 Xiaoli Li
Xiaoli Li Jian Shen*
Jian Shen*
- Department of Pediatrics, Affiliated Hangzhou First People’s Hospital, Zhejiang University School of Medicine, Hangzhou, China
Background: Carnitine-acylcarnitine translocase deficiency (CACT deficiency) is a rare and life-threatening autosomal recessive disorder of mitochondrial fatty acid oxidation caused by variant of