
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 04 November 2022
Sec. Pediatric Rheumatology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1039291
This article is part of the Research TopicCase Reports in Pediatric Rheumatology 2022View all 12 articles
 Ana Catarina Lunz Macedo1,2*
Ana Catarina Lunz Macedo1,2* Lazara Elena Santisteban Lores2
Lazara Elena Santisteban Lores2 José Antonio Tavares Albuquerque2Nilo José Coelho Duarte3
José Antonio Tavares Albuquerque2Nilo José Coelho Duarte3 Paschoalina Romano3Persio Almeida Rezende Ebner3Vinicius Marcondes Rezende3Clovis A. Silva4Luís Eduardo Coelho Andrade5
Paschoalina Romano3Persio Almeida Rezende Ebner3Vinicius Marcondes Rezende3Clovis A. Silva4Luís Eduardo Coelho Andrade5 Dewton Moraes Vasconcelos6
Dewton Moraes Vasconcelos6 Lourdes Isaac2
Lourdes Isaac2
Factor H (FH) is one of the most important regulatory proteins of the alternative pathway of the complement system. FH deficiency is a rare condition that causes unregulated C3 consumption, leading to an increased susceptibility to infections and glomerulopathies. Our previous studies have demonstrated a FH deficient patient carrying a c.452G > A, p.R127H FH mutation which leads to a misfolded protein and its retention in the endoplasmic reticulum. In his cultured fibroblasts, FH-delayed secretion was partially rescued when treated with curcumin, and once secreted, exhibited normal regulatory function. Here, we report a childhood-onset systemic lupus erythematosus (cSLE) in this FH deficient patient and the results of experimental treatment with curcumin aiming to rescue FH secretion and regulatory activity.
Factor H (FH) is a central regulator of the complement system that inhibits excessive alternative pathway activation by acting as a cofactor for Factor I in the cleavage of C3b into iC3b. FH also competes with Factor B (FB) for binding to C3b, resulting in the dissociation of Bb from C3- and C5-convertases. FH deficiency is related to C3 deficiency owing to the acceleration of C3 consumption. Complete homozygous FH deficiency is a rare condition, and to date a few cases have been described (1, 2). In humans, a single gene on chromosome 1q32 codes for FH. Mutations in the human FH gene can result in the lack of protein in the plasma due to misfolding and impaired protein secretion or in normal range detection but with defective regulatory functions (3). FH deficiency is mainly associated with an increased susceptibility to infections, and patient's defective regulatory functions can lead to C3-glomerulopathy, atypical haemolytic uraemic syndrome, and age-related macular degeneration (1–5). FH- and C3-deficient patients are treated constantly with antibiotics and when in critical conditions, they are plasma infused to replace complement proteins levels. Clearly, new options of treatment are required. In experimental models, the use of mini-FH (containing domains 1–4 and 19–20) offer promising results in the regulation of the alternative pathway (6). However, so far this has not been tested in FH-deficient patients.
We have previously described the case of a patient with FH deficiency diagnosed after two episodes of complicated pneumonia (7). The homozygous variant c.452G > A in CFH determining p.R127H was found in the patient by Sanger sequencing analysis (7). In vitro, the patient's fibroblasts retain FH in the endoplasmic reticulum, resulting in delayed secretion. These cells, when treated with curcumin, showed increased secretion of the mutant FH, which once transported, had normal regulatory function in the alternative pathway of the complement system (8).
Curcumin is a hydrophobic polyphenol derived from the rhizome of the Curcuma longa plant with a broad spectrum of antioxidant, anti-inflammatory, antimicrobial, and anticancer effects. It is well accepted that curcumin has low bioavailability in humans, chemical instability, and rapid metabolism, even when administered orally at doses as high as 12 g/day (9). A formulation of curcumin in nanoparticles (Theracurmin®) has been used in clinical trials, presenting better absorption and bioavailability (10, 11). Based on these observations, we hypothesised that Theracurmin® could be used to treat FH deficiency in vivo.
Before the experimental treatment, the patient presented unexpected signs and symptoms of childhood-onset systemic lupus erythematosus (cSLE). SLE is a rare autoimmune disorder that affects multiple organs and systems (12–14). Deficiencies in the first components of the classical pathway C1q/r/s, C4, and C2 are frequently associated with early onset SLE or lupus-like disease (15). However, deficiency of alternative pathway proteins has rarely been associated with SLE development (1).
In this case report, we discuss the diagnosis of cSLE in this patient and the results of treatment with Theracurmin® for his FH deficiency.
