclampsia and FGR (216, 217). Second, there is a typical deviation of the renin-angiotensin-aldosterone system with reduced plasma renin activity and the development of circulatory volume in preeclampsia. Characterization of this pathway identified a pathognomonic circulatory protein in preeclamptic women: angiotensin II type 1 agonistic receptor autoantibody (AT1-AA), inhibiting trophoblastic invasiveness by stimulating the expression of sFlt-1 and sEng (218, 219).
Over the past ten years, several studies investigated the imbalance of angiogenic factors in placental dysfunction, demonstrating increased concentrations of sFLT-1 and sENG and contextual reduction of PlGF in the plasma of women with preeclampsia and/or FGR compared to healthy controls (143, 220, 221).
Maternal plasma concentration of sFlt-1 was correlated with doppler abnormalities of the uterine and umbilical arteries on prenatal ultrasound evaluations in women with PE or FGR (222). In addition, a low plasma angiogenic index-1 (PlGF/sFLT-1 ratio) at 20–23 weeks of gestation identified patients with high maternal vascular underperfusion lesions and a higher incidence of preterm delivery (223). Moreover, the reduced placental angiogenic index-1, measured by enzyme-linked immunosorbent assays, strongly increased the risk of fetal death (224). Likewise, the increased sFlt-1/PlGF ratio correlated with fetal death, FGR, HELLP, and placental abruption in PE with high oxidative stress (225). These findings confirm that maternal endothelial dysfunction severity influences fetal and neonatal outcomes.
Angiogenic biomarkers in feto-placental circulation are less studied. However, sFLT-1 appears to increase in preeclamptic women's neonates, while data on VEGF levels are conflicting (211, 226–231). In FGR, an overall reduction in proangiogenic factors is established (226, 229). A low circulating level of insulin-like growth factor-1 (IGF-1), an anabolic hormone with proangiogenic effects, is among the documented metabolic problems of infants with a history of FGR (232).
An extensive analysis of angiogenic factors in a large cohort of infants with both endotypes of prematurity evaluated the correlation between plasma angiogenic factors at birth and placental histology (152). Two findings of this study are particularly relevant: (1) VEGF and PlGF were decreased in infants from pregnancies with maternal vascular underperfusion lesions; (2) more than 50% of severe maternal vascular underperfusion lesions were associated with FGR, while acute inflammation was associated with no or mild maternal vascular underperfusion lesions. Therefore, although preclinical and clinical data showed a reduction of proangiogenic factors even in the infectious/inflammatory endotype, a severe antiangiogenic imbalance is characteristic of chronic inflammation induced by dysfunctional placentation and should be considered in FGR (233, 234).
The consequences of angiogenic inhibition are significant. Pulmonary vasculature immaturity and poor lung growth secondary to FGR increase respiratory complications in extreme prematurity, causing high mortality in this group of infants (235). BPD development is extensively established in FGR, particularly when BPD is associated with pulmonary hypertension (11, 145, 152, 236–238). Conversely, preserving vascular growth with an sFlt-1 inhibitor or recombinant IGF-1 promotes alveolarization and sustains the architecture of the distal airspace in preclinical models (239–241). In this regard, the disruption of angiogenesis appears to be a connecting bridge between dysfunctional placentation and BPD. Additionally, the prenatal impairment of VEGF, PlGF, and IGF-1 pathways, associated with the increase in oxidative stress, mimics the pathogenesis of the first stage of ROP, suggesting a direct association between ROP and dysfunctional placentation (242, 243).
Placental dysfunction is accompanied by marked alterations in vasoactive mediators, particularly gasotransmitters and endothelin-1 (ET-1) (244–246). Gasotransmitters, such as NO, hydrogen sulfide (H2S), and carbon monoxide (CO), are endogenously-produced, volatile molecules characterized by a high reactivity and free diffusion through cell membranes (247). Gasotransmitters exert a synergistic action of smooth muscle relaxation: NO and CO through an increase of cyclic guanosine monophosphate (cGMP) and H2S through KATP channels mediated hyperpolarization (247, 248).
In particular, NO is the main mediator of endothelium-dependent vasodilation, which plays a pivotal role in maintaining vascular tone (160, 249–251). It is generated via L-arginine oxidation by a family of NO synthase (NOS) enzymes (252). All isoforms of NOS can be endogenously inhibited by asymmetric dimethylarginine (ADMA), a methylated product of L-arginine synthesized by protein arginine methyltransferase (PRMT) and degraded by dimethylarginine dimethylaminohydrolase (DDAH) (226, 253).
Under physiological conditions, maternal gasotransmitters concentrations are significantly higher throughout pregnancy than in non-pregnant women (246, 254). They participate in trophoblast invasion and apoptosis, regulate placental blood pressure, exert antiplatelet properties in the intervillous space, enhance VEGF, and reduce sFLT-1 expression (246, 255, 256). Conversely, their reduced bioavailability, especially of NO, is one of the leading causes of abnormal placentation and endothelial dysfunction in preeclampsia (254).
ET-1 concentrations are also pathological in dysfunctional placentation (257). ET-1 is a vasoconstrictor and mitogenic peptide constitutively synthesized by endothelial cells and syncytiotrophoblasts. It can be overexpressed, mobilizing its stores in the Weibel-Palade bodies by several proinflammatory stimuli, such as hypoxia, cytokines, ROS, and shear stress (258). ET-1 induces cell proliferation and vasoconstriction binding ET-A and ET-B membrane receptors on vascular smooth muscle cells, which can persist for several hours, despite the short plasma half-life of ET-1 (257). Differently, endothelial ET-B receptors lead to NO-mediated vasodilation (259).
In general, gasotransmitters production and bioactivity are significantly reduced in preeclamptic women (246). However, there are important differences in the studies that focus on NO levels and endothelial NOS (eNOS) expression in preeclampsia (260). Moreover, nitrate and nitrite used to evaluate NO production are affected by diet and plasma clearance (244). Nevertheless, a meta-analysis showed an overall reduction in maternal serum NO level in preeclampsia and an increased risk of the condition related to genetic variations of eNOS (261). Additionally, ADMA and cGMP levels are consistently higher and lower, respectively, before and during the onset of preeclampsia, indicating decreased maternal NO bioactivity (255). These findings are supported by experimental evidence of impaired NO-mediated relaxation in vessels exposed to plasma of preeclamptic subjects and by the occurrence of FGR in pregnant rats following NOS inhibition (262–266).
Besides maternal NO levels, fetal and neonatal levels are reduced. A study by Aikio et al. found perinatal undetectable nitrate and nitrite values on airway specimens in a group of very preterm infants affected by maternal preeclampsia compared to higher values in infants with infectious/inflammatory endotype (267). Moreover, recent clinical data show higher levels of ADMA and PRMT-1 in the placenta and cord blood of FGR patients, associated with a decrease in ADMA's metabolizer DDAH-1, NO, and eNOS (226).
In addition, ex vivo experiments on HUVECs showed a reduction in angiogenesis with an increase in sFlt-1 after stimulation with high doses of ADMA (226). Interestingly, the link between NO bioavailability and vascular growth restriction is confirmed in cases of PROM-associated oligohydramnios. Although PROM is classified in the other endotype, it has pathophysiological overlaps with these processes. When oligohydramnios lasts more than a week, it can lead to lung growth restriction, locally simulating the lung condition of FGR (268). In the same study by Aikio et al., a group of very preterm infants affected by PROM with hypoxic respiratory failure showed undetectable perinatal nitrate and nitrite levels on airway specimens and pulmonary hypertension responsive to inhaled NO. For these reasons, the pulmonary endothelial consequences discussed in this endotype should be considered when approaching a newborn affected by long-lasting oligohydramnios (267).
In contrast to the vasodilatory gasotransmitters, ET-1 levels are increased in preeclampsia in correlation with the severity of the condition (257). Furthermore, ET-1 levels correlate with increased levels of sFLT-1 and sENG, and HIF-1 appears to regulate its synthesis (245). ET-1 physiologically contributes to high fetal pulmonary vascular resistance. Preclinical models of chronic fetal pulmonary hypertension showed a 3-fold higher ET-1, decreased ET-B receptor-mediated vasodilation, enhanced ET-A receptor-mediated vasoconstriction, and impaired angiogenesis (269, 270). Conversely, chronic intrauterine antagonism of the ET-A receptor reduces fetal pulmonary artery pressure, right ventricular hypertrophy, and distal muscularization of small pulmonary arteries, reducing pulmonary vascular resistance at birth and confirming the link between ET-1 and FGR (271).
Finally, increasing evidence indicates epigenetic pathways regulating the alterations in endogenous vasoactive mediators in the fetus (272). Histone modifications regulate eNOS expression in human umbilical artery endothelial cells (HUAEC) in patients with placental insufficiency and lead to an ET-1 increase in pulmonary vascular endothelial cells with persistent effects weeks after birth in a rat model of FGR (273, 274). In turn, the placental nitrergic system controls epigenetic mechanisms, including the function of histone deacetylase (256). Experimental data suggest that NO, ADMA, and PRMT imbalances could participate in fetal epigenetic reprogramming, providing a biological link between dysfunctional placentation and adult cardiovascular disease in FGR infants (256). This evidence may explain the long-term cardiovascular effects typical of the placental dysfunction endotype and strengthens maternal treatment as the best preventive strategy. However, research in the neonatal period is also necessary to design appropriate preventive and/or therapeutic strategies for endothelial dysfunction. Identifying epigenetic biomarkers, such as plasma microRNAs, would allow early screening and follow-up for high-risk individuals, avoiding long-term complications (275).
Preterm birth syndrome impairs numerous endothelial pathways with potentially severe early and late neonatal adverse outcomes. Endotyping prematurity is a pathophysiological-based approach useful for neonatologists to predict the main endothelial features of each preterm infant, frame its early and late cardiovascular risk factors, anticipate and prevent associated hemodynamic complications, and design personalized therapeutic strategies.
Of note, the classification into endotypes is a dichotomous model and, therefore, has inherent limits to consider. First, in endothelial phenotypes, elements common to both endotypes coexist. Any cause of premature birth inevitably leads to an overlap of intrauterine inflammatory pathways, making them characteristic and not exclusive to a single endotype. On the other hand, each endotype has an inter-individual variability depending on trigger severity, timing, fetal development, and genetic background. Lastly, there is always the superimposition of postnatal “hits”, such as acidosis, hypoxia, hyperglycemia, mechanical ventilation, and sepsis, which further compromise endothelial function (60, 76, 236, 276). Hence, identifying, understanding, and quantifying the contribution of pre- and postnatal processes to endothelial dysfunction is challenging and requires an early endothelial evaluation to limit confounding factors (103). Although many fetal processes are not fully understood, the available endothelial biomarkers to perform this evaluation in NICU are numerous (76, 103, 152, 175, 260). They await further studies to validate their diagnostic and therapeutic potential. We believe that advancing endothelial characterization could be a promising way to provide patient-tailored care to the most vulnerable newborns.
