 Seoyun Jang
Seoyun Jang Yo Han Ahn
Yo Han Ahn Jung Min Ko1,2,4
Jung Min Ko1,2,4 Jae Sung Ko
Jae Sung Ko Sojung Lim
Sojung Lim Hee Gyung Kang
Hee Gyung Kang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 31 October 2022
Sec. Pediatric Nephrology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1032653
Atypical progeroid syndrome (APS) is a rare type of progeroid syndrome mainly caused by heterozygous missense mutations in the LMNA (MIM 150330) gene. APS has heterogeneous clinical manifestations, and its kidney manifestations, particularly in children, are rarely documented. Here, we report the first pediatric case of APS with focal segmental glomerulosclerosis (FSGS). A 10-year-old boy with progeroid features was referred to the nephrology clinic because of hyperuricemia. He had dark skin, protruding eyes, and beaked nose and was very thin, suggesting lipodystrophy. He had been treated for recurrent urinary tract infection during infancy, and liver biopsy for persisting hepatitis showed steatohepatitis. He also had hypertrophic cardiomyopathy (HCMP) with mitral and tricuspid valve regurgitation. Genetic studies were performed considering his multisystem symptoms, and he was diagnosed as having APS according to exome sequencing findings (c.898G > C, p.Asp300His of LMNA). During the first visit to the nephrology clinic, he had minimal proteinuria (urine protein/creatinine ratio of 0.23 mg/mg), which worsened during follow-up. In three years, his urine protein/creatinine ratio and N-acetyl-b-D-glucosaminidase/creatinine ratio increased to 1.52 and 18.7, respectively. The kidney biopsy result was consistent with findings of FSGS, peri-hilar type, showing segmental sclerosis of 1 (5%) glomerulus out of 21 glomeruli. An angiotensin receptor blocker was added to manage his proteinuria. This is the first pediatric report of FSGS in an APS patient with confirmed LMNA defect, who manifested progeroid features, lipodystrophy, HCMP with heart valve dysfunction, and steatohepatitis. Our case suggests that screening for proteinuric nephropathy is essential for managing APS patients since childhood.
Progeroid syndromes are a group of rare genetic disorders characterized by clinical features that mimic physiologic aging. Progeroid syndromes share similar clinical features such as hair loss, short stature, skin tightness, cardiovascular diseases, and osteoporosis. However, the underlying mechanism can vary according to the causative gene (1). The progeroid syndrome can be classified into two groups according to its molecular pathophysiology: alterations in components of the nuclear envelope and mutations in genes involved in DNA-repair pathways (1, 2). The nuclear envelope component involved in progeroid syndrome is the nuclear lamina, a thin protein meshwork between chromatin and the inner nuclear membrane (3, 4), composed of lamins. Lamins contribute to the maintenance of nuclear shape and structure, chromatin organization, and other aspects of nuclear metabolism by interacting with regulatory molecules (5–8). Lamin A and lamin C (encoded by LMNA, MIM 150330) are widely expressed in somatic cells, and diverse LMNA mutations cause various disorders, laminopathies, including diseases affecting striated and cardiac muscle, lipodystrophy syndromes such as familial partial lipodystrophy (FPLD), peripheral neuropathy, and premature aging (progeroid syndromes) (9, 10).
Among the laminopathies, FPLD encompasses abnormal fat distribution and insulin resistance disorders. FPLD type 2 (Dunnigan-type, MIM 151660) is known to be caused by LMNA mutation and is characterized by the progressive lipoatrophy of the limbs, buttocks, and trunk sparing the neck and face. Metabolic alterations are common and cardiovascular comorbidities, and hepatic steatosis is often reported (11). Hutchinson-Gilford progeria syndrome (HGPS, MIM 176670) is another form of laminopathy, an early-onset premature aging disorder. It typically presents at 1–2 years of age with severe growth retardation, lipodystrophy, and skeletal and cardiovascular features (12, 13). The average life span is 13 years (12, 14, 15). Typical HGPS is caused by de novo heterozygous silent mutation in the LMNA gene c.1924C > T (p.Gly608Gly) (16, 17). This activates a cryptic splicing site and results in abnormal splicing of the prelamin A, producing a truncated protein called progerin, which is known to cause toxic effects when accumulated (18–21). Atypical progeroid syndrome (APS) is another type of progeria from LMNA mutation. APS is very rare, and until now, 69 patients with LMNA mutation have been reported worldwide (22–33). It is characterized by not having an accumulation of the Lamin A precursors (14, 15) and is caused by heterozygous LMNA mutations other than c.1924C > T. The onset of APS symptoms is relatively late, and the life span of APS patients is generally longer than those of HGPS (12, 14, 15). Though its clinical phenotypes have not been well established, typically, affected patients have growth retardation, joint contractures, and progeroid features, including a prominent nose with beaking, partial alopecia, dental crowding, and skin anomalies (14, 15, 34). Also, most of the patients suffered from marked metabolic abnormalities such as insulin resistance, diabetes mellitus, hypertriglyceridemia, steatohepatitis, various degrees of lipodystrophy, and cardiomyopathy (15, 35, 36).
Kidney involvement as proteinuria has been seldomly described in APS (26), and the pathophysiology of kidney manifestations remains unclear. Here, we report a case of a clinical presentation of an APS patient who presented with proteinuria and pathologically confirmed focal segmental glomerulosclerosis (FSGS).