A 2-year-old Brazilian boy, the second child of first-degree cousins with Japanese ascendance, was hospitalised in the intensive care unit because of complicated pneumonia with bilateral pleural effusion. The patient was treated with antibiotic therapy and bilateral thoracic drainage with complete resolution. At the age of 3 years, he presented with a new episode of pneumonia that required invasive ventilation and antibiotic therapy, and was discharged after three weeks. He was fully vaccinated according to the public health schedule available when he was a child, which at that time did not include pneumococcal vaccine. After hospitalisation, prophylactic amoxicillin was prescribed. Immunological evaluation revealed FH deficiency, associated with reduced levels of C9, C3, and FB owing to the lack of regulation of the alternative pathway. The variant c.452G > A in CFH, which determines p.R127H, was found in the homozygous patient by Sanger sequencing analysis, and this variant was found in both parents in heterozygosis (7). He had chickenpox with no complications at 6 years of age, despite not vaccinated. At 15 years of age, he presented with a malar rash and photosensitivity, with periods of improvement and worsening. One year later, the patient developed persistent rashes on the posterior cervical and thoracic region. At the age of 17, he reported episodes of acute and painful oligoarthritis involving the right elbow, right ankle, and right fourth finger (proximal interphalangeal joint). These episodes lasted two to four days, and improved after a short course of non-steroidal anti-inflammatory drugs.
At 18 years of age, when he was invited to participate in the experimental treatment protocol, he had a malar rash, photosensitivity, and posterior cervical and thoracic rash, and had reported episodes of oligoarthritis. Ultrasound examination performed during a new episode of acute arthritis revealed normal radiographic findings and small joint effusion with synovial thickening in the olecranial fossae of the elbow. Despite the distribution of the rashes in the posterior cervical and thoracic region, the lesions were not compatible with dermatomyositis, and skin biopsies confirmed the immunofluorescence pattern characteristic of the skin lesion in SLE. Thoracic lesion skin biopsy showed lupus band test with immunofluorescence of continuous cross-linked IgM and focal granular IgG, with the absence of IgA and C3 along the dermoepidermal junction. IgG fluorescence was also observed in the keratinocyte nuclei. Ophthalmic exam did not reveal any abnormalities or pigmentary changes in the retina. The laboratory findings showed haemoglobin, 14.6 g/dl; white blood cell count, 6,560/mm³ (47% neutrophils, 32% lymphocytes, 5% eosinophils, 1% basophils, and 15% monocytes); and platelet count 300,000/mm³. C reactive protein was 2.18 mg/dl, C3 < 4 mg/dl (reference 67–149 mg/dl), C4 26,7 mg/dl (reference 10–38 mg/dl), C1q 389 mg/dl (reference 100–250 mg/dl), and C2 14,1 (reference 14–25 mg/dl). Anti-C1q 32 U/ml (reference <9 U/ml). Immunological tests showed antinuclear antibodies on HEp-2 cells (HEp-2 ANA) 1:1,280 thick speckled pattern, positive ENA (anti-Sm 42,8 U/ml, anti-RNP >200 U/ml, anti-Ro/SSA >200 U/ml, anti-La/SSB 35,92 U/ml—reference values <15 U/ml) IgG 21.58 GPL/ml anticardiolipin (reference <10 GPL/ml) and IgM anticardiolipin 26.11 MPL/ml (reference <7 MPL/ml) autoantibodies. Rheumatoid factor was positive, with 219.2 U/ml (reference value <14 U/ml). Anti-double-stranded DNA (anti-dsDNA) antibodies, ANCA (antineutrophil cytoplasmic antibodies), anti-JO1 (anti-histidyl T-RNA synthetase), and anti SCL-70 (anti-topoisomerase I) were negative. Urinalysis did not indicate the presence of leukocytes or erythrocytes and the albumin/creatinine ratio was 9.2 mg/g. The serum urea and creatinine levels were 18 mg/dl and 0.79 mg/dl, respectively.
Theracurmin® was kindly provided by Theravalues Corporation (Tokyo, Japan). After obtaining informed consent and local ethical committee approval, we initiated the regimen of a 2-g dose of Theracurmin® powder (containing 200 mg curcumin), diluted in water, orally every 12 h for 106 days. The dosage was subsequently increased to a 4-g dose of Theracurmin® every 12 h for an additional 29 days. As indicated in Figure 1, during treatment with Theracurmin®, the patient was not treated with standard treatment for SLE. He first received Theracurmin® alone and, after the washout, started standard treatment with hydroxychloroquine. During treatment, the patient was closely observed for any worsening in his condition, which would lead to an immediate discontinuation of the protocol.