GSA, LP, GR, IA, SG, FG, EV, and GC contributed to the study's conception and design. GSA, LP, VC, FMa, MT, GCe, AT, VP, and GC contributed to the study's methodology, investigation, and data curation. GSA, LP, GR, IA, SG, and GC wrote the original draft preparation of the manuscript. LP, GSA, GR, IA, SG, AT, VC, FM, FG, GCe, EV, and GC wrote, reviewed, and edited the manuscript. GSA, EV, LP, GR, and GC contributed to designing Figures 1, 2. FMo and GC contributed equally to the visualization of the manuscript. GCa contributed to the supervision and project administration of the study. All authors contributed to the article and approved the submitted version.
This study was (partially) funded by the Italian Ministry of Health – Current Research IRCCS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Walani SR. Global burden of preterm birth. Int J Gynaecol Obstet. (2020) 150(1):31–3. doi: 10.1002/ijgo.13195
2. Lee AC, Blencowe H, Lawn JE. Small babies, big numbers: global estimates of preterm birth. Lancet Glob Health. (2019) 7(1):e2–3. doi: 10.1016/S2214-109X(18)30484-4
3. Purisch SE, Gyamfi-Bannerman C. Epidemiology of preterm birth. Semin Perinatol. (2017) 41(7):387–91. doi: 10.1053/j.semperi.2017.07.009
4. Di Renzo GC, Tosto V, Giardina I. The biological basis and prevention of preterm birth. Best Pract Res Clin Obstet Gynaecol. (2018) 52:13–22. doi: 10.1016/j.bpobgyn.2018.01.022
5. Menon R. Spontaneous preterm birth, a clinical dilemma: etiologic, pathophysiologic and genetic heterogeneities and racial disparity. Acta Obstet Gynecol Scand. (2008) 87(6):590–600. doi: 10.1080/00016340802005126
6. Tikkanen M. Placental abruption: epidemiology, risk factors and consequences. Acta Obstet Gynecol Scand. (2011) 90(2):140–9. doi: 10.1111/j.1600-0412.2010.01030.x
7. Ananth CV, Oyelese Y, Srinivas N, Yeo L, Vintzileos AM. Preterm premature rupture of membranes, intrauterine infection, and oligohydramnios: risk factors for placental abruption. Obstet Gynecol. (2004) 104(1):71–7. doi: 10.1097/01.AOG.0000128172.71408.a0
8. Gagliardi L, Rusconi F, Bellu R, Zanini R, Italian Neonatal N. Association of maternal hypertension and chorioamnionitis with preterm outcomes. Pediatrics. (2014) 134(1):e154–61. doi: 10.1542/peds.2013-3898
9. Gagliardi L, Rusconi F, Da Fre M, Mello G, Carnielli V, Di Lallo D, et al. Pregnancy disorders leading to very preterm birth influence neonatal outcomes: results of the population-based ACTION cohort study. Pediatr Res. (2013) 73(6):794–801. doi: 10.1038/pr.2013.52
10. McElrath TF, Hecht JL, Dammann O, Boggess K, Onderdonk A, Markenson G, et al. Pregnancy disorders that lead to delivery before the 28th week of gestation: an epidemiologic approach to classification. Am J Epidemiol. (2008) 168(9):980–9. doi: 10.1093/aje/kwn202
11. Pierro M, Villamor-Martinez E, van Westering-Kroon E, Alvarez-Fuente M, Abman SH, Villamor E. Association of the dysfunctional placentation endotype of prematurity with bronchopulmonary dysplasia: a systematic review, meta-analysis and meta-regression. Thorax. (2022) 77(3):268–75. doi: 10.1136/thoraxjnl-2020-21648534301740
12. Pierro M, Van Mechelen K, van Westering-Kroon E, Villamor-Martínez E, Villamor E. Endotypes of prematurity and phenotypes of bronchopulmonary dysplasia: toward personalized neonatology. J Pers Med. (2022) 12(5):687. doi: 10.3390/jpm12050687
13. Lotvall J, Akdis CA, Bacharier LB, Bjermer L, Casale TB, Custovic A, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol. (2011) 127(2):355–60. doi: 10.1016/j.jaci.2010.11.037
14. Hamilos DL. Chronic rhinosinusitis endotyping: sharpening the focus on inflammation. J Allergy Clin Immunol. (2016) 137(5):1457–9. doi: 10.1016/j.jaci.2016.03.011
15. Battaglia M, Ahmed S, Anderson MS, Atkinson MA, Becker D, Bingley PJ, et al. Introducing the endotype concept to address the challenge of disease heterogeneity in type 1 diabetes. Diabetes Care. (2020) 43(1):5–12. doi: 10.2337/dc19-0880
16. Malhotra A, Mesarwi O, Pepin J-L, Owens RL. Endotypes and phenotypes in obstructive sleep apnea. Curr Opin Pulm Med. (2020) 26(6):609. doi: 10.1097/MCP.0000000000000724
17. Cardell L-O, Stjärne P, Jonstam K, Bachert C. Endotypes of chronic rhinosinusitis: impact on management. J Allergy Clin Immunol. (2020) 145(3):752–6. doi: 10.1016/j.jaci.2020.01.019
18. Neyton LP, Zheng X, Skouras C, Doeschl-Wilson A, Gutmann MU, Uings I, et al. Molecular patterns in acute pancreatitis reflect generalizable endotypes of the host response to systemic injury in humans. Ann Surg. (2022) 275(2), e453–e62. doi: 10.1097/SLA.0000000000003974
19. Wong HR, Sweeney TE, Lindsell CJ. Simplification of a septic shock endotyping strategy for clinical application. Am J Respir Crit Care Med. (2017) 195(2):263–5. doi: 10.1164/rccm.201607-1535LE
20. Bavineni M, Wassenaar TM, Agnihotri K, Ussery DW, Luscher TF, Mehta JL. Mechanisms linking preterm birth to onset of cardiovascular disease later in adulthood. Eur Heart J. (2019) 40(14):1107–12. doi: 10.1093/eurheartj/ehz025
21. Krüger-Genge A, Blocki A, Franke R-P, Jung F. Vascular endothelial cell biology: an update. Int J Mol Sci. (2019) 20(18):4411. doi: 10.3390/ijms20184411
22. Fiedler U, Augustin HG. Angiopoietins: a link between angiogenesis and inflammation. Trends Immunol. (2006) 27(12):552–8. doi: 10.1016/j.it.2006.10.004
23. Jeon BH. Endothelial dysfunction: from pathophysiology to novel therapeutic approaches. Biomedicines. (2021) 9(11):1571. doi: 10.3390/biomedicines9111571
24. Granger DN, Kubes P. The microcirculation and inflammation: modulation of leukocyte-endothelial cell adhesion. J Leukoc Biol. (1994) 55(5):662–75. doi: 10.1002/jlb.55.5.662
25. Burger D, Touyz RM. Cellular biomarkers of endothelial health: microparticles, endothelial progenitor cells, and circulating endothelial cells. J Am Soc Hypertens. (2012) 6(2):85–99. doi: 10.1016/j.jash.2011.11.003
26. Sankar MJ, Sankar J, Chandra P. Anti-vascular endothelial growth factor (VEGF) drugs for treatment of retinopathy of prematurity. Cochrane Database Syst Rev. (2018) 1:CD009734. doi: 10.1002/14651858.CD009734.pub3
27. Bassareo PP, Fanos V, Puddu M, Demuru P, Cadeddu F, Balzarini M, et al. Reduced brachial flow-mediated vasodilation in young adult ex extremely low birth weight preterm: a condition predictive of increased cardiovascular risk? J Matern Fetal Neonatal Med. (2010) 23(Suppl 3):121–4. doi: 10.3109/14767058.2010.506811
28. Franco MC, Christofalo DM, Sawaya AL, Ajzen SA, Sesso R. Effects of low birth weight in 8- to 13-year-old children: implications in endothelial function and uric acid levels. Hypertension. (2006) 48(1):45–50. doi: 10.1161/01.HYP.0000223446.49596.3a
29. Skilton MR, Viikari JS, Juonala M, Laitinen T, Lehtimaki T, Taittonen L, et al. Fetal growth and preterm birth influence cardiovascular risk factors and arterial health in young adults: the cardiovascular risk in young Finns study. Arterioscler Thromb Vasc Biol. (2011) 31(12):2975–81. doi: 10.1161/ATVBAHA.111.234757
30. Jung E, Romero R, Yeo L, Diaz-Primera R, Marin-Concha J, Para R, et al. The fetal inflammatory response syndrome: the origins of a concept, pathophysiology, diagnosis, and obstetrical implications. Semin Fetal Neonatal Med. (2020) 25(4):101146. doi: 10.1016/j.siny.2020.101146
31. Helmo FR, Lopes AMM, Carneiro A, Campos CG, Silva PB, Monteiro MLGdR, et al. Angiogenic and antiangiogenic factors in preeclampsia. Pathol Res Pract. (2018) 214(1):7–14. doi: 10.1016/j.prp.2017.10.021
32. Okuda KS, Hogan BM. Endothelial cell dynamics in vascular development: insights from live-imaging in zebrafish. Front Physiol. (2020) 11:842. doi: 10.3389/fphys.2020.00842
33. Parker TA, le Cras TD, Kinsella JP, Abman SH. Developmental changes in endothelial nitric oxide synthase expression and activity in ovine fetal lung. Am J Physiol Lung Cell Mol Physiol. (2000) 278(1):L202–8. doi: 10.1152/ajplung.2000.278.1.L202
34. McCafferty C, Busuttil-Crellin X, Cai T, Monagle P, Goldenberg NA, Ignjatovic V. Plasma proteomic analysis reveals age-specific changes in platelet- and endothelial cell-derived proteins and regulators of plasma coagulation and fibrinolysis. J Pediatr. (2020) 221S:S29–36. doi: 10.1016/j.jpeds.2020.01.051
35. Jaffray J, Young G. Developmental hemostasis: clinical implications from the fetus to the adolescent. Pediatr Clin North Am. (2013) 60(6):1407–17. doi: 10.1016/j.pcl.2013.08.003
36. Ygberg S, Nilsson A. The developing immune system - from foetus to toddler. Acta Paediatr. (2012) 101(2):120–7. doi: 10.1111/j.1651-2227.2011.02494.x
37. Brown R, Gagnon R, Delisle M-F. No. 373-cervical insufficiency and cervical cerclage. J Obstet Gynaecol Can. (2019) 41(2):233–47. doi: 10.1016/j.jogc.2018.08.009
38. Oh KJ, Romero R, Park JY, Lee J, Conde-Agudelo A, Hong J-S, et al. Evidence that antibiotic administration is effective in the treatment of a subset of patients with intra-amniotic infection/inflammation presenting with cervical insufficiency. Am J Obstet Gynecol. (2019) 221(2):140.e1–e18. doi: 10.1016/j.ajog.2019.03.017