A 10-year-old boy visited the nephrology clinic for further evaluation of incidentally found hyperuricemia. Previously, his perinatal medical history was unremarkable, with full-term vaginal delivery and birth weight of 3.64 kg, but he had four episodes of urinary tract infection during infancy. Bilateral vesicoureteral reflux (left grade 4 and right grade 1) was identified at three months, and he was given nitrofurantoin prophylaxis until 22 months. It was discontinued as there was no further urinary tract infection for a year. After the discontinuation, his vesicourethrogram at the age of 28 months showed no reflux. He was the only child, devoid of any remarkable family history of diseases. At 2 years of age, he complained of discoloration of extremities after exposure to cold, but evaluations for autoimmune disease and arteriography of upper and lower extremities did not reveal any abnormal findings. At 7 years of age, he was noticed to have waddling gait and knee and hip flexion limitations. Two years later, marked coxa valga with relative coxa magna with a small pelvis were found on orthopedic evaluation. He visited a genetic specialist at 9 years of age. While he had normal growth with a height of 143 cm (88.6 percentile) and weight of 33 kg (47.5 percentile), with normal development, decreased subcutaneous fat tissue in his trunk and both extremities with pale and dark skin were noted. He also had acanthosis nigricans at his neck and both axillary areas, a short neck, protruding eyes, and a beaked nose. Raynaud's phenomenon was still present. Liver function abnormality was found with elevated liver transaminases (AST 102 IU/L, normal range 15–50IU/L; ALT 248 IU/L, normal range 5–45 IU/L) and cholesterol (LDL-cholesterol 186 mg/dl, normal range 60–140 mg/dl, triglyceride 305 mg/dl, normal range 31–108 mg/dl; HDL-cholesterol 42 mg/dl, normal range: ≥40 mg/dl). His HbA1c was 6.5% (normal range: 4.0–6.4%), with a fasting blood glucose level of 83 mg/dl (normal range: 70–99 mg/dl), and increased insulin (94 μIU/ml, normal range 1.9–15.97 μIU/ml). He was referred to the endocrinologist for elevated HbA1c and dyslipidemia, however, his fasting glucose was lower than 100 mg/dl, but HbA1c and insulin were still elevated (6.1%, 55.7 μIU/ml) showing insulin resistance. Liver biopsy at 10 years showed a fatty change of hepatocytes and portal and periportal fibrosis. Mild mitral and tricuspid valve regurgitation was noted, which worsened during follow-up along with the development of hypertrophic cardiomyopathy (HCMP), requiring mechanical mitral valve replacement at 13 years of age. His diagnosis remained elusive despite multisystem symptoms. Various studies targeting progressive storage disorders, including glycogen storage disease, were performed, with no meaningful results. Eventually, he was found to have a de novo heterozygous mutation c.898G > C (p.Asp300His) of LMNA, genetically diagnosing the patient as APS. During regular follow-up for the steatohepatitis, hyperuricemia (serum uric acid 7.6 mg/dl, normal range: 3.0–7.0 mg/dl) was noticed, and he was referred to the nephrology clinic.
His hyperuricemia was managed well with benzbromarone. Initially, minimal proteinuria [urine protein/creatinine (Cr) ratio of 0.23 mg/mg] was present, which gradually worsened during follow-up. At 13, his urine protein/Cr ratio (normal range: 0–0.2 mg/mg) and N-acetyl-b-D-glucosaminidase/Cr ratio (normal range: 0–5.6 IU/gCr) increased to 1.52 and 18.7, respectively. Urine ß2 microglobulin was 0.33 ug/ml, and serum uric acid level was 12.4 mg/dl. However, he was normotensive with a height of 160.4 cm (50–75 percentile), a weight of 37.7 kg (5–10 percentile), and a BMI of 14.65 kg/m2, with normal serum albumin (4.4 g/dl, normal range: 3.3–5.2 g/dl) and serum Cr (0.71 mg/dl, normal range: 0.31–0.88 mg/dl). Doppler kidney sonography was unremarkable with normal size, parenchymal echogenicity, and intact internal perfusion. His kidney biopsy revealed FSGS, peri-hilar type, showing segmental sclerosis of 1 (5%) glomerulus out of 21 glomeruli. In microscopic findings, glomerular size was mildly increased, and there was focal mild hypercellularity involving mesangial and endothelial cells (Figure 1). Tubules were slightly atrophic, and there was no interstitial fibrosis, with mild focal infiltration of mononuclear cells. Immunofluorescence staining for immunoglobulins (IgG, IgM, and IgA), Kappa light chains, Lambda light chains, complement C3 and C1q, were all negative. Electron microscopy revealed a normal glomerular basement membrane, and effacement of the foot process was mild. There were no electron-dense deposits.