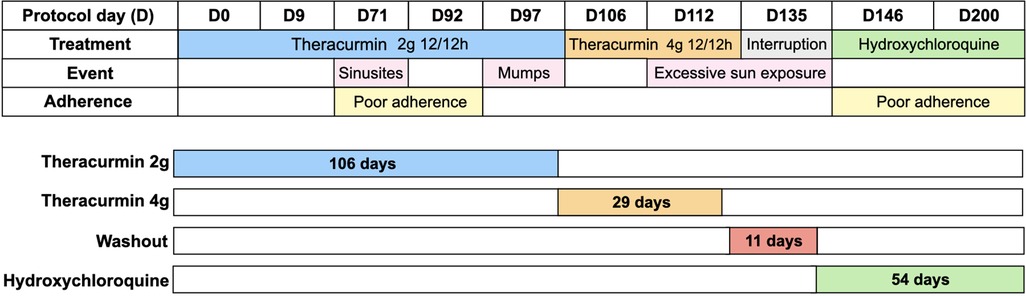
Figure 1. Protocol design, evaluation time, therapeutic intervention, events and report of treatment adherence.
Plasma levels of FH and factors B and C3 were assessed before and throughout the treatment. Plasma levels of IL-1β, IL-6, IL-8, IL-10, IL-12p70, and TNF-α were quantified before the first dose (D0), and on D9 (Table 1).

Table 1. Concentrations of the cytokines IL-1ß, IL-6, IL-8, IL-10, IL-12p70 and TNF-α were assessed pre-treatment at D0 and D9 in regular use of Theracurmin® 2-g every 12 h.
Tolerance and side effects were assessed using clinical and laboratory parameters throughout the protocol. Lupus activity was measured using SLEDAI-2 K (16). The patient's quality of life was assessed using the Paediatric Quality of Life Inventory 4.0 - PedsQL 4.0 - (17), covering four domains: health and activities; emotional aspects; social aspects; and school performance before treatment (D0), D135, and D200. During the protocol, the patient had sinusitis, mumps despite being vaccinated, and self-limiting episodes of arthritis. Our patient reported poor compliance at two time points during the treatment. These events are illustrated in Figure 1.
To confirm the diagnosis of SLE, clinical and laboratory findings were correlated with the scoring criteria from the American College of Rheumatology (ACR) (18), Systemic Lupus International Collaborating Clinics (SLICC) (19), and the EULAR/ACR classification (EULAR) (20). Although low levels of C3 are primarily caused by FH deficiency, SLE was diagnosed in this patient by all three classifications. Since the patient developed clinical signs of lupus before the age of 18, cSLE was confirmed.
The patient presented with more than 6 of the 11 points of ACR classification. The sensitivity and specificity of the 1982 ACR criteria for paediatric lupus are 96% and 100%, respectively (21). Without considering the low level of C3, the patient met two clinical and two immunological criteria of the SLICC classification. In the EULAR/ACR classification, the patient scored more than 10 points. Although the characteristic skin biopsy is not scored as a criterion, immunofluorescence performed on biopsy reinforces lupus diagnosis.
To confirm that cSLE was absent at 3 years of age, when he was diagnosed with primary immunodeficiency, the same autoantibodies were also tested in the patient's frozen serum samples. The following auto antibodies were analysed: HEp-2 ANA, anti-DNA, ANCA, anti-JO1, anti SCL-70, RNP-SM, anti-SM, anti-Ro, anti-La, anticardiolipin IgG, and anticardiolipin IgM. All titres were negative, except for HEp-2 ANA (1:320). Autoantibodies can be present many years before the diagnosis of SLE (22), but for this patient, the retrospective assessment for lupus was negative at 3-year-old, confirming that cSLE occurred after the primary immunodeficiency.
Case reports that relate autoimmune diseases to the complement system are often related to the deficiency of components of the classical pathway and this association between immunodeficiency and autoimmune diseases has been well-documented in the literature (23). Turley et al. (2015) studied 77 complement-deficient patients and observed that 37% of those presented defects in the classical pathway had SLE-like disease (24). The most remarkable genetic association with SLE is the high frequency of deficiencies in early classical pathway components, mainly C1q (90%–93%), C1r/C1s (50%–57%), C4 (75%), and C2 (10%). These patients usually present with SLE at an early age, with severe symptoms and poor prognosis (15). However, the association between SLE and deficiencies in the components of the alternative pathway is unusual. Homozygous FH deficiency is a rare phenomenon and only a few cases have been reported (2). There are only two cases in the literature of concomitant FH deficiency and SLE (25, 26). In one case, a Caucasian daughter of non-consanguineous parents presented with subacute cutaneous lupus, with ANA and anti-DNA positivity, FH deficiency, and undetectable C3 serum levels. C2 levels were reduced, with normal serum C4 and C1q levels. Until the period when the case was published at age 59, this patient did not present with glomerulonephritis (25). In another case, a Caucasian daughter of consanguineous first-degree cousins had arthritis, fever, erythema malar, anaemia, nephritis, ANA positivity, and intense fluorescence for IgG in the nuclei of keratinocytes in the skin biopsy. The patient had undetectable serum concentrations of FH, C3, and FB; low concentrations of C2; and low levels of C1q and C4, with normal C1-INH (26). These two cases reported FH deficiency and lupus. They also had deficiencies in the initial components of the classical pathway, which could justify their association with lupus. Here, we describe a rare association of a homozygous deficiency in FH, a regulatory protein of the alternative pathway, and cSLE. To the best of our knowledge, there is no previous case of a patient who initially presented with FH deficiency at an early age, with normal components of the classical pathway, and cSLE development during adolescence. To date, we cannot speculate which pathophysiological mechanisms could explain a possible association between FH deficiency and SLE. Possibly, there are underlying genetic variants that may predispose to SLE, which we have not been able to explore until now.