39. Heymans C, de Lange IH, Hutten MC, Lenaerts K, de Ruijter NJE, Kessels L, et al. Chronic intra-uterine Ureaplasma parvum infection induces injury of the enteric nervous system in ovine fetuses. Front Immunol. (2020) 11:189. doi: 10.3389/fimmu.2020.00189
40. Romero R, Gomez-Lopez N, Winters AD, Jung E, Shaman M, Bieda J, et al. Evidence that intra-amniotic infections are often the result of an ascending invasion–a molecular microbiological study. J. Perinat. Med. (2019) 47(9):915–31. doi: 10.1515/jpm-2019-0297
41. Kim CJ, Romero R, Chaemsaithong P, Chaiyasit N, Yoon BH, Kim YM. Acute chorioamnionitis and funisitis: definition, pathologic features, and clinical significance. Am J Obstet Gynecol. (2015) 213(4 Suppl):S29–52. doi: 10.1016/j.ajog.2015.08.040
42. Villamor-Martinez E, Lubach GA, Rahim OM, Degraeuwe P, Zimmermann LJ, Kramer BW, et al. Association of histological and clinical chorioamnionitis with neonatal sepsis among preterm infants: a systematic review, meta-analysis, and meta-regression. Front Immunol. (2020) 11:972. doi: 10.3389/fimmu.2020.00972
43. Been JV, Lievense S, Zimmermann LJ, Kramer BW, Wolfs TG. Chorioamnionitis as a risk factor for necrotizing enterocolitis: a systematic review and meta-analysis. J Pediatr. (2013) 162(2):236–42.e2. doi: 10.1016/j.jpeds.2012.07.012
44. Toti P, De Felice C. Chorioamnionitis and fetal/neonatal brain injury. Biol Neonate. (2001) 79(3–4):201–4. doi: 10.1159/000047091
45. Abiramalatha T, Bandyopadhyay T, Ramaswamy VV, Shaik NB, Thanigainathan S, Pullattayil AK, et al. Risk factors for periventricular leukomalacia in preterm infants: a systematic review, meta-analysis, and GRADE-based assessment of certainty of evidence. Pediatr Neurol. (2021) 124:51–71. doi: 10.1016/j.pediatrneurol.2021.08.003
46. Villamor-Martinez E, Álvarez-Fuente M, Ghazi AM, Degraeuwe P, Zimmermann LJ, Kramer BW, et al. Association of chorioamnionitis with bronchopulmonary dysplasia among preterm infants: a systematic review, meta-analysis, and metaregression. JAMA Network Open. (2019) 2(11):e1914611. doi: 10.1001/jamanetworkopen.2019.14611
47. Villamor-Martinez E, Cavallaro G, Raffaeli G, Mohammed Rahim OM, Gulden S, Ghazi AM, et al. Chorioamnionitis as a risk factor for retinopathy of prematurity: an updated systematic review and meta-analysis. PLoS One. (2018) 13(10):e0205838. doi: 10.1371/journal.pone.0205838
48. Villamor-Martinez E, Fumagalli M, Mohammed Rahim O, Passera S, Cavallaro G, Degraeuwe P, et al. Chorioamnionitis is a risk factor for intraventricular hemorrhage in preterm infants: a systematic review and meta-analysis. Front Physiol. (2018) 9:1253. doi: 10.3389/fphys.2018.01253
49. Behbodi E, Villamor-Martínez E, Degraeuwe PL, Villamor E. Chorioamnionitis appears not to be a risk factor for patent ductus arteriosus in preterm infants: a systematic review and meta-analysis. Sci Rep. (2016) 6(1):1–10. doi: 10.1038/srep37967
50. Cavallaro G, Villamor-Martínez E, Filippi L, Mosca F, Villamor E. Probiotic supplementation in preterm infants does not affect the risk of retinopathy of prematurity: a meta-analysis of randomized controlled trials. Sci Rep. (2017) 7(1):13014. doi: 10.1038/s41598-017-13465-2
51. Villamor-Martínez E, Pierro M, Cavallaro G, Mosca F, Kramer B, Villamor E. Probiotic supplementation in preterm infants does not affect the risk of bronchopulmonary dysplasia: a meta-analysis of randomized controlled trials. Nutrients. (2017) 9(11):1197. doi: 10.3390/nu9111197
52. Yoon BH, Romero R, Kim CJ, Jun JK, Gomez R, Choi JH, et al. Amniotic fluid interleukin-6: a sensitive test for antenatal diagnosis of acute inflammatory lesions of preterm placenta and prediction of perinatal morbidity. Am J Obstet Gynecol. (1995) 172(3):960–70. doi: 10.1016/0002-9378(95)90028-4
53. Romero R, Mazor M, Brandt F, Sepulveda W, Avila C, Cotton DB, et al. Interleukin-1 alpha and interleukin-1 beta in preterm and term human parturition. Am J Reprod Immunol. (1992) 27(3-4):117–23. doi: 10.1111/j.1600-0897.1992.tb00737.x
54. Gomez R, Romero R, Ghezzi F, Yoon BH, Mazor M, Berry SM. The fetal inflammatory response syndrome. Am J Obstet Gynecol. (1998) 179(1):194–202. doi: 10.1016/S0002-9378(98)70272-8
55. Pugni L, Pietrasanta C, Acaia B, Merlo D, Ronchi A, Ossola MW, et al. Chorioamnionitis and neonatal outcome in preterm infants: a clinical overview. J Matern Fetal Neonatal Med. (2016) 29(9):1525–9. doi: 10.3109/14767058.2015.1053862
56. Van Teeffelen JW, Brands J, Stroes ES, Vink H. Endothelial glycocalyx: sweet shield of blood vessels. Trends Cardiovasc Med. (2007) 17(3):101–5. doi: 10.1016/j.tcm.2007.02.002
57. Alphonsus CS, Rodseth RN. The endothelial glycocalyx: a review of the vascular barrier. Anaesthesia. (2014) 69(7):777–84. doi: 10.1111/anae.12661
58. Johansson PI, Stensballe J, Rasmussen LS, Ostrowski SR. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann Surg. (2011) 254(2):194–200. doi: 10.1097/SLA.0b013e318226113d
59. Reitsma S, Slaaf DW, Vink H. van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. (2007) 454(3):345–59. doi: 10.1007/s00424-007-0212-8
60. Richter RP, Payne GA, Ambalavanan N, Gaggar A, Richter JR. The endothelial glycocalyx in critical illness: a pediatric perspective. Matrix Biol Plus. (2022) 14:100106. doi: 10.1016/j.mbplus.2022.100106
61. Dogne S, Flamion B. Endothelial glycocalyx impairment in disease: focus on hyaluronan shedding. Am J Pathol. (2020) 190(4):768–80. doi: 10.1016/j.ajpath.2019.11.016
62. Huang X, Han S, Liu X, Wang T, Xu H, Xia B, et al. Both UFH and NAH alleviate shedding of endothelial glycocalyx and coagulopathy in LPS-induced sepsis. Exp Ther Med. (2020) 19(2):913–22. doi: 10.3892/etm.2019.8285
63. Iba T, Levy JH. Derangement of the endothelial glycocalyx in sepsis. J Thromb Haemost. (2019) 17(2):283–94. doi: 10.1111/jth.14371
64. Qu J, Cheng Y, Wu W, Yuan L, Liu X. Glycocalyx impairment in vascular disease: focus on inflammation. Front Cell Dev Biol. (2021) 9:730621. doi: 10.3389/fcell.2021.730621
65. Noguer O, Villena J, Lorita J, Vilaro S, Reina M. Syndecan-2 downregulation impairs angiogenesis in human microvascular endothelial cells. Exp Cell Res. (2009) 315(5):795–808. doi: 10.1016/j.yexcr.2008.11.016
66. Harfouche R, Hentschel DM, Piecewicz S, Basu S, Print C, Eavarone D, et al. Glycome and transcriptome regulation of vasculogenesis. Circulation. (2009) 120(19):1883–92. doi: 10.1161/CIRCULATIONAHA.108.837724
67. Klewer SE, Yatskievych T, Pogreba K, Stevens MV, Antin PB, Camenisch TD. Has2 expression in heart forming regions is independent of BMP signaling. Gene Expr Patterns. (2006) 6(5):462–70. doi: 10.1016/j.modgep.2005.11.005
68. Henderson-Toth CE, Jahnsen ED, Jamarani R, Al-Roubaie S, Jones EA. The glycocalyx is present as soon as blood flow is initiated and is required for normal vascular development. Dev Biol. (2012) 369(2):330–9. doi: 10.1016/j.ydbio.2012.07.009
69. Puchwein-Schwepcke A, Artmann S, Rajwich L, Genzel-Boroviczeny O, Nussbaum C. Effect of gestational age and postnatal age on the endothelial glycocalyx in neonates. Sci Rep. (2021) 11(1):3133. doi: 10.1038/s41598-021-81847-8
70. Puchwein-Schwepcke A, Genzel-Boroviczeny O, Nussbaum C. The endothelial glycocalyx: physiology and pathology in neonates, infants and children. Front Cell Dev Biol. (2021) 9:733557. doi: 10.3389/fcell.2021.733557
71. Pesonen E, Keski-Nisula J, Andersson S, Palo R, Salminen J, Suominen PK. High-dose methylprednisolone and endothelial glycocalyx in paediatric heart surgery. Acta Anaesthesiol Scand. (2016) 60(10):1386–94. doi: 10.1111/aas.12785
72. Chappell D, Hofmann-Kiefer K, Jacob M, Rehm M, Briegel J, Welsch U, et al. TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res Cardiol. (2009) 104(1):78–89. doi: 10.1007/s00395-008-0749-5
73. Chappell D, Jacob M, Hofmann-Kiefer K, Bruegger D, Rehm M, Conzen P, et al. Hydrocortisone preserves the vascular barrier by protecting the endothelial glycocalyx. Anesthesiology. (2007) 107(5):776–84. doi: 10.1097/01.anes.0000286984.39328.96
74. Voyvodic PL, Min D, Liu R, Williams E, Chitalia V, Dunn AK, et al. Loss of syndecan-1 induces a pro-inflammatory phenotype in endothelial cells with a dysregulated response to atheroprotective flow. J Biol Chem. (2014) 289(14):9547–59. doi: 10.1074/jbc.M113.541573
75. Martin L, Peters C, Schmitz S, Moellmann J, Martincuks A, Heussen N, et al. Soluble heparan sulfate in Serum of septic shock patients induces mitochondrial dysfunction in murine cardiomyocytes. Shock. (2015) 44(6):569–77. doi: 10.1097/SHK.0000000000000462
76. Pietrasanta C, Pugni L, Ronchi A, Bottino I, Ghirardi B, Sanchez-Schmitz G, et al. Vascular endothelium in neonatal sepsis: basic mechanisms and translational opportunities. Front Pediatr. (2019) 7:340. doi: 10.3389/fped.2019.00340
77. Dempsey EM, Barrington KJ, Marlow N, O'Donnell CPF, Miletin J, Naulaers G, et al. Hypotension in preterm infants (HIP) randomised trial. Arch Dis Child Fetal Neonatal Ed. (2021) 106(4):398–403. doi: 10.1136/archdischild-2020-320241
78. Johansson PI, Stensballe J, Ostrowski SR. Shock induced endotheliopathy (SHINE) in acute critical illness - a unifying pathophysiologic mechanism. Crit Care. (2017) 21(1):25. doi: 10.1186/s13054-017-1605-5