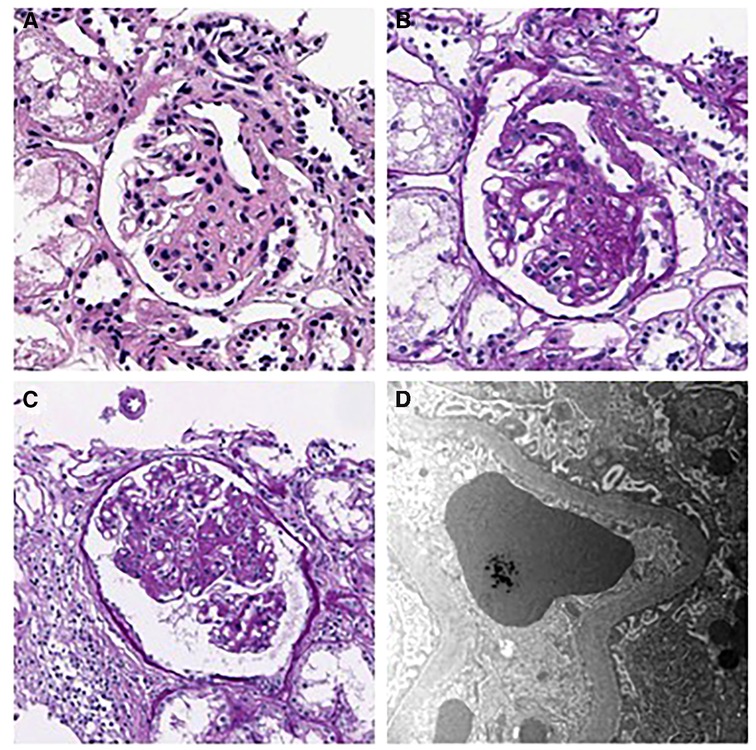
Figure 1. Kidney biopsy findings. (A–B) H&E and PAS staining show segmental sclerosis at the vascular pole of the glomerulus. The patient was diagnosed as having focal segmental glomerulosclerosis, perihilar variant. (A) H&E stain, × 400, (B) PAS stain, × 400). (C) Mild endocapillary hypercellularity and glomerular enlargement was observed in other glomeruli, with a maximum diameter of 302 μm. (PAS stain, × 250). (D) Electron microscopy showing mild effacement of foot processes. (EM, × 12,000).
An Angiotensin receptor blocker (Losartan) was added to manage his proteinuria (0.7 mg/kg), but it was difficult to continue due to dizziness. Since his proteinuria worsened during follow-up (urine protein/Cr ratio: 3.7 mg/mg, serum Cr 0.69 mg/dl, eGFR 98.22 ml/min/1.73 m2), losartan was restarted. His proteinuria waxed and waned with his cardiac condition, and the dosage was adjusted according to the symptoms (up to 1.2 mg/kg) (Figure 2). While taking losartan, his proteinuria did not aggravate, and his kidney function stayed stationary (urine protein/Cr ratio: 1.34 mg/mg, serum Cr 0.83 mg/dl, eGFR 82.45 ml/min/1.73 m2). During his last follow-up at 15 years of age, he did well without complaints and worked out daily. He has been prescribed benzbromarone, losartan, and sodium bicarbonate at the nephrology clinic (serum uric acid 6.8 mg/dl, serum Cr 0.79 mg/dl, cystatin C 1.74 mg/l, eGFR 84.43 ml/min/1.73 m2, and urine protein/Cr ratio 0.83 mg/mg). He has taken warfarin for mitral valve replacement and a pacemaker for postoperative sinus node dysfunction. He was also taking a beta-blocker, diuretic, and amlodipine for HCMP and omega-3 for hyperlipidemia.
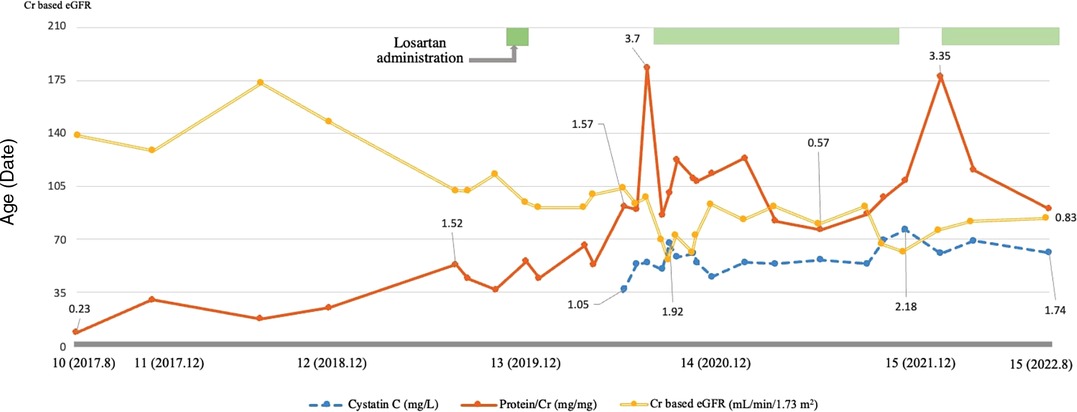
Figure 2. Characteristics of the patient during the follow-up. This graph shows the patient's trend of proteinuria and creatinine-based estimated glomerular filtration rate (eGFR) and cystatin C levels during the follow-up. Proteinuria is indicated using a solid line, cystatin C using a dashed line, and creatinine-based eGFR using a double line. The patient's proteinuria and kidney function waxed and waned during the follow-up, and following the administration of losartan, his proteinuria and renal function improved and remains stationary. Creatinine based eGFR (ml/min/1.73 m2), Cystatin C (mg/L), Protein/Cr (mg/mg).