Patient plasma concentrations of curcumin were assessed at the beginning of the treatments with 2-g and 4-g doses of Theracurmin®. Blood samples were collected immediately before and at 30 min, 60 min, 90 min, 2 h, 4 h, 6 h, 8 h, 12 h, 24 h after the administration of the first 2-g dose of Theracurmin®. The same protocol was performed with the first 4-g dose Theracurmin® (there was no washout period during treatment).
There are studies using doses as low as 500 mg up to 12 g with good tolerance and minor adverse events. The different curcumin treatment regimens and formulations available in the literature are not comparable with each other and there is no consensus on their use. We designed this specific protocol with Theracurmin® because its better absorption might be an advantage for its clinical function (27–29).
It was possible to identify through HPLC-MS/MS that the highest plasma concentration of curcumin throughout the treatment was 432 ng/ml (1,17 µM), which was reached immediately after the first dose. The plasma curcumin concentration after the very first 2-g dose Theracurmin® was consistently higher than the results obtained after chronic use, even with a 4-g dose as well as throughout the treatment. This finding of lower plasma curcumin concentrations in chronic use despite the increased dose of Theracurmin® has not been previously explained in the literature.
A previous report assessed plasma curcumin levels in healthy volunteers after a single oral 150-mg dose followed by a 210-mg dose of Theracurmin®, separated by a washout period of two weeks between dose escalation. They demonstrated an increase in plasma curcumin levels in a dose-dependent manner (10).
Oral curcumin is known to have low bioavailability owing to its low absorption by the small intestine, coupled with extensive reductive and conjugative metabolism in the liver and elimination through the gall bladder. After ingestion, curcumin is subsequently reduced in enterocytes and hepatocytes by a reductase to dihydrocurcumin, tetrahydrocurcumin, hexa-hydrocurcumin, and octahydrocurcumin, which can be found in free forms or as glucuronides (9). Several studies have been performed using oral curcumin powder supplements to investigate their absorption. Degradation reactions change the structure and properties of curcumin, thereby affecting its pharmacokinetic and pharmacodynamic behaviour (9).
We speculate that our finding of lower plasma curcumin levels despite increasing (doubling) the dose of Theracurmin® in a continuous treatment could be an effect of enzymatic induction and tolerance with acceleration of the drug's metabolism over time (30–33). Both curcumin and its major reduced forms (dihydrocurcumin, tetrahydrocurcumin, and hexahydrocurcumin) can undergo glucuronidation. To clarify these questions, plasma samples were treated with glucuronidase, and the amounts of curcumin and tetrahydrocurcumin were assessed using HPLC-MS/MS for comparative analysis. This additional evaluation showed high concentrations of plasma curcumin after the first 2-g dose of Theracurmin®, whereas high levels of its metabolite, tetrahydrocurcumin, were observed after continuous use, assessed with 4-g dose of Theracurmin® (Figure 2).
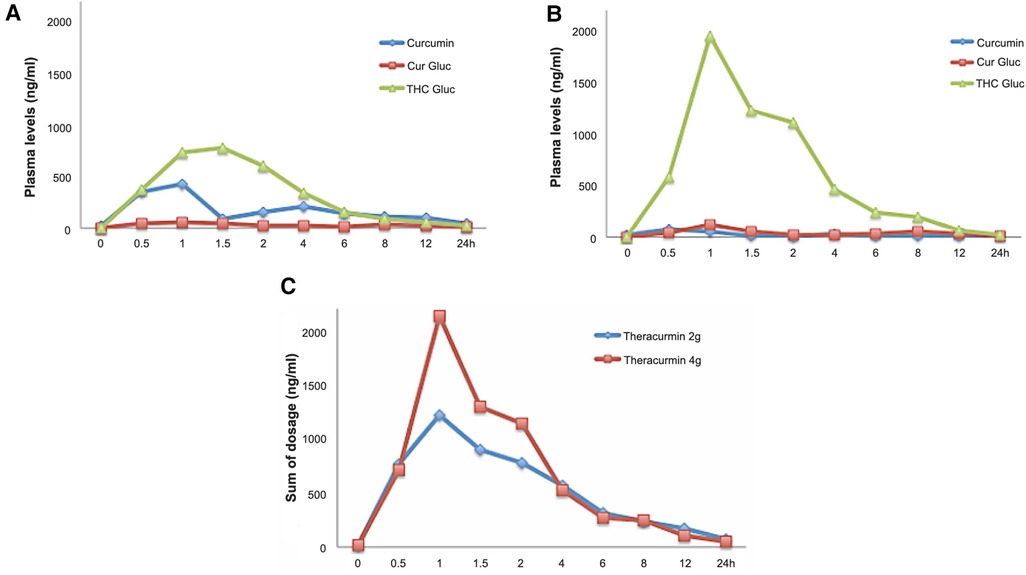
Figure 2. Curcumin and metabolites levels in patients's plasma. (A) 2-g Theracurmin®; (B) 4-g Theracurmin®; (C) Sum comparison of all curcumin fractions and metabolites with 2-g and 4-g Theracurmin®. Cur Gluc: Curcumin Glucuronidase; THC Gluc: Tetrahydrocurcumin Glucorunidase.