79. Damiani E, Ince C, Orlando F, Pierpaoli E, Cirioni O, Giacometti A, et al. Effects of the infusion of 4% or 20% human Serum albumin on the skeletal muscle microcirculation in endotoxemic rats. PLoS One. (2016) 11(3):e0151005. doi: 10.1371/journal.pone.0151005
80. Horstick G, Lauterbach M, Kempf T, Bhakdi S, Heimann A, Horstick M, et al. Early albumin infusion improves global and local hemodynamics and reduces inflammatory response in hemorrhagic shock. Crit Care Med. (2002) 30(4):851–5. doi: 10.1097/00003246-200204000-00023
81. Hoffmann JN, Vollmar B, Laschke MW, Inthorn D, Schildberg FW, Menger MD. Hydroxyethyl starch (130 kD), but not crystalloid volume support, improves microcirculation during normotensive endotoxemia. J Am Soc Anesthesiol. (2002) 97(2):460–70. doi: 10.1097/00000542-200208000-00025
82. de Carvalho H, Dorigo D, Bouskela E. Effects of ringer-acetate and ringer-dextran solutions on the microcirculation after LPS challenge: observations in the hamster cheek pouch. Shock. (2001) 15(2):157–62. doi: 10.1097/00024382-200115020-00013
83. Steinbauer M, Guba M, Büchner M, Farkas S, Anthuber M, Jauch K-W. Impact of polynitroxylated albumin (PNA) and tempol on ischemia/reperfusion injury: intravital microscopic study in the dorsal skinfold chamber of the Syrian golden hamster. Shock. (2000) 14(2):163–8. doi: 10.1097/00024382-200014020-00015
84. Vincent JL, De Backer D, Wiedermann CJ. Fluid management in sepsis: the potential beneficial effects of albumin. J Crit Care. (2016) 35:161–7. doi: 10.1016/j.jcrc.2016.04.019
85. Weiss SL, Peters MJ, Alhazzani W, Agus MSD, Flori HR, Inwald DP, et al. Surviving sepsis campaign international guidelines for the management of septic shock and sepsis-associated organ dysfunction in children. Intensive Care Med. (2020) 46(Suppl 1):10–67. doi: 10.1007/s00134-019-05878-6
86. Kayal S, Jais JP, Aguini N, Chaudiere J, Labrousse J. Elevated circulating E-selectin, intercellular adhesion molecule 1, and von Willebrand factor in patients with severe infection. Am J Respir Crit Care Med. (1998) 157(3 Pt 1):776–84. doi: 10.1164/ajrccm.157.3.9705034
87. Shuvaev VV, Tliba S, Nakada M, Albelda SM, Muzykantov VR. Platelet-endothelial cell adhesion molecule-1-directed endothelial targeting of superoxide dismutase alleviates oxidative stress caused by either extracellular or intracellular superoxide. J Pharmacol Exp Ther. (2007) 323(2):450–7. doi: 10.1124/jpet.107.127126
88. Henninger DD, Panes J, Eppihimer M, Russell J, Gerritsen M, Anderson DC, et al. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J Immunol. (1997) 158(4):1825–32.9029122
89. Granger DN, Senchenkova E. Inflammation and the microcirculation. In: Colloquium Series on Integrated Systems Physiology: From Molecule to Function. San Rafael (CA): Morgan & Claypool Life Sciences; (2010) 2(1): 1–87. doi: 10.4199/C00013ED1V01Y201006ISP008
90. Cook-Mills JM, Marchese ME, Abdala-Valencia H. Vascular cell adhesion molecule-1 expression and signaling during disease: regulation by reactive oxygen species and antioxidants. Antioxid Redox Signal. (2011) 15(6):1607–38. doi: 10.1089/ars.2010.3522
91. Smith CW. Endothelial adhesion molecules and their role in inflammation. Can J Physiol Pharmacol. (1993) 71(1):76–87. doi: 10.1139/y93-012
92. Smith CW. 3. Adhesion molecules and receptors. J Allergy Clin Immunol. (2008) 121(2 Suppl):S375–9; quiz S414. doi: 10.1016/j.jaci.2007.07.030
93. Woodfin A, Voisin MB, Nourshargh S. PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler Thromb Vasc Biol. (2007) 27(12):2514–23. doi: 10.1161/ATVBAHA.107.151456
94. Wisgrill L, Muck M, Wessely I, Berger A, Spittler A, Forster-Waldl E, et al. Endothelial cells of extremely premature infants display impaired immune response after proinflammatory stimulation. Pediatr Res. (2018) 83(1-1):128–34. doi: 10.1038/pr.2017.202
95. Kuypers E, Willems MG, Jellema RK, Kemp MW, Newnham JP, Delhaas T, et al. Responses of the spleen to intraamniotic lipopolysaccharide exposure in fetal sheep. Pediatr Res. (2015) 77(1):29–35. doi: 10.1038/pr.2014.152
96. Auvinen K, Jalkanen S, Salmi M. Expression and function of endothelial selectins during human development. Immunology. (2014) 143(3):406–15. doi: 10.1111/imm.12318
97. D'Alquen D, Kramer BW, Seidenspinner S, Marx A, Berg D, Groneck P, et al. Activation of umbilical cord endothelial cells and fetal inflammatory response in preterm infants with chorioamnionitis and funisitis. Pediatr Res. (2005) 57(2):263–9. doi: 10.1203/01.PDR.0000148713.48218.86
98. Hansen AB, Verder H, Staun-Olsen P. Soluble intercellular adhesion molecule and C-reactive protein as early markers of infection in newborns. J Perinat Med. (2000) 28(2):97–103. doi: 10.1515/JPM.2000.012
99. Zonneveld R, Jongman RM, Juliana A, Molema G, van Meurs M, Plotz FB. Serum concentrations of endothelial cell adhesion molecules and their shedding enzymes and early onset sepsis in newborns in Suriname. BMJ Paediatr Open. (2018) 2(1):e000312. doi: 10.1136/bmjpo-2018-000312
100. Austgulen R, Arntzen KJ, Haereid PE, Aag S, Dollner H. Infections in neonates delivered at term are associated with increased serum levels of ICAM-1 and E-selectin. Acta Paediatr. (1997) 86(3):274–80. doi: 10.1111/j.1651-2227.1997.tb08889.x
101. Salmi M, Jalkanen S. Developmental regulation of the adhesive and enzymatic activity of vascular adhesion protein-1 (VAP-1) in humans. Blood. (2006) 108(5):1555–61. doi: 10.1182/blood-2005-11-4599
102. Lorant DE, Li W, Tabatabaei N, Garver MK, Albertine KH. P-selectin expression by endothelial cells is decreased in neonatal rats and human premature infants. Blood. (1999) 94(2):600–9. doi: 10.1182/blood.V94.2.600
103. Amelio GS, Provitera L, Raffaeli G, Amodeo I, Gulden S, Cortesi V, et al. Proinflammatory endothelial phenotype in very preterm infants: a pilot study. Biomedicines. (2022) 10(5):1185. doi: 10.3390/biomedicines10051185
104. Rehm M, Zahler S, Lotsch M, Welsch U, Conzen P, Jacob M, et al. Endothelial glycocalyx as an additional barrier determining extravasation of 6% hydroxyethyl starch or 5% albumin solutions in the coronary vascular bed. Anesthesiology. (2004) 100(5):1211–23. doi: 10.1097/00000542-200405000-00025
105. Lukasz A, Hillgruber C, Oberleithner H, Kusche-Vihrog K, Pavenstadt H, Rovas A, et al. Endothelial glycocalyx breakdown is mediated by angiopoietin-2. Cardiovasc Res. (2017) 113(6):671–80. doi: 10.1093/cvr/cvx023
106. Muradashvili N, Tyagi R, Lominadze D. A dual-tracer method for differentiating transendothelial transport from paracellular leakage in vivo and in vitro. Front Physiol. (2012) 3:166. doi: 10.3389/fphys.2012.00166
107. Vestweber D, Winderlich M, Cagna G, Nottebaum AF. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol. (2009) 19(1):8–15. doi: 10.1016/j.tcb.2008.10.001
108. Levick JR, Michel CC. Microvascular fluid exchange and the revised starling principle. Cardiovasc Res. (2010) 87(2):198–210. doi: 10.1093/cvr/cvq062
109. Chen W, Gassner B, Borner S, Nikolaev VO, Schlegel N, Waschke J, et al. Atrial natriuretic peptide enhances microvascular albumin permeability by the caveolae-mediated transcellular pathway. Cardiovasc Res. (2012) 93(1):141–51. doi: 10.1093/cvr/cvr279
110. Ning W, Li S, Yang W, Yang B, Xin C, Ping X, et al. Blocking exosomal miRNA-153-3p derived from bone marrow mesenchymal stem cells ameliorates hypoxia-induced myocardial and microvascular damage by targeting the ANGPT1-mediated VEGF/PI3k/Akt/eNOS pathway. Cell Signal. (2021) 77:109812. doi: 10.1016/j.cellsig.2020.109812
111. Koh GY. Orchestral actions of angiopoietin-1 in vascular regeneration. Trends Mol Med. (2013) 19(1):31–9. doi: 10.1016/j.molmed.2012.10.010
112. Reis LA, Chiu LL, Wu J, Feric N, Laschinger C, Momen A, et al. Hydrogels with integrin-binding angiopoietin-1-derived peptide, QHREDGS, for treatment of acute myocardial infarction. Circ Heart Fail. (2015) 8(2):333–41. doi: 10.1161/CIRCHEARTFAILURE.114.001881
113. Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. (1997) 277(5322):55–60. doi: 10.1126/science.277.5322.55
114. Han S, Lee SJ, Kim KE, Lee HS, Oh N, Park I, et al. Amelioration of sepsis by TIE2 activation-induced vascular protection. Sci Transl Med. (2016) 8(335):335ra55. doi: 10.1126/scitranslmed.aad9260
115. Lee HJ, Cho CH, Hwang SJ, Choi HH, Kim KT, Ahn SY, et al. Biological characterization of angiopoietin-3 and angiopoietin-4. FASEB J. (2004) 18(11):1200–8. doi: 10.1096/fj.03-1466com
116. Heier JS, Singh RP, Wykoff CC, Csaky KG, Lai TYY, Loewenstein A, et al. The angiopoietin/tie pathway in retinal vascular diseases: a review. Retina. (2021) 41(1):1–19. doi: 10.1097/IAE.0000000000003003
117. Richter RP, Russell RT, Hu PJ, Uhlich RM, Swain TA, Kerby JD, et al. Plasma angiopoietin-2/-1 ratio is elevated and angiopoietin-2 levels correlate with plasma syndecan-1 following pediatric trauma. Shock. (2019) 52(3):340–6. doi: 10.1097/SHK.0000000000001267
118. Wright JK, Hayford K, Tran V, Al Kibria GM, Baqui A, Manajjir A, et al. Biomarkers of endothelial dysfunction predict sepsis mortality in young infants: a matched case-control study. BMC Pediatr. (2018) 18(1):118. doi: 10.1186/s12887-018-1087-x