This is the first report of FSGS in a pediatric APS patient with confirmed LMNA defect, who manifested progeroid features, lipodystrophy, HCMP with heart valve dysfunction, and steatohepatitis. APS is an extremely rare disease, and kidney involvement in APS is not a typical finding. Previous reports of kidney manifestation showed proteinuria, including the nephrotic range (26). While the mutation of our case, c.898G > C (p.Asp300His) in LMNA (Figure 3), was previously reported in two cases (25, 29), kidney involvement was not described. It is not a novel mutation, as the mutation was reported previously (25, 29). However, it is an extremely rare mutation, so its frequency has not been reported in genomic databases, including GNOMAD or Clinvar. One case with this variant was a 24-year-old Chinese man with multiple vascular lesions, progeroid features, hypertension, numerous intracranial calcifications, peripheral artery disease, and dyslipidemia (29). The other case is a 23-year-old woman from Myanmar with progeroid features, including short stature, thin scalp hair, absent eyebrow and eyelashes, and a beaked nose. She suffered from hypertension, secondary amenorrhea, generalized lipodystrophy, bilateral carotid artery stenosis, and left ventricular hypertrophy (25). Another missense variant of the same nucleotide locus (c.898G > A, p.Asp300Asn) was found in a 31-year-old French man with progeroid features, osteoporosis, premature atheromatosis, lipoatrophy, and cerebral ischemic disease (28). APS with cardiovascular diseases and dyslipidemia were common in all these patients, including our patient, but proteinuria was not described. Therefore, it is unclear if the FSGS of our case is a true manifestation of his APS.
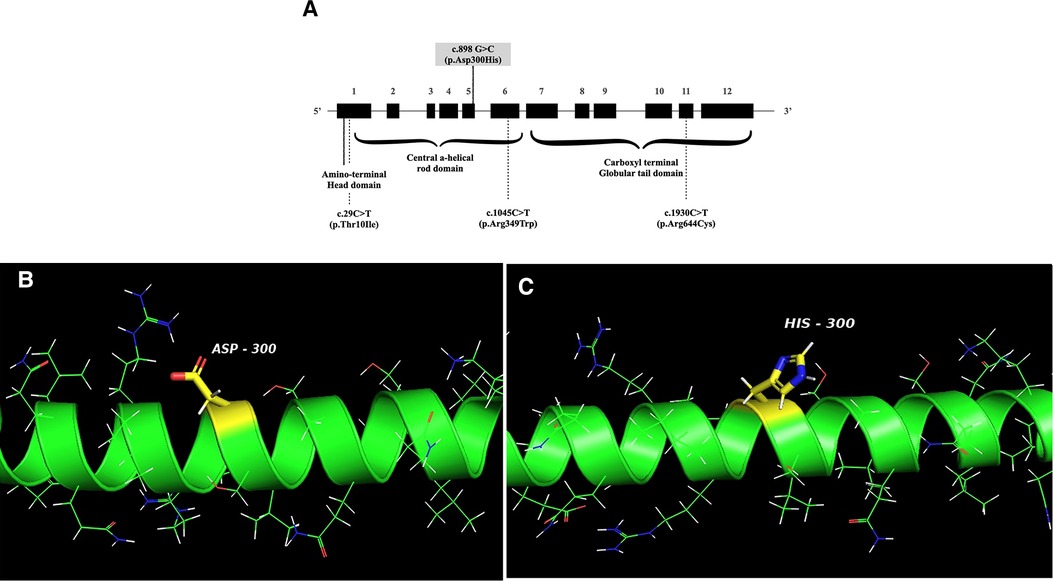
Figure 3. (A) Schematic structure of the LMNA gene. The mutation of the patient is indicated with a solid line, and the locations of mutations associated with FSGS are shown in dotted lines. FSGS: focal segmental glomerular sclerosis. (B,C) The crystal structure of wild-type human lamin A coil 2B domain and a model of proposed p.Asp300His mutant. I-TASSER software and PyMol were used to model the wild-type human lamin A and the p.Asp300His mutant. The peptide chain of the human lamin A coil 2B domain (aa. 290–310 from left to right) is shown as an alpha-helical structure (green). Side chains are shown as sticks. The carbon atoms in the mutation site are highlighted in yellow and other carbons are in green. Nitrogen atoms are labeled blue, oxygen atoms are red, hydrogen atoms are white, and sulfur atoms are shown orange.
However, proteinuria has been reported in several cases with LMNA mutations, and to our knowledge, nine patients were confirmed with FSGS by kidney biopsy (23, 24, 26, 37–41) (Table 1). The primary diagnosis of these cases was lipodystrophy with or without APS. Their presentations of proteinuria were all during adulthood, and two of them with APS features required kidney replacement therapy (23, 26). Since the others with the same mutation (p.Arg349Trp) had normal to impaired kidney function, the kidney outcome seems variable (23, 26, 37, 41), and proteinuria might have been present since their childhood as in our case, but not detected earlier. Therefore, if possible, our patient also needs careful follow-up and intervention since some required dialysis in their 30s, although their variant site was different from ours.