Albuquerque et al. (2012) used 2 µM pure curcumin (Sigma-Aldrich) in the patient's fibroblast culture (8). The in vitro concentration of curcumin in that experiment was high, and this was not achievable in the in vivo treatment protocol because curcumin is unstable under physiological conditions and rapidly metabolised to other products.
Plasma levels of FH, FB and C3 were assessed before and throughout the treatment. Despite the levels of curcumin and its metabolites in the patient's plasma, the treatment did not affect the concentration of FH, FB and C3.
The plasma concentrations of IL-1β, IL-6, IL-8, IL-10, IL-12p70, and TNF-α were determined before treatment (D0) and on D9 under regular use of Theracurmin® 2 g every 12 h (Table 1). Considering the cytokine response to treatment, there was a marked drop in the plasma concentration of IL-6, and a reduction of approximately 20% in IL-8 levels. However, IL-1β increased more than 50%, and slight increases in TNF-α, IL-10, and IL-12p70 levels were observed. A meta-analysis of nine randomised controlled studies demonstrated that IL-6 levels were reduced by treatment with curcumin, and this effect was more evident in more intense inflammatory states. Our results only partially agree with the literature, which describes the reduction in serum concentrations of IL-1β, IL-6, IL-8, IL-12, and TNF-α with the use of curcumin (34).
To ensure the safety of the treatment, the patient was closely monitored and lupus activity was measured using SLEDAI-2K throughout the treatment. SLEDAI-2K remained stable, as did haematologic, renal, and hepatic parameters (Table 2). Since the low level of C3 is secondary to FH deficiency, we did not consider C3 in the SLEDAI-2K; however, even if it were considered, it would have remained at 4 (a low activity index) throughout the protocol. We concluded that neither the introduction nor interruption of curcumin treatment interfered with lupus activity.
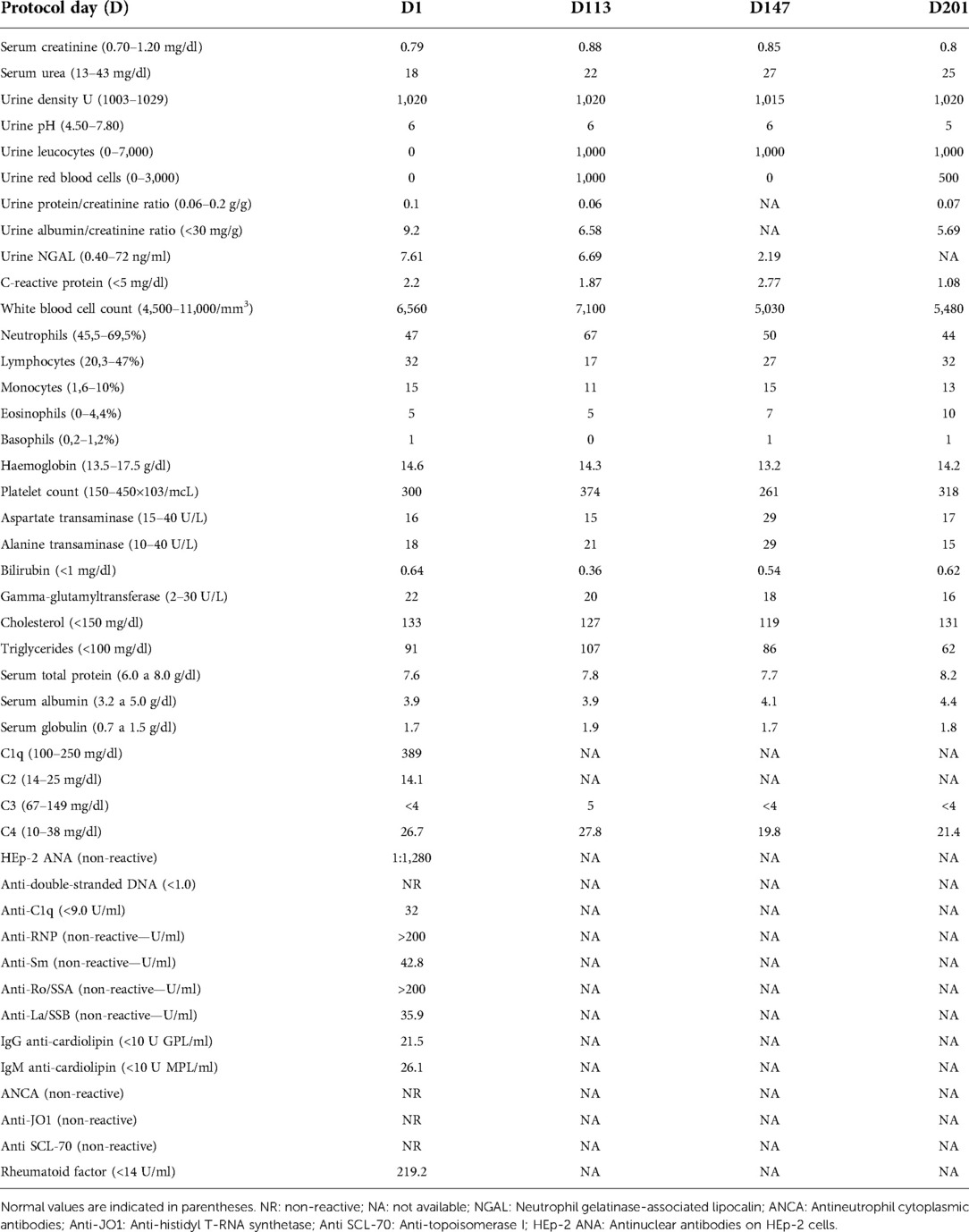
Table 2. Laboratorial tests assessed pre-treatment at D0, D113 in regular use of Theracurmin® 4 g every 12 h, D147 after 11 days washout, and D201 in regular use of Hydroxychloroquine 400 mg.
We conclude that the patient showed good tolerance to both doses of Theracurmin® without adverse symptoms or side effects, as reported in previous publications.