119. Zonneveld R, Jongman R, Juliana A, Zijlmans W, Plotz F, Molema G, et al. Low Serum angiopoietin-1, high serum angiopoietin-2, and high Ang-2/Ang-1 protein ratio are associated with early onset sepsis in Surinamese newborns. Shock. (2017) 48(6):638–43. doi: 10.1097/SHK.0000000000000903
120. Calfee CS, Gallagher D, Abbott J, Thompson BT, Matthay MA, Network NA. Plasma angiopoietin-2 in clinical acute lung injury: prognostic and pathogenetic significance. Crit Care Med. (2012) 40(6):1731–7. doi: 10.1097/CCM.0b013e3182451c87
121. Kramer BW, Kaemmerer U, Kapp M, Herbst D, Marx A, Berg D, et al. Decreased expression of angiogenic factors in placentas with chorioamnionitis after preterm birth. Pediatr Res. (2005) 58(3):607–12. doi: 10.1203/01.PDR.0000175641.39056.7A
122. Pacora P, Romero R, Chaiworapongsa T, Kusanovic JP, Erez O, Vaisbuch E, et al. Amniotic fluid angiopoietin-2 in term and preterm parturition, and intra-amniotic infection/inflammation. J Perinat Med. (2009) 37(5):503–11. doi: 10.1515/JPM.2009.093
123. Yap V, Perlman JM. Mechanisms of brain injury in newborn infants associated with the fetal inflammatory response syndrome. Semin Fetal Neonatal Med. (2020) 25(4):101110. doi: 10.1016/j.siny.2020.101110
124. Gurnik S, Devraj K, Macas J, Yamaji M, Starke J, Scholz A, et al. Angiopoietin-2-induced blood-brain barrier compromise and increased stroke size are rescued by VE-PTP-dependent restoration of Tie2 signaling. Acta Neuropathol. (2016) 131(5):753–73. doi: 10.1007/s00401-016-1551-3
125. Levi M, van der Poll T. Coagulation and sepsis. Thromb Res. (2017) 149:38–44. doi: 10.1016/j.thromres.2016.11.007
126. Grover SP, Mackman N. Neutrophils, NETs, and immunothrombosis. Blood. (2018) 132(13):1360–1. doi: 10.1182/blood-2018-08-868067
127. Zhou W, Zhou W, Bai J, Ma S, Liu Q, Ma X. TEG In the monitoring of coagulation changes in patients with sepsis and the clinical significance. Exp Ther Med. (2019) 17(5):3373–82. doi: 10.3892/etm.2019.7342
128. Raffaeli G, Tripodi A, Cavallaro G, Cortesi V, Scalambrino E, Pesenti N, et al. Thromboelastographic profiles of healthy very low birthweight infants serially during their first month. Arch Dis Child Fetal Neonatal Ed. (2020) 105(4):412–8. doi: 10.1136/archdischild-2019-317860
129. Amelio GS, Raffaeli G, Amodeo I, Gulden S, Cortesi V, Manzoni F, et al. Hemostatic evaluation with viscoelastic coagulation monitor: a nicu experience. Front Pediatr. (2022) 10:910646. doi: 10.3389/fped.2022.910646
130. Sokou R, Giallouros G, Konstantinidi A, Pantavou K, Nikolopoulos G, Bonovas S, et al. Thromboelastometry for diagnosis of neonatal sepsis-associated coagulopathy: an observational study. Eur J Pediatr. (2018) 177(3):355–62. doi: 10.1007/s00431-017-3072-z
131. Grant HW, Hadley GP. Prediction of neonatal sepsis by thromboelastography. Pediatr Surg Int. (1997) 12(4):289–92. doi: 10.1007/BF01372152
132. Cortesi V, Raffaeli G, Amelio GS, Amodeo I, Gulden S, Manzoni F, et al. Hemostasis in neonatal ECMO. Front Pediatr. (2022) 10:988681. doi: 10.3389/fped.2022.988681
133. Raffaeli G, Ghirardello S, Passera S, Mosca F, Cavallaro G. Oxidative stress and neonatal respiratory extracorporeal membrane oxygenation. Front Physiol. (2018) 9:1739. doi: 10.3389/fphys.2018.01739
134. Tripodi A, Raffaeli G, Scalambrino E, Padovan L, Clerici M, Chantarangkul V, et al. Procoagulant imbalance in preterm neonates detected by thrombin generation procedures. Thromb Res. (2020) 185:96–101. doi: 10.1016/j.thromres.2019.11.013
135. Ghirardello S, Raffaeli G, Crippa BL, Gulden S, Amodeo I, Consonni D, et al. The thromboelastographic profile at birth in very preterm newborns with patent ductus arteriosus. Neonatology. (2020) 117(3):316–23. doi: 10.1159/000507553
136. Raffaeli G, Pesenti N, Cavallaro G, Cortesi V, Manzoni F, Amelio GS, et al. Optimizing fresh-frozen plasma transfusion in surgical neonates through thromboelastography: a quality improvement study. Eur J Pediatr. (2022) 181(5):2173–82. doi: 10.1007/s00431-022-04427-6
137. Scarlatescu E, Tomescu D, Arama SS. Sepsis-Associated coagulopathy. J Crit Care Med (Targu Mures). (2016) 2(4):156–63. doi: 10.1515/jccm-2016-0024
138. Puhlmann M, Weinreich DM, Farma JM, Carroll NM, Turner EM, Alexander HR Jr. Interleukin-1beta induced vascular permeability is dependent on induction of endothelial tissue factor (TF) activity. J Transl Med. (2005) 3:37. doi: 10.1186/1479-5876-3-37
139. Nadir Y, Brenner B, Zetser A, Ilan N, Shafat I, Zcharia E, et al. Heparanase induces tissue factor expression in vascular endothelial and cancer cells. J Thromb Haemost. (2006) 4(11):2443–51. doi: 10.1111/j.1538-7836.2006.02212.x
140. Maltepe E, Fisher SJ. Placenta: the forgotten organ. Annu Rev Cell Dev Biol. (2015) 31:523–52. doi: 10.1146/annurev-cellbio-100814-125620
141. Sun C, Groom KM, Oyston C, Chamley LW, Clark AR, James JL. The placenta in fetal growth restriction: what is going wrong? Placenta. (2020) 96:10–8. doi: 10.1016/j.placenta.2020.05.003
142. Qu H, Khalil RA. Vascular mechanisms and molecular targets in hypertensive pregnancy and preeclampsia. Am J Physiol Heart Circ Physiol. (2020) 319(3):H661–H81. doi: 10.1152/ajpheart.00202.2020
143. Kwiatkowski S, Dolegowska B, Kwiatkowska E, Rzepka R, Marczuk N, Loj B, et al. Maternal endothelial damage as a disorder shared by early preeclampsia, late preeclampsia and intrauterine growth restriction. J Perinat Med. (2017) 45(7):793–802. doi: 10.1515/jpm-2016-0178
144. Gardosi J, Kady SM, McGeown P, Francis A, Tonks A. Classification of stillbirth by relevant condition at death (ReCoDe): population based cohort study. Br Med J. (2005) 331(7525):1113–7. doi: 10.1136/bmj.38629.587639.7C
145. Mestan KK, Check J, Minturn L, Yallapragada S, Farrow KN, Liu X, et al. Placental pathologic changes of maternal vascular underperfusion in bronchopulmonary dysplasia and pulmonary hypertension. Placenta. (2014) 35(8):570–4. doi: 10.1016/j.placenta.2014.05.003
146. Chu A, Dhindsa Y, Sim MS, Altendahl M, Tsui I. Prenatal intrauterine growth restriction and risk of retinopathy of prematurity. Sci Rep. (2020) 10(1):17591. doi: 10.1038/s41598-020-74600-0
147. Filippi L, Cavallaro G, Berti E, Padrini L, Araimo G, Regiroli G, et al. Propranolol 0.2% eye micro-drops for retinopathy of prematurity: a prospective phase IIB study. Front Pediatr. (2019) 7:180. doi: 10.3389/fped.2019.00180
148. Filippi L, Cavallaro G, Bagnoli P, Dal Monte M, Fiorini P, Berti E, et al. Propranolol 0.1% eye micro-drops in newborns with retinopathy of prematurity: a pilot clinical trial. Pediatr Res. (2017) 81(2):307–14. doi: 10.1038/pr.2016.230
149. Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. (2010) 376(9741):631–44. doi: 10.1016/S0140-6736(10)60279-6
150. Burton GJ, Jauniaux E. Pathophysiology of placental-derived fetal growth restriction. Am J Obstet Gynecol. (2018) 218(2S):S745–S61. doi: 10.1016/j.ajog.2017.11.577
151. Chaiworapongsa T, Chaemsaithong P, Yeo L, Romero R. Pre-eclampsia part 1: current understanding of its pathophysiology. Nat Rev Nephrol. (2014) 10(8):466–80. doi: 10.1038/nrneph.2014.102
152. Mestan KK, Gotteiner N, Porta N, Grobman W, Su EJ, Ernst LM. Cord blood biomarkers of placental maternal vascular underperfusion predict bronchopulmonary dysplasia-associated pulmonary hypertension. J Pediatr. (2017) 185:33–41. doi: 10.1016/j.jpeds.2017.01.015
153. Lash GE, Otun HA, Innes BA, Kirkley M, De Oliveira L, Searle RF, et al. Interferon-gamma inhibits extravillous trophoblast cell invasion by a mechanism that involves both changes in apoptosis and protease levels. FASEB J. (2006) 20(14):2512–8. doi: 10.1096/fj.06-6616com
154. Maynard SE, Karumanchi SA. Angiogenic factors and preeclampsia. Semin Nephrol. (2011) 31(1):33–46. doi: 10.1016/j.semnephrol.2010.10.004
155. Tosun M, Celik H, Avci B, Yavuz E, Alper T, Malatyalioglu E. Maternal and umbilical serum levels of interleukin-6, interleukin-8, and tumor necrosis factor-alpha in Normal pregnancies and in pregnancies complicated by preeclampsia. J Matern Fetal Neonatal Med. (2010) 23(8):880–6. doi: 10.3109/14767051003774942
156. Catarino C, Santos-Silva A, Belo L, Rocha-Pereira P, Rocha S, Patricio B, et al. Inflammatory disturbances in preeclampsia: relationship between maternal and umbilical cord blood. J Pregnancy. (2012) 2012:684384. doi: 10.1155/2012/684384
157. Malhotra A, Allison BJ, Castillo-Melendez M, Jenkin G, Polglase GR, Miller SL. Neonatal morbidities of fetal growth restriction: pathophysiology and impact. Front Endocrinol. (2019) 10:55. doi: 10.3389/fendo.2019.00055
158. Burton GJ. Oxygen, the Janus gas; its effects on human placental development and function. J Anat. (2009) 215(1):27–35. doi: 10.1111/j.1469-7580.2008.00978.x
159. Droge W. Free radicals in the physiological control of cell function. Physiol Rev. (2002) 82(1):47–95. doi: 10.1152/physrev.00018.2001
160. Villamor E, Moreno L, Mohammed R, Pérez-Vizcaíno F, Cogolludo A. Reactive oxygen species as mediators of oxygen signaling during fetal-to-neonatal circulatory transition. Free Radic Biol Med. (2019) 142:82–96. doi: 10.1016/j.freeradbiomed.2019.04.008