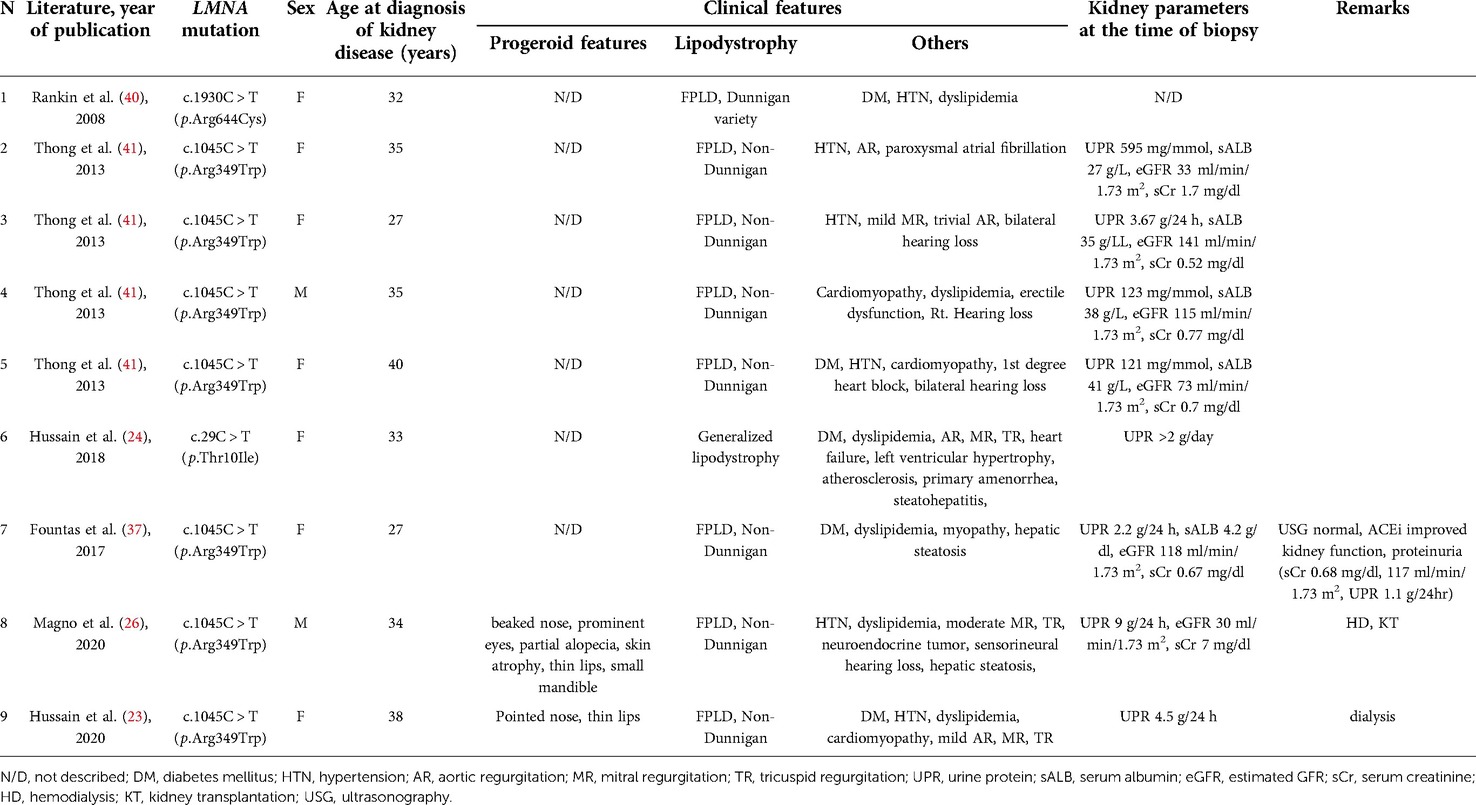
Table 1. LMNA mutations associated with pathologic findings of focal segmental glomerulosclerosis.
However, the pathophysiology of FSGS in LMNA mutations remains unclear. Transforming growth factor-beta1 (TGFβ1), which is activated in lipodystrophy in laminopathies (42, 43), might play a role in the disease mechanism. It is well known that activation of TGFβ1, the central regulator of fibrotic responses (44, 45), leads to mesangial cell matrix overproduction and glomerulosclerosis (46, 47) in diseased glomeruli. Interestingly, lamin A or C was essential for inhibiting fibroblast proliferation by TGFβ1 (48). Therefore, LMNA mutation might be linked to FSGS. Also, metabolic alteration of laminopathies, including our patient, might contribute to podocyte injury leading to kidney damage (49, 50, 51). Our patient also had a history of urinary tract infection and vesicoureteral reflux; FSGS might come from reflux nephropathy. However, his kidney pathology was incompatible with typical pathologic findings of reflux nephropathy, such as interstitial scarring, tubular atrophy, or loss of nephron mass.
Regarding the treatment, as TGFβ1 plays a crucial role in pathogenesis, targeting this cytokine appears promising. However, its therapeutic application is held back because of its multifunctional and pleiotropic actions. Fresolimumab, a human monoclonal antibody neutralizing human isoforms of TGFβ, was proven ineffective in clinical trials in FSGS (52, 53). Other approaches, including decreasing the production of prelamin A or clearing progerin (54) showed limited effect (55), as APS is not associated with the accumulation of lamin A precursors (15, 56). Therefore, so far, early recognition and treatment of the manifestations is the mainstay of treatment, which makes identifying the phenotypes of the disease more crucial. Therefore, in this case, early recognition and intervention might improve kidney outcomes.