The perception of quality of life by PedsQL 4.0 was assessed before treatment on D0, on D135 (during treatment with Theracurmin® 4 g every 12 h) and on D200 (after discontinuation of Theracurmin® and under treatment with hydroxychloroquine 400 mg). In the PedsQL 4.0, the best quality of life was related to the lowest score. In the patient's opinion, the scores were 11, 15, and 4 at the respective three time points. Parents' views on adolescents’ quality of life were 17, 19, and 2 at the same time points. Considering the four domains, there was a significant improvement in quality of life, mainly in health and activities, in the third assessment under use of hydroxychloroquine.
We have reported a rare case of FH deficiency and cSLE. Oral curcumin supplementation for FH deficiency was well tolerated with no adverse effects. Despite the previous success of in vitro treatment of patient fibroblasts, in vivo treatment did not result in an increase in the plasma levels of FH, C3, and FB. Although the plasma concentrations of IL-6 and IL-8 declined and IL-1β increased after the treatment, no improvement or burden in the clinical and laboratory cSLE parameters were observed.
It is well established that curcumin has low bioavailability and that it can be ameliorated by new formulations. In our case, using a nanoparticle formulation (Theracurmin®), the plasma levels of curcumin rapidly decreased with continuous use, whereas the level of tetrahidrocurcumin increased. These results could be due to enzymatic induction and tolerance with acceleration of the drug's metabolism over time and might justify the absence of an effect of curcumin treatment for this patient.
For this patient, personalised translational medicine showed that previous successful in vitro treatment could not be assumed as a beneficial long-term treatment for FH deficiency. More efforts are needed to clarify curcumin bioavailability, absorption, reductive, and conjugative metabolism during short-term and long-term treatment.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by local institutional review board (CEP 1151183). The patient and the legal guardian provided their written informed consent to participate in this study.
LI, JATA, and ACLM conceptualised and designed the study. ACLM, JATA, and LESL performed the initial analyses and drafted the initial manuscript. CAS, LECA, DMV, and LI critically reviewed this manuscript. NJCD, PR, PARE, and VMR provided curcumin and metabolite dosages throughout the treatment. ACLM and DMV were responsible for the patient treatment. All authors contributed to the article and approved the submitted version.