161. Adams L, Franco MC, Estevez AG. Reactive nitrogen species in cellular signaling. Exp Biol Med. (2015) 240(6):711–7. doi: 10.1177/1535370215581314
162. Joo EH, Kim YR, Kim N, Jung JE, Han SH, Cho HY. Effect of endogenic and exogenic oxidative stress triggers on adverse pregnancy outcomes: preeclampsia, fetal growth restriction, gestational diabetes mellitus and preterm birth. Int J Mol Sci. (2021) 22(18):10122. doi: 10.3390/ijms221810122
163. Martínez MC, Andriantsitohaina R. Reactive nitrogen Species: molecular mechanisms and potential significance in health and disease. Antioxid Redox Signaling. (2009) 11(3):669–702. doi: 10.1089/ars.2007.1993
164. Myatt L. Review: reactive oxygen and nitrogen species and functional adaptation of the placenta. Placenta. (2010) 31:S66–S9. doi: 10.1016/j.placenta.2009.12.021
165. Myatt L, Rosenfield RB, Eis AL, Brockman DE, Greer I, Lyall F. Nitrotyrosine residues in placenta: evidence of peroxynitrite formation and action. Hypertension. (1996) 28(3):488–93. doi: 10.1161/01.HYP.28.3.488
166. Kurutas EB. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr J. (2015) 15(1):1–22. doi: 10.1186/s12937-016-0186-5
167. Raffaeli G, Manzoni F, Cortesi V, Cavallaro G, Mosca F, Ghirardello S. Iron homeostasis disruption and oxidative stress in preterm newborns. Nutrients. (2020) 12(6):1554. doi: 10.3390/nu12061554
168. Huizing MJ, Cavallaro G, Moonen RM, Gonzalez-Luis GE, Mosca F, Vento M, et al. Is the C242T polymorphism of the CYBA gene linked with oxidative stress-associated complications of prematurity? Antioxid Redox Signal. (2017) 27(17):1432–8. doi: 10.1089/ars.2017.7042
169. Bernardi F, Guolo F, Bortolin T, Petronilho F, Dal-Pizzol F. Oxidative stress and inflammatory markers in Normal pregnancy and preeclampsia. J Obstet Gynaecol Res. (2008) 34(6):948–51. doi: 10.1111/j.1447-0756.2008.00803.x
170. Schoots MH, Gordijn SJ, Scherjon SA, van Goor H, Hillebrands J-L. Oxidative stress in placental pathology. Placenta. (2018) 69:153–61. doi: 10.1016/j.placenta.2018.03.003
171. Zygula A, Kosinski P, Wroczynski P, Makarewicz-Wujec M, Pietrzak B, Wielgos M, et al. Oxidative stress markers differ in two placental dysfunction pathologies: pregnancy-induced hypertension and intrauterine growth restriction. Oxid Med Cell Longev. (2020) 2020:1323891. doi: 10.1155/2020/1323891
172. Raijmakers MT, Dechend R, Poston L. Oxidative stress and preeclampsia: rationale for antioxidant clinical trials. Hypertension. (2004) 44(4):374–80. doi: 10.1161/01.HYP.0000141085.98320.01
173. Ortega MA, Fraile-Martínez O, García-Montero C, Sáez MA, Álvarez-Mon MA, Torres-Carranza D, et al. The pivotal role of the placenta in normal and pathological pregnancies: a focus on preeclampsia, fetal growth restriction, and maternal chronic venous disease. Cells. (2022) 11(3):568. doi: 10.3390/cells11030568
174. Mehendale S, Kilari A, Dangat K, Taralekar V, Mahadik S, Joshi S. Fatty acids, antioxidants, and oxidative stress in pre-eclampsia. Int J Gynaecol Obstet. (2008) 100(3):234–8. doi: 10.1016/j.ijgo.2007.08.011
175. Fragoso MBT, Ferreira RC, Tenorio M, Moura FA, de Araujo ORP, Bueno NB, et al. Biomarkers of inflammation and redox imbalance in umbilical cord in pregnancies with and without preeclampsia and consequent perinatal outcomes. Oxid Med Cell Longev. (2021) 2021:9970627. doi: 10.1155/2021/9970627
176. Howlader MZ, Parveen S, Tamanna S, Khan TA, Begum F. Oxidative stress and antioxidant status in neonates born to pre-eclamptic mother. J Trop Pediatr. (2009) 55(6):363–7. doi: 10.1093/tropej/fmp025
177. Tastekin A, Ors R, Demircan B, Saricam Z, Ingec M, Akcay F. Oxidative stress in infants born to preeclamptic mothers. Pediatr Int. (2005) 47(6):658–62. doi: 10.1111/j.1442-200x.2005.02146.x
178. Bharadwaj SK, Vishnu Bhat B, Vickneswaran V, Adhisivam B, Bobby Z, Habeebullah S. Oxidative stress, antioxidant status and neurodevelopmental outcome in neonates born to pre-eclamptic mothers. Indian J Pediatr. (2018) 85(5):351–7. doi: 10.1007/s12098-017-2560-5
179. Bharadwaj S, Bhat VB, Vickneswaran V, Adhisivam B, Zachariah B, Habeebullah S. Oxidative stress in preeclamptic mother – newborn dyads and its correlation with early neonatal outcome – a case control study. J Matern Fetal Neonatal Med. (2018) 31(12):1548–53. doi: 10.1080/14767058.2017.1319933
180. Namdev S, Bhat V, Adhisivam B, Zachariah B. Oxidative stress and antioxidant status among neonates born to mothers with pre-eclampsia and their early outcome. J Matern Fetal Neonatal Med. (2014) 27(14):1481–4. doi: 10.3109/14767058.2013.860521
181. Bowen RS, Moodley J, Dutton MF, Theron AJ. Oxidative stress in pre-eclampsia. Acta Obstet Gynecol Scand. (2001) 80(8):719–25. doi: 10.1034/j.1600-0412.2001.080008719.x
182. Braekke K, Harsem NK, Staff AC. Oxidative stress and antioxidant status in fetal circulation in preeclampsia. Pediatr Res. (2006) 60(5):560–4. doi: 10.1203/01.pdr.0000242299.01219.6a
183. Altunhan H, Annagur A, Kurban S, Ertugrul S, Konak M, Ors R. Total oxidant, antioxidant, and paraoxonase levels in babies born to pre-eclamptic mothers. J Obstet Gynaecol Res. (2013) 39(5):898–904. doi: 10.1111/jog.12026
184. Allison BJ, Brain KL, Niu Y, Kane AD, Herrera EA, Thakor AS, et al. Fetal in vivo continuous cardiovascular function during chronic hypoxia. J Physiol. (2016) 594(5):1247–64. doi: 10.1113/JP271091
185. Torrance HL, Krediet TG, Vreman HJ, Visser GH, van Bel F. Oxidative stress and proinflammatory cytokine levels are increased in premature neonates of preeclamptic mothers with HELLP syndrome. Neonatology. (2008) 94(2):138–42. doi: 10.1159/000119724
186. Abdel Ghany EA, Alsharany W, Ali AA, Youness ER, Hussein JS. Anti-oxidant profiles and markers of oxidative stress in preterm neonates. Paediatr Int Child Health. (2016) 36(2):134–40. doi: 10.1179/2046905515Y.0000000017
187. Lee YS, Chou YH. Antioxidant profiles in full term and preterm neonates. Chang Gung Med J. (2005) 28(12):846–51.16515018
188. Ferencz A, Orvos H, Hermesz E. Major differences in the levels of redox status and antioxidant defence markers in the erythrocytes of pre- and full-term neonates with intrauterine growth restriction. Reprod Toxicol. (2015) 53:10–4. doi: 10.1016/j.reprotox.2015.02.008
189. Mirzarahimi M, Ahadi A, Bohlooli S, Namakikhalajan E, Barak M. Antioxidant levels in cord blood of term neonates and its association with birth weight. Iran J Child Neurol. (2016) 10(1):31–4.27057185
190. Madoglio RJ, Rugolo LM, Kurokawa CS, Sa MP, Lyra JC, Antunes LC. Inflammatory and oxidative stress airway markers in premature newborns of hypertensive mothers. Braz J Med Biol Res. (2016) 49(9):e5160. doi: 10.1590/1414-431x20165160
191. Mansur RB, Cunha GR, Asevedo E, Zugman A, Rios AC, Salum GA, et al. Perinatal complications, lipid peroxidation, and mental health problems in a large community pediatric sample. Eur Child Adolesc Psychiatry. (2017) 26(5):521–9. doi: 10.1007/s00787-016-0914-6
192. Peterside IE, Selak MA, Simmons RA. Impaired oxidative phosphorylation in hepatic mitochondria in growth-retarded rats. Am J Physiol Endocrinol Metab. (2003) 285(6):E1258–66. doi: 10.1152/ajpendo.00437.2002
193. Selak MA, Storey BT, Peterside I, Simmons RA. Impaired oxidative phosphorylation in skeletal muscle of intrauterine growth-retarded rats. Am J Physiol Endocrinol Metab. (2003) 285(1):E130–7. doi: 10.1152/ajpendo.00322.2002
194. Oke SL, Hardy DB. The role of cellular stress in intrauterine growth restriction and postnatal dysmetabolism. Int J Mol Sci. (2021) 22(13):6986. doi: 10.3390/ijms22136986
195. Burton GJ, Fowden AL, Thornburg KL. Placental origins of chronic disease. Physiol Rev. (2016) 96(4):1509–65. doi: 10.1152/physrev.00029.2015
196. Miller SL, Wallace EM, Walker DW. Antioxidant therapies: a potential role in perinatal medicine. Neuroendocrinology. (2012) 96(1):13–23. doi: 10.1159/000336378
197. Di Fabrizio C, Giorgione V, Khalil A, Murdoch CE. Antioxidants in pregnancy: do we really need more trials? Antioxidants. (2022) 11(5):812. doi: 10.3390/antiox11050812
198. Gao Q, Zhong C, Zhou X, Chen R, Xiong T, Hong M, et al. The association between intake of dietary lycopene and other carotenoids and gestational diabetes mellitus risk during mid-trimester: a cross-sectional study. Br J Nutr. (2019) 121(12):1405–12. doi: 10.1017/S0007114519000606
199. Hobson SR, Gurusinghe S, Lim R, Alers NO, Miller SL, Kingdom JC, et al. Melatonin improves endothelial function in vitro and prolongs pregnancy in women with early-onset preeclampsia. J Pineal Res. (2018) 65(3):e12508. doi: 10.1111/jpi.12508
200. Holmquist E, Brantsæter AL, Meltzer HM, Jacobsson B, Barman M, Sengpiel V. Maternal selenium intake and selenium status during pregnancy in relation to preeclampsia and pregnancy-induced hypertension in a large Norwegian pregnancy cohort study. Sci Total Environ. (2021) 798:149271. doi: 10.1016/j.scitotenv.2021.149271
201. McCarthy C, Kenny LC. Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci Rep. (2016) 6:32683. doi: 10.1038/srep32683