In conclusion, this is the first pediatric APS patient with FSGS. Though kidney manifestation of the disease has not been emphasized before, accompanying proteinuria and FSGS might further deteriorate the prognosis, especially when detected belatedly after the advancement of sclerosis. Therefore, screening for proteinuria and kidney function should be considered when managing patients with APS. Further studies are needed for novel treatment strategies.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by Institutional Review Board of Seoul National University Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
SJ and HGK drafted the initial manuscript and reviewed and revised the manuscript. JSK, JMK, and YHA reviewed the manuscript for important content. SL reviewed the pathologic findings of the patient. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Carrero D, Soria-Valles C, López-Otín C. Hallmarks of progeroid syndromes: lessons from mice and reprogrammed cells. Dis Model Mech. (2016) 9:719–35. doi: 10.1242/dmm.024711
2. Navarro CL, Cau P, Lévy N. Molecular bases of progeroid syndromes. Hum Mol Genet. (2006) 15(Spec No 2):R151–61. doi: 10.1093/hmg/ddl214
3. Dwyer N, Blobel G. A modified procedure for the isolation of a pore complex-lamina fraction from rat liver nuclei. J Cell Biol. (1976) 70:581–91. doi: 10.1083/jcb.70.3.581
4. Gerace L, Huber MD. Nuclear lamina at the crossroads of the cytoplasm and nucleus. J Struct Biol. (2012) 177:24–31. doi: 10.1016/j.jsb.2011.11.007
5. Burke B, Stewart CL. The nuclear lamins: flexibility in function. Nat Rev Mol Cell Biol. (2013) 14:13–24. doi: 10.1038/nrm3488
6. Han X, Feng X, Rattner JB, Smith H, Bose P, Suzuki K, et al. Tethering by lamin A stabilizes and targets the ING1 tumour suppressor. Nat Cell Biol. (2008) 10:1333–40. doi: 10.1038/ncb1792
7. Ivorra C, Kubicek M, González JM, Sanz-González SM, Alvarez-Barrientos A, O’Connor JE, et al. A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev. (2006) 20:307–20. doi: 10.1101/gad.349506
8. Wilson KL, Foisner R. Lamin-binding proteins. Cold Spring Harb Perspect Biol. (2010) 2:a000554. doi: 10.1101/cshperspect.a000554
9. Worman HJ, Ostlund C, Wang Y. Diseases of the nuclear envelope. Cold Spring Harb Perspect Biol. (2010) 2:a000760. doi: 10.1101/cshperspect.a000760
10. Worman HJ, Bonne G. “Laminopathies”: a wide spectrum of human diseases. Exp Cell Res. (2007) 313:2121–33. doi: 10.1016/j.yexcr.2007.03.028
11. Fernandez-Pombo A, Sanchez-Iglesias S, Araujo-Vilar D, Guillin-amarelle C. Lipodystrophic laminopathies diagn clues. Nucleus. (2018) 9:249–60. doi: 10.1080/19491034.2018.1454167
12. Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. (2006) 140:2603–24. doi: 10.1002/ajmg.a.31346
13. Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. (2008) 358:592–604. doi: 10.1056/NEJMoa0706898
14. Doubaj Y, De Sandre-Giovannoli A, Vera EV, Navarro CL, Elalaoui SC, Tajir M, et al. An inherited LMNA gene mutation in atypical progeria syndrome. Am J Med Genet A. (2012) 158A:2881–7. doi: 10.1002/ajmg.a.35557
15. Garg A, Subramanyam L, Agarwal AK, Simha V, Levine B, D’Apice MR, et al. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J Clin Endocrinol Metab. (2009) 94:4971–83. doi: 10.1210/jc.2009-0472
16. De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. (2003) 300:2055. doi: 10.1126/science.1084125
17. Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. (2003) 423:293–8. doi: 10.1038/nature01629
18. Dechat T, Shimi T, Adam SA, Rusinol AE, Andres DA, Spielmann HP, et al. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc Natl Acad Sci. (2007) 104:4955–60. doi: 10.1073/pnas.0700854104
19. Delbarre E, Tramier M, Coppey-Moisan M, Gaillard C, Courvalin JC, Buendia B. The truncated prelamin A in Hutchinson-Gilford progeria syndrome alters segregation of A-type and B-type lamin homopolymers. Hum Mol Genet. (2006) 15:1113–22. doi: 10.1093/hmg/ddl026
20. Liu Y, Rusinol A, Sinensky M, Wang Y, Zou Y. DNA Damage responses in progeroid syndromes arise from defective maturation of prelamin A. J Cell Sci. (2006) 119:4644–9. doi: 10.1242/jcs.03263
21. Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci. (2006) 103:8703–8. doi: 10.1073/pnas.0602569103