This study was supported by grants from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP #2010/50043-0 and FAPESP #2017/12924-3) and Conselho Nacional de Pesquisa e Desenvolvimento (CNPq 421474/2016-5 and 312654/2021-9).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Reis ES, Falcão DA, Isaac L. Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H. Scand J Immunol. (2006) 63:155–68. doi: 10.1111/j.1365-3083.2006.01729.x
2. Pickering MC, Cook HT. Translational Mini-Review Series on Complement Factor H: renal diseases associated with complement factor H: novel insights from humans and animals. Clin Exp Immunol. (2008) 151:210–30. doi: 10.1111/j.1365-2249.2007.03574.x
3. Zipfel PF, Skerka C, Chen Q, Wiech T, Goodship T, Johnson S, et al. The role of complement in C3 glomerulopathy. Mol Immunol. (2015) 67(1):21–30. doi: 10.1016/j.molimm.2015.03.012
4. Dragon-Durey MA, Frémeaux-Bacchi V, Loirat C, Blouin J, Niaudet P, Deschenes G, et al. Heterozygous and homozygous factor H deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: report and genetic analysis of 16 cases. J Am Soc Nephrol. (2004) 15(3):787–95. doi: 10.1097/01.ASN.0000115702.28859.A7
5. Teixeira AG, Silva AS, Lin FLH, Velletri R, Bavia L, Jr BR, et al. Association of complement factor H Y402H polymorphism and age-related macular degeneration in Brazilian patients. Acta Ophthalmol. (2010) 88:e165–9. doi: 10.1111/j.1755-3768.2010.01932.x
6. de Boer ECW, van Mourik AG, Jongerius I. Therapeutic lessons to be learned from the role of complement regulators as double-edged sword in health and disease. Front Immunol. (2020) 11:578069. doi: 10.3389/fimmu.2020.578069
7. Falcão DA, Reis ES, Paixão-Cavalcante D, Amano MT, Delcolli MIMV, Florido MPC, et al. Deficiency of the human complement regulatory protein factor H associated with low levels of component C9. Scand J Immunol. (2008) 68:445–55. doi: 10.1111/j.1365-3083.2008.02152.x
8. Albuquerque JAT, Lamers ML, Castiblanco-Valencia MM, Santos M, Isaac L. Chemical chaperones curcumin and 4-phenylbutyric acid improve secretion of mutant factor H R127H by fibroblasts from a factor H-deficient patient. J Immunol. (2012) 189:3242–8. doi: 10.4049/jimmunol.1201418
9. Cas M D, Ghidoni R. Dietary curcumin: correlation between bioavailability and health potential. Nutrients. (2019) 11(9):2147. doi: 10.3390/nu11092147
10. Kanai M, Imaizumi A, Otsuka Y, Sasaki H, Hashiguchi M, Tsujiko K, et al. Dose-escalation and pharmacokinetic study of nanoparticle curcumin, a potential anticancer agent with improved bioavailability, in healthy human volunteers. Cancer Chemother Pharmacol. (2012) 69:65–70. doi: 10.1007/s00280-011-1673-1
11. Kanai M, Otsuka Y, Otsuka K, Sato M, Nishimura T, Mori Y, et al. A phase I study investigating the safety and pharmacokinetics of highly bioavailable curcumin (Theracurmin) in cancer patients. Cancer Chemother Pharmacol. (2013) 71:1521–30. doi: 10.1007/s00280-013-2151-8
12. Carneiro-Sampaio M, Liphaus BL, Jesus AA, Silva CAA, Oliveira JB, Kiss MH. Understanding systemic lupus erythematosus physiopathology in the light of primary immunodeficiencies. J Clin Immunol. (2008) 28(1):S34–41. doi: 10.1007/s10875-008-9187-2
13. Silva CA. Childhood-onset systemic lupus erythematosus: early disease manifestations that the paediatrician must know. Expert Rev Clin Immunol. (2016 Sep) 12(9):907–10. doi: 10.1080/1744666X.2016.1195685
14. Silva CA, Aikawa NE, Pereira RM, Campos LM. Management considerations for childhood-onset systemic lupus erythematosus patients and implications on therapy. Expert Rev Clin Immunol. (2016) 12(3):301–13. doi: 10.1586/1744666X.2016.1123621
15. Macedo ACL, Isaac L. Systemic lupus erythematosus and deficiencies of early components of the complement classical pathway. Front Immunol. (2016) 7:55. doi: 10.3389/fimmu.2016.00055
16. Gladman DD, Ibañez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol. (2002 Feb) 29(2):288–91. PMID: 11838846
17. Klatchoian DA, Len CA, Terreri MT, Silva M, Itamoto C, Ciconelli RM, et al. Quality of life of children and adolescents from São Paulo: reliability and validity of the Brazilian version of the Pediatric Quality of Life Inventory version 4.0 Generic Core Scales. J Pediatr (Rio J). (2008) 84(4):308–15. doi: 10.2223/JPED.1788
18. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. (1997) 40(9):1725. doi: 10.1002/art.1780400928
19. Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of systemic lupus international collaborating clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. (2012) 64(8):2677–86. doi: 10.1002/art.34473
20. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. 2019 European league against rheumatism/American college of rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. (2019) 71(9 ):1400–12. doi: 10.1002/art.40930
21. Ferraz MB, Goldenberg J, Hilario MO, Bastos WA, Oliveira SK, Azevedo EC, et al. Evaluation of the 1982 ARA lupus criteria data set in pediatric patients. Committees of Pediatric Rheumatology of the Brazilian Society of Pediatrics and the Brazilian Society of Rheumatology. Clin Exp Rheumatol. (1994) 12(1):83–7. PMID: 7741825
22. Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. (2003 Oct 16) 349(16):1526–33. doi: 10.1056/NEJMoa021933
23. Bussone G, Mouthon L. Autoimmune manifestations in primary immune deficiencies. Autoimmun Rev. (2009) 8:332–6. doi: 10.1016/j.autrev.2008.11.004
24. Turley AJ, Gathmann B, Bangs C, Bradbury M, Seneviratne S, Gonzalez-Granado LI, et al. Spectrum and management of complement immunodeficiencies (excluding hereditary angioedema) across Europe. J Clin Immunol. (2015) 35(2):199–205. doi: 10.1007/s10875-015-0137-5
25. Fijen CA, Kuijper EJ, Te Bulte M, van de Heuvel MM, Holdrinet AC, Sim RB, et al. Heterozygous and homozygous factor H deficiency states in a Dutch family. Clin Exp Immunol. (1996) 105:511–6. doi: 10.1046/j.1365-2249.1996.d01-777.x
26. Brai M, Misiano G, Maringhini S, Cutaja I, Hauptmann G. Combined homozygous factor H and heterozygous C2 deficiency in an Italian family. J Clin Immunol. (1988) 8:51–6. doi: 10.1007/BF00915156
27. Carroll RE, Benya RV, Turgeon DK, Vareed S, Neuman M, Rodriguez L, et al. Phase IIa clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer Prev Res. (2011) 4:354–64. doi: 10.1158/1940-6207.CAPR-10-0098
28. Lao CD, Ruffin MT 4th, Normolle D, Heath DD, Murray SI, Bailey JM, et al. Dose escalation of a curcuminoid formulation. BMC Complement Altern Med. (2006) 6:6–10. doi: 10.1186/1472-6882-6-6
29. Vareed SK, Kakarala M, Ruffin MT, Crowell JA, Normolle DP, Djuric Z, et al. Pharmacokinetics of curcumin conjugate metabolites in healthy human subjects. Cancer Epidemiol Biomarkers Prev. (2008) 17:1411–7. doi: 10.1158/1055-9965.EPI-07-2693
30. Hoehle SI, Pfeiffer E, Slyom AM, Metzler M. Metabolism of curcuminoids in tissue slices and subcellular fractions from rat liver. J Agric Food Chem. (2006) 54(3):756–64. doi: 10.1021/jf058146a
31. Sueth-Santiago V, Mendes-Silva GP, Decoté-Ricardo D, Lima MEF. Curcumina, o pó dourado do açafrão-da-terra: introspecções sobre química e atividades biológicas. Quim Nova. (2015) 38:538–52. doi: 10.5935/0100-4042.20150035
32. Douglass BJ, Clouatre DL. Beyond yellow curry: assessing commercial curcumin absorption technologies. J Am Coll Nutr. (2015) 34(4):347–58. doi: 10.1080/07315724.2014.950392
33. Kurita T, Makino Y. Novel curcumin oral delivery systems. Anticancer Res. (2013) 33:2807–22. PMID: 23780965
Keywords: FH deficiency, lupus, curcumin, experimental treatment, translational medicine, case report
Citation: Lunz Macedo AC, Santisteban Lores LE, Albuquerque José Antonio Tavares de, Duarte NJC, Romano P, Ebner Persio de Almeida Rezende, Rezende VM, Silva CA, Andrade LEC, Vasconcelos Dewton de Moraes and Isaac L (2022) A rare association between factor H deficiency and lupus: Case report and experimental treatment with curcumin. Front. Pediatr. 10:1039291. doi: 10.3389/fped.2022.1039291
Received: 8 September 2022; Accepted: 4 October 2022;
Published: 4 November 2022.
Edited by:
Vahid Ziaee, Tehran University of Medical Sciences, IranReviewed by:
Mario Sestan, University of Zagreb, Croatia© 2022 Lunz Macedo, Santisteban Lores, Albuquerque, Duarte, Romano, Ebner, Rezende, Silva, Andrade, Vasconcelos and Isaac. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Catarina Lunz Macedo YW5hLm1hY2Vkb0BoYy5mbS51c3AuYnI=
Specialty Section: This article was submitted to Pediatric Rheumatology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.