202. Panagodage S, Yong HE, Costa FDS, Borg AJ, Kalionis B, Brennecke SP, et al. Low-dose acetylsalicylic acid treatment modulates the production of cytokines and improves trophoblast function in an in vitro model of early-onset preeclampsia. Am J Pathol. (2016) 186(12):3217–24. doi: 10.1016/j.ajpath.2016.08.010
203. Paul BD. Ergothioneine: a stress vitamin with antiaging, vascular, and neuroprotective roles? Antioxid Redox Signaling. (2022) 36(16–18):1306–17. doi: 10.1089/ars.2021.0043
204. Rumbold A, Ota E, Nagata C, Shahrook S, Crowther CA. Vitamin C supplementation in pregnancy. Cochrane Database Syst Rev. (2015) 9:CD004072. doi: 10.1002/14651858.CD004072.pub3
205. Tenório M, Ferreira R, Moura F, Bueno N, Goulart M, Oliveira A. Oral antioxidant therapy for prevention and treatment of preeclampsia: meta-analysis of randomized controlled trials. Nutr Metab Cardiovasc Dis. (2018) 28(9):865–76. doi: 10.1016/j.numecd.2018.06.002
206. Vaka R, Deer E, LaMarca B. Is mitochondrial oxidative stress a viable therapeutic target in preeclampsia? Antioxidants (Basel). (2022) 11(2):210. doi: 10.3390/antiox11020210
207. Williamson RD, McCarthy FP, Manna S, Groarke E, Kell DB, Kenny LC, et al. L-(+)-ergothioneine significantly improves the clinical characteristics of preeclampsia in the reduced uterine perfusion pressure rat model. Hypertension. (2020) 75(2):561–8. doi: 10.1161/HYPERTENSIONAHA.119.13929
208. Xiong W, Wang Y, Zhou X. Low-dose aspirin might alleviate the symptoms of preeclampsia by increasing the expression of antioxidative enzymes. Exp Ther Med. (2021) 22(6):1418. doi: 10.3892/etm.2021.10853
209. Yang Y, Xu P, Zhu F, Liao J, Wu Y, Hu M, et al. The potent antioxidant MitoQ protects against preeclampsia during late gestation but increases the risk of preeclampsia when administered in early pregnancy. Antioxid Redox Signal. (2021) 34(2):118–36. doi: 10.1089/ars.2019.7891
210. Hansell JA, Richter HG, Camm EJ, Herrera EA, Blanco CE, Villamor E, et al. Maternal melatonin: effective intervention against developmental programming of cardiovascular dysfunction in adult offspring of complicated pregnancy. J Pineal Res. (2022) 72(1):e12766. doi: 10.1111/jpi.12766
211. Kulkarni AV, Mehendale SS, Yadav HR, Kilari AS, Taralekar VS, Joshi SR. Circulating angiogenic factors and their association with birth outcomes in preeclampsia. Hypertens Res. (2010) 33(6):561–7. doi: 10.1038/hr.2010.31
212. Bridges JP, Gilbert JS, Colson D, Gilbert SA, Dukes MP, Ryan MJ, et al. Oxidative stress contributes to soluble fms-like tyrosine kinase-1 induced vascular dysfunction in pregnant rats. Am J Hypertens. (2009) 22(5):564–8. doi: 10.1038/ajh.2009.24
213. Choudhry H, Harris AL. Advances in hypoxia-inducible factor biology. Cell Metab. (2018) 27(2):281–98. doi: 10.1016/j.cmet.2017.10.005
214. Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. (2003) 111(5):649–58. doi: 10.1172/JCI17189
215. Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. (2006) 26(22):8336–46. doi: 10.1128/MCB.00425-06
216. Ashur-Fabian O, Yerushalmi GM, Mazaki-Tovi S, Steinberg DM, Goldshtein I, Yackobovitch-Gavan M, et al. Cell free expression of hif1alpha and p21 in maternal peripheral blood as a marker for preeclampsia and fetal growth restriction. PLoS One. (2012) 7(5):e37273. doi: 10.1371/journal.pone.0037273
217. Caniggia I, Winter JL. Adriana and luisa castellucci award lecture 2001. Hypoxia inducible factor-1: oxygen regulation of trophoblast differentiation in Normal and pre-eclamptic pregnancies – a review. Placenta. (2002) 23(Suppl A):S47–57. doi: 10.1053/plac.2002.0815
218. Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. (1999) 103(7):945–52. doi: 10.1172/JCI4106
219. Cunningham MW Jr., Castillo J, Ibrahim T, Cornelius DC, Campbell N, Amaral L, et al. AT1-AA (angiotensin II type 1 receptor agonistic autoantibody) blockade prevents preeclamptic symptoms in placental ischemic rats. Hypertension. (2018) 71(5):886–93. doi: 10.1161/HYPERTENSIONAHA.117.10681
220. Aggarwal PK, Chandel N, Jain V, Jha V. The relationship between circulating endothelin-1, soluble fms-like tyrosine kinase-1 and soluble endoglin in preeclampsia. J Hum Hypertens. (2012) 26(4):236–41. doi: 10.1038/jhh.2011.29
221. Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. (2006) 12(6):642–9. doi: 10.1038/nm1429
222. Chaiworapongsa T, Espinoza J, Gotsch F, Kim YM, Kim GJ, Goncalves LF, et al. The maternal plasma soluble vascular endothelial growth factor receptor-1 concentration is elevated in SGA and the magnitude of the increase relates to Doppler abnormalities in the maternal and fetal circulation. J Matern Fetal Neonatal Med. (2008) 21(1):25–40. doi: 10.1080/14767050701832833
223. Korzeniewski SJ, Romero R, Chaiworapongsa T, Chaemsaithong P, Kim CJ, Kim YM, et al. Maternal plasma angiogenic index-1 (placental growth factor/soluble vascular endothelial growth factor receptor-1) is a biomarker for the burden of placental lesions consistent with uteroplacental underperfusion: a longitudinal case-cohort study. Am J Obstet Gynecol. (2016) 214(5):629.e1–e17. doi: 10.1016/j.ajog.2015.11.015
224. Chaiworapongsa T, Romero R, Erez O, Tarca AL, Conde-Agudelo A, Chaemsaithong P, et al. The prediction of fetal death with a simple maternal blood test at 20-24 weeks: a role for angiogenic index-1 (PlGF/sVEGFR-1 ratio). Am J Obstet Gynecol. (2017) 217(6):682.e1–e13. doi: 10.1016/j.ajog.2017.10.001
225. Turpin CA, Sakyi SA, Owiredu WK, Ephraim RK, Anto EO. Association between adverse pregnancy outcome and imbalance in angiogenic regulators and oxidative stress biomarkers in gestational hypertension and preeclampsia. BMC Pregnancy Childbirth. (2015) 15:189. doi: 10.1186/s12884-015-0624-y
226. Dai Y, Zhang J, Liu R, Xu N, Yan SB, Chen Y, et al. The role and mechanism of asymmetric dimethylarginine in fetal growth restriction via interference with endothelial function and angiogenesis. J Assist Reprod Genet. (2020) 37(5):1083–95. doi: 10.1007/s10815-020-01750-5
227. Tsao PN, Wei SC, Su YN, Chou HC, Chen CY, Hsieh WS. Excess soluble fms-like tyrosine kinase 1 and low platelet counts in premature neonates of preeclamptic mothers. Pediatrics. (2005) 116(2):468–72. doi: 10.1542/peds.2004-2240
228. Schlembach D, Wallner W, Sengenberger R, Stiegler E, Mortl M, Beckmann MW, et al. Angiogenic growth factor levels in maternal and fetal blood: correlation with Doppler ultrasound parameters in pregnancies complicated by pre-eclampsia and intrauterine growth restriction. Ultrasound Obstet Gynecol. (2007) 29(4):407–13. doi: 10.1002/uog.3930
229. Wallner W, Sengenberger R, Strick R, Strissel PL, Meurer B, Beckmann MW, et al. Angiogenic growth factors in maternal and fetal serum in pregnancies complicated by intrauterine growth restriction. Clin Sci. (2007) 112(1):51–7. doi: 10.1042/CS20060161
230. Kwon JY, Maeng YS, Kwon YG, Kim YH, Kang MH, Park YW. Decreased endothelial progenitor cells in umbilical cord blood in severe preeclampsia. Gynecol Obstet Invest. (2007) 64(2):103–8. doi: 10.1159/000100081
231. Galazios G, Papazoglou D, Giagloglou K, Vassaras G, Koutlaki N, Maltezos E. Umbilical cord serum vascular endothelial growth factor (VEGF) levels in Normal pregnancies and in pregnancies complicated by preterm delivery or pre-eclampsia. Int J Gynaecol Obstet. (2004) 85(1):6–11. doi: 10.1016/j.ijgo.2003.08.009
232. Martin-Estal I, de la Garza RG, Castilla-Cortazar I. Intrauterine growth retardation (IUGR) as a novel condition of insulin-like growth factor-1 (IGF-1) deficiency. Rev Physiol Biochem Pharmacol. (2016) 170:1–35. doi: 10.1007/112_2015_5001
233. Kallapur SG, Bachurski CJ, Le Cras TD, Joshi SN, Ikegami M, Jobe AH. Vascular changes after intra-amniotic endotoxin in preterm lamb lungs. Am J Physiol Lung Cell Mol Physiol. (2004) 287(6):L1178–85. doi: 10.1152/ajplung.00049.2004
234. Thomas W, Seidenspinner S, Kramer BW, Kawczynska-Leda N, Chmielnicka-Kopaczyk M, Marx A, et al. Airway concentrations of angiopoietin-1 and endostatin in ventilated extremely premature infants are decreased after funisitis and unbalanced with bronchopulmonary dysplasia/death. Pediatr Res. (2009) 65(4):468–73. doi: 10.1203/PDR.0b013e3181991f35
235. Harkin P, Marttila R, Pokka T, Saarela T, Hallman M. Survival analysis of a cohort of extremely preterm infants born in Finland during 2005-2013. J Matern Fetal Neonatal Med. (2021) 34(15):2506–12. doi: 10.1080/14767058.2019.1668925
236. Baker CD, Abman SH. Impaired pulmonary vascular development in bronchopulmonary dysplasia. Neonatology. (2015) 107(4):344–51. doi: 10.1159/000381129
237. Kim DH, Kim HS. Serial changes of serum endostatin and angiopoietin-1 levels in preterm infants with severe bronchopulmonary dysplasia and subsequent pulmonary artery hypertension. Neonatology. (2014) 106(1):55–61. doi: 10.1159/000358374
238. Mohamed WA, Niyazy WH, Mahfouz AA. Angiopoietin-1 and endostatin levels in cord plasma predict the development of bronchopulmonary dysplasia in preterm infants. J Trop Pediatr. (2011) 57(5):385–8. doi: 10.1093/tropej/fmq112
239. Wallace B, Peisl A, Seedorf G, Nowlin T, Kim C, Bosco J, et al. Anti-sFlt-1 therapy preserves lung alveolar and vascular growth in antenatal models of bronchopulmonary dysplasia. Am J Respir Crit Care Med. (2018) 197(6):776–87. doi: 10.1164/rccm.201707-1371OC