22. Ajluni N, Meral R, Neidert AH, Brady GF, Buras E, McKenna B, et al. Spectrum of disease associated with partial lipodystrophy: lessons from a trial cohort. Clin Endocrinol (Oxf). (2017) 86:698–707. doi: 10.1111/cen.13311
23. Hussain I, Jin RR, Baum HBA, Greenfield JR, Devery S, Xing C, et al. Multisystem progeroid syndrome with lipodystrophy, cardiomyopathy, and nephropathy due to an LMNA p.R349W variant. J Endocr Soc. (2020) 4:bvaa104. doi: 10.1210/jendso/bvaa104
24. Hussain I, Patni N, Ueda M, Sorkina E, Valerio CM, Cochran E, et al. A novel generalized lipodystrophy-associated progeroid syndrome due to recurrent heterozygous LMNA p.T10I mutation. J Clin Endocrinol Metab. (2018) 103:1005–14. doi: 10.1210/jc.2017-02078
25. Kandhaya-Pillai R, Hisama FM, Bucks SA, Yarzar S, Korovou H, Martin GM, et al. Novel LMNA mutations in Greek and Myanmar patients with progeroid features and cardiac manifestations. Aging Pathobiol Ther. (2020) 2:101–5. doi: 10.31491/apt.2020.06.021
26. Magno S, Ceccarini G, Pelosini C, Ferrari F, Prodam F, Gilio D, et al. Atypical progeroid syndrome and partial lipodystrophy due to LMNA gene p.R349W mutation. J Endocr Soc. (2020) 4:bvaa108. doi: 10.1210/jendso/bvaa108
27. Motegi S, Yokoyama Y, Uchiyama A, Ogino S, Takeuchi Y, Yamada K, et al. First Japanese case of atypical progeroid syndrome/atypical Werner syndrome with heterozygous LMNA mutation. J Dermatol. (2014) 41:1047–52. doi: 10.1111/1346-8138.12657
28. Renard D, Fourcade G, Milhaud D, Bessis D, Esteves-Vieira V, Boyer A, et al. Novel LMNA mutation in atypical werner syndrome presenting with ischemic disease. Stroke. (2009) 40:e11–4. doi: 10.1161/STROKEAHA.108.531780
29. Yanhua X, Suxian Z. Cerebral haemorrhage in a young patient with atypical Werner syndrome due to mutations in LMNA. Front Endocrinol (Lausanne). (2018) 9:433. doi: 10.3389/fendo.2018.00433
30. Jiajue R, Feng K, Wang R, Xia W. Recurrent femoral fractures in a boy with an atypical progeroid syndrome: a case report. Calcif Tissue Int. (2020) 106:325–30. doi: 10.1007/s00223-019-00639-5
31. Schultz B, Miller DD, Maguiness S. Diffuse, mottled hyperpigmentation and mutations in LMNA gene in a 5-year-old boy, his mother, and his grandmother: atypical progeroid syndrome. Pediatr Dermatol. (2019) 36:913–7. doi: 10.1111/pde.13917
32. Yukina M, Nuralieva N, Sorkina E, Troshina E, Tiulpakov A, Belaya Z, et al. Atypical progeroid syndrome (p.E262K LMNA mutation): a rare cause of short stature and osteoporosis. Endocrinol Diabetes Metab Case Rep. (2021) 2021:20-0188. doi: 10.1530/EDM-20-0188
33. Lee S, Park SM, Kim HJ, Kim JW, Yu DS, Lee YB. Genomic diagnosis by whole genome sequencing in a Korean family with atypical progeroid syndrome. J Dermatol. (2015) 42:1149–52. doi: 10.1111/1346-8138.13005
34. Liang L, Zhang H, Gu X. Homozygous LMNA mutation R527C in atypical Hutchinson-Gilford progeria syndrome: evidence for autosomal recessive inheritance. Acta Paediatr. (2009) 98:1365–8. doi: 10.1111/j.1651-2227.2009.01324.x
35. Doh YJ, Kim HK, Jung ED, Choi SH, Kim JG, Kim BW, et al. Novel LMNA gene mutation in a patient with atypical Werner's Syndrome. Korean J Intern Med. (2009) 24:68–72. doi: 10.3904/kjim.2009.24.1.68
36. Mahdi L, Kahn A, Dhamija R, Vargas HE. Hepatic steatosis resulting from LMNA-associated familial lipodystrophy. ACG Case Rep J. (2020) 7:e00375. doi: 10.14309/crj.0000000000000375
37. Fountas A, Giotaki Z, Dounousi E, Liapis G, Bargiota A, Tsatsoulis A, et al. Familial partial lipodystrophy and proteinuric renal disease due to a missense c.1045C > T LMNA mutation. Endocrinol Diabetes Metab Case Rep. (2017) 2017:17-0049. doi: 10.1530/EDM-17-0049
38. Imachi H, Murao K, Ohtsuka S, Fujiwara M, Muraoka T, Hosokawa H, et al. A case of Dunnigan-type familial partial lipodystrophy (FPLD) due to lamin A/C (LMNA) mutations complicated by end-stage renal disease. Endocrine. (2009) 35:18–21. doi: 10.1007/s12020-008-9127-1
39. Owen KR, Donohoe M, Ellard S, Clarke TJ, Nicholls AJ, Hattersley AT, et al. Mesangiocapillary glomerulonephritis type 2 associated with familial partial lipodystrophy (Dunnigan-Kobberling syndrome). Nephron Clin Pract. (2004) 96:c35–8. doi: 10.1159/000076396
40. Rankin J, Auer-Grumbach M, Bagg W, Colclough K, Nguyen TD, Fenton-May J, et al. Extreme phenotypic diversity and nonpenetrance in families with the LMNA gene mutation R644C. Am J Med Genet A. (2008) 146A:1530–42. doi: 10.1002/ajmg.a.32331
41. Thong KM, Xu Y, Cook J, Takou A, Wagner B, Kawar B, et al. Cosegregation of focal segmental glomerulosclerosis in a family with familial partial lipodystrophy due to a mutation in LMNA. Nephron Clin Pract. (2013) 124:31–7. doi: 10.1159/000354716
42. Clouthier DE, Comerford SA, Hammer RE. Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like syndrome in PEPCK-TGF-beta1 transgenic mice. J Clin Invest. (1997) 100:2697–713. doi: 10.1172/JCI119815
43. Le Dour C, Wu W, Béréziat V, Capeau J, Vigouroux C, Worman HJ. Extracellular matrix remodeling and transforming growth factor-beta signaling abnormalities induced by lamin A/C variants that cause lipodystrophy. J Lipid Res. (2017) 58:151–63. doi: 10.1194/jlr.M071381
44. Frangogiannis N. Transforming growth factor-beta in tissue fibrosis. J Exp Med. (2020) 217:e20190103. doi: 10.1084/jem.20190103
45. Baek HS. Mechanism, clinical consequences, and management of dyslipidemia in children with nephrotic syndrome. Child Kidney Dis. (2022) 26(1):25–30. doi: 10.3339/ckd.22.020
46. Kim JH, Kim BK, Moon KC, Hong HK, Lee HS. Activation of the TGF-beta/smad signaling pathway in focal segmental glomerulosclerosis. Kidney Int. (2003) 64:1715–21. doi: 10.1046/j.1523-1755.2003.00288.x
47. Lee HS, Song CY. Effects of TGF-beta on podocyte growth and disease progression in proliferative podocytopathies. Kidney Blood Press Res. (2010) 33:24–9. doi: 10.1159/000285844
48. Van Berlo JH, Voncken JW, Kubben N, Broers JL, Duisters R, van Leeuwen RE, et al. A-type lamins are essential for TGF-beta1 induced PP2A to dephosphorylate transcription factors. Hum Mol Genet. (2005) 14:2839–49. doi: 10.1093/hmg/ddi316
49. Chen RX, Zhang L, Ye W, Wen YB, Si N, Li H, et al. The renal manifestations of type 4 familial partial lipodystrophy: a case report and review of literature. BMC Nephrol. (2018) 19:111. doi: 10.1186/s12882-018-0913-6
50. Zhang LH, Zhu XY, Eirin A, Nargesi AA, Woollard JR, Santelli A, et al. Early podocyte injury and elevated levels of urinary podocyte-derived extracellular vesicles in swine with metabolic syndrome: role of podocyte mitochondria. Am J Physiol Ren Physiol. (2019) 317:F12–22. doi: 10.1152/ajprenal.00399.2018
51. Lan HY. Smads as therapeutic targets for chronic kidney disease. Kidney Res Clin Pract. (2012) 31(1):4–11. doi: 10.1016/j.krcp.2011.12.001
52. Trachtman H, Fervenza FC, Gipson DS, Heering P, Jayne DR, Peters H, et al. A phase 1, single-dose study of fresolimumab, an anti-TGF-beta antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. (2011) 79:1236–43. doi: 10.1038/ki.2011.33
53. Vincenti F, Fervenza FC, Campbell KN, Diaz M, Gesualdo L, Nelson P, et al. A phase 2, double-blind, placebo-controlled, randomized study of fresolimumab in patients with steroid-resistant primary focal segmental glomerulosclerosis. Kidney Int Rep. (2017) 2:800–10. doi: 10.1016/j.ekir.2017.03.011
54. Harhouri K, Frankel D, Bartoli C, Roll P, De Sandre-Giovannoli A, Lévy N. An overview of treatment strategies for Hutchinson-Gilford progeria syndrome. Nucleus. (2018) 9:246–57. doi: 10.1080/19491034.2018.1460045
55. Varela I, Pereira S, Ugalde AP, Navarro CL, Suárez MF, Cau P, et al. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med. (2008) 14:767–72. doi: 10.1038/nm1786
Keywords: focal segmental glomerular sclerosis (FSGS), atypical progeroid syndrome, lipodystrophy, LMNA, TGF - β1
Citation: Jang S, Ahn YH, Ko JM, Ko JS, Lim S and Kang HG (2022) Case report: Focal segmental glomerulosclerosis in a pediatric atypical progeroid syndrome. Front. Pediatr. 10:1032653. doi: 10.3389/fped.2022.1032653
Received: 31 August 2022; Accepted: 3 October 2022;
Published: 31 October 2022.
Edited by:
Tracy E. Hunley, Monroe Carell Jr. Children's Hospital at Vanderbilt, United StatesReviewed by:
Gentzon Hall, Duke University, United States© 2022 Jang, Ahn, Ko, Ko, Lim and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hee Gyung Kang a2FuZ2hnQHNudS5hYy5rcg==
Specialty Section: This article was submitted to Pediatric Nephrology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.