240. Seedorf G, Kim C, Wallace B, Mandell EW, Nowlin T, Shepherd D, et al. rhIGF-1/BP3 preserves lung growth and prevents pulmonary hypertension in experimental bronchopulmonary dysplasia. Am J Respir Crit Care Med. (2020) 201(9):1120–34. doi: 10.1164/rccm.201910-1975OC
241. Thebaud B, Abman SH. Bronchopulmonary dysplasia: where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med. (2007) 175(10):978–85. doi: 10.1164/rccm.200611-1660PP
242. Cavallaro G, Filippi L, Bagnoli P, La Marca G, Cristofori G, Raffaeli G, et al. The pathophysiology of retinopathy of prematurity: an update of previous and recent knowledge. Acta Ophthalmol. (2014) 92(1):2–20. doi: 10.1111/aos.12049
243. Zayed MA, Uppal A, Hartnett ME. New-onset maternal gestational hypertension and risk of retinopathy of prematurity. Invest Ophthalmol Vis Sci. (2010) 51(10):4983–8. doi: 10.1167/iovs.10-5283
244. Dymara-Konopka W, Laskowska M. The role of nitric oxide, ADMA, and homocysteine in the etiopathogenesis of preeclampsia-review. Int J Mol Sci. (2019) 20(11):2757. doi: 10.3390/ijms20112757
245. Tal R. The role of hypoxia and hypoxia-inducible factor-1alpha in preeclampsia pathogenesis. Biol Reprod. (2012) 87(6):134. doi: 10.1095/biolreprod.112.102723
246. Rengarajan A, Mauro AK, Boeldt DS. Maternal disease and gasotransmitters. Nitric Oxide. (2020) 96:1–12. doi: 10.1016/j.niox.2020.01.001
247. Liu T, Mukosera GT, Blood AB. The role of gasotransmitters in neonatal physiology. Nitric Oxide. (2020) 95:29–44. doi: 10.1016/j.niox.2019.12.002
248. van der Sterren S, Kleikers P, Zimmermann LJ, Villamor E. Vasoactivity of the gasotransmitters hydrogen sulfide and carbon monoxide in the chicken ductus arteriosus. Am J Physiol Regul Integr Comp Physiol. (2011) 301(4):R1186–98. doi: 10.1152/ajpregu.00729.2010
249. Vallance P, Collier J, Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. (1989) 2(8670):997–1000. doi: 10.1016/S0140-6736(89)91013-1
250. Provitera L, Amelio GS, Tripodi M, Raffaeli G, Macchini F, Amodeo I, et al. Veno-Arterial extracorporeal membrane oxygenation (ECMO) impairs bradykinin-induced relaxation in neonatal porcine coronary arteries. Biomedicines. (2022) 10(9):2083. doi: 10.3390/biomedicines10092083
251. Provitera L, Cavallaro G, Griggio A, Raffaeli G, Amodeo I, Gulden S, et al. Cyclic nucleotide-dependent relaxation in human umbilical vessels. J Physiol Pharmacol. (2019) 70(4):619–30. doi: 10.26402/jpp.2019.4.13
252. Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. (1992) 258(5090):1898–902. doi: 10.1126/science.1281928
253. Moonen RM, Huizing MJ, Cavallaro G, González-Luis GE, Bas-Suárez P, Bakker JA, et al. Plasma levels of dimethylarginines in preterm very low birth weight neonates: its relation with perinatal factors and short-term outcome. Int J Mol Sci. (2014) 16(1):19–39. doi: 10.3390/ijms16010019
254. Sutton EF, Gemmel M, Powers RW. Nitric oxide signaling in pregnancy and preeclampsia. Nitric Oxide. (2020) 95:55–62. doi: 10.1016/j.niox.2019.11.006
255. Holwerda KM, Faas MM, van Goor H, Lely AT. Gasotransmitters: a solution for the therapeutic dilemma in preeclampsia? Hypertension. (2013) 62(4):653–9. doi: 10.1161/HYPERTENSIONAHA.113.01625
256. Huang LT, Hsieh CS, Chang KA, Tain YL. Roles of nitric oxide and asymmetric dimethylarginine in pregnancy and fetal programming. Int J Mol Sci. (2012) 13(11):14606–22. doi: 10.3390/ijms131114606
257. Saleh L, Verdonk K, Visser W, van den Meiracker AH, Danser AH. The emerging role of endothelin-1 in the pathogenesis of pre-eclampsia. Ther Adv Cardiovasc Dis. (2016) 10(5):282–93. doi: 10.1177/1753944715624853
258. Rubanyi GM, Polokoff MA. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol Rev. (1994) 46(3):325–415.7831383
259. Enevoldsen FC, Sahana J, Wehland M, Grimm D, Infanger M, Krüger M. Endothelin receptor antagonists: status quo and future perspectives for targeted therapy. J Clin Med. (2020) 9(3):824. doi: 10.3390/jcm9030824
260. Tashie W, Fondjo LA, Owiredu W, Ephraim RKD, Asare L, Adu-Gyamfi EA, et al. Altered bioavailability of nitric oxide and L-arginine is a key determinant of endothelial dysfunction in preeclampsia. Biomed Res Int. (2020) 2020:3251956. doi: 10.1155/2020/3251956
261. Dai B, Liu T, Zhang B, Zhang X, Wang Z. The polymorphism for endothelial nitric oxide synthase gene, the level of nitric oxide and the risk for pre-eclampsia: a meta-analysis. Gene. (2013) 519(1):187–93. doi: 10.1016/j.gene.2013.01.004
262. Walsh SK, English FA, Johns EJ, Kenny LC. Plasma-mediated vascular dysfunction in the reduced uterine perfusion pressure model of preeclampsia: a microvascular characterization. Hypertension. (2009) 54(2):345–51. doi: 10.1161/HYPERTENSIONAHA.109.132191
263. McCarthy AL, Woolfson RG, Raju SK, Poston L. Abnormal endothelial cell function of resistance arteries from women with preeclampsia. Am J Obstet Gynecol. (1993) 168(4):1323–30. doi: 10.1016/0002-9378(93)90389-Z
264. Pascoal IF, Lindheimer MD, Nalbantian-Brandt C, Umans JG. Preeclampsia selectively impairs endothelium-dependent relaxation and leads to oscillatory activity in small omental arteries. J Clin Invest. (1998) 101(2):464–70. doi: 10.1172/JCI557
265. Aalkjaer C, Danielsen H, Johannesen P, Pedersen EB, Rasmussen A, Mulvany MJ. Abnormal vascular function and morphology in pre-eclampsia: a study of isolated resistance vessels. Clin Sci. (1985) 69(4):477–82. doi: 10.1042/cs0690477
266. Diaz V, Lebras-Isabet MN, Denjean A. Effect of nomega-nitro-L-arginine methyl ester-induced intrauterine growth restriction on postnatal lung growth in rats. Pediatr Res. (2005) 58(3):557–61. doi: 10.1203/01.PDR.0000179398.62365.43
267. Aikio O, Metsola J, Vuolteenaho R, Perhomaa M, Hallman M. Transient defect in nitric oxide generation after rupture of fetal membranes and responsiveness to inhaled nitric oxide in very preterm infants with hypoxic respiratory failure. J Pediatr. (2012) 161(3):397–403.e1. doi: 10.1016/j.jpeds.2012.03.008
268. Hallman M, Ronkainen E, Saarela TV, Marttila RH. Management practices during perinatal respiratory transition of very premature infants. Front Pediatr. (2022) 10:862038. doi: 10.3389/fped.2022.862038
269. Ivy DD, Ziegler JW, Dubus MF, Fox JJ, Kinsella JP, Abman SH. Chronic intrauterine pulmonary hypertension alters endothelin receptor activity in the ovine fetal lung. Pediatr Res. (1996) 39(3):435–42. doi: 10.1203/00006450-199603000-00010
270. Gien J, Tseng N, Seedorf G, Roe G, Abman SH. Endothelin-1 impairs angiogenesis in vitro through rho-kinase activation after chronic intrauterine pulmonary hypertension in fetal sheep. Pediatr Res. (2013) 73(3):252–62. doi: 10.1038/pr.2012.177
271. Ivy DD, Parker TA, Ziegler JW, Galan HL, Kinsella JP, Tuder RM, et al. Prolonged endothelin A receptor blockade attenuates chronic pulmonary hypertension in the ovine fetus. J Clin Invest. (1997) 99(6):1179–86. doi: 10.1172/JCI119274
272. Krause BJ, Costello PM, Munoz-Urrutia E, Lillycrop KA, Hanson MA, Casanello P. Role of DNA methyltransferase 1 on the altered eNOS expression in human umbilical endothelium from intrauterine growth restricted fetuses. Epigenetics. (2013) 8(9):944–52. doi: 10.4161/epi.25579
273. Postberg J, Kanders M, Forcob S, Willems R, Orth V, Hensel KO, et al. Cpg signalling, H2A.Z/H3 acetylation and microRNA-mediated deferred self-attenuation orchestrate foetal NOS3 expression. Clin Epigenetics. (2015) 7:9. doi: 10.1186/s13148-014-0042-4
274. Xu XF, Xu SS, Fu LC, Hu QY, Lv Y, Du LZ. Epigenetic changes in peripheral leucocytes as biomarkers in intrauterine growth retardation rat. Biomed Rep. (2016) 5(5):548–52. doi: 10.3892/br.2016.775
275. Yu GZ, Reilly S, Lewandowski AJ, Aye CYL, Simpson LJ, Newton L, et al. Neonatal micro-RNA profile determines endothelial function in offspring of hypertensive pregnancies. Hypertension. (2018) 72(4):937–45. doi: 10.1161/HYPERTENSIONAHA.118.11343
Keywords: endothelial dysfunction, endothelium, glycocalyx, preterm infant, preterm birth, chorioamnionitis, dysfunctional placentation, fetal growth restriction
Citation: Amelio GS, Provitera L, Raffaeli G, Tripodi M, Amodeo I, Gulden S, Cortesi V, Manzoni F, Cervellini G, Tomaselli A, Pravatà V, Garrido F, Villamor E, Mosca F and Cavallaro G (2022) Endothelial dysfunction in preterm infants: The hidden legacy of uteroplacental pathologies. Front. Pediatr. 10:1041919. doi: 10.3389/fped.2022.1041919
Received: 11 September 2022; Accepted: 14 October 2022;
Published: 4 November 2022.
Edited by:
Mikko Hallman, University of Oulu, FinlandReviewed by:
Gaston Ofman, University of Oklahoma Health Sciences Center, United States© 2022 Amelio, Provitera, Raffaeli, Tripodi, Amodeo, Gulden, Cortesi, Manzoni, Cervellini, Tomaselli, Pravatà, Garrido, Villamor, Mosca and Cavallaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giacomo Cavallaro Z2lhY29tby5jYXZhbGxhcnJvQHBvbGljbGluaWNvLm1pLml0 Genny Raffaeli Z2VubnkucmFmZmFlbGlAdW5pbWkuaXQ=
Specialty Section: This article was submitted to Neonatology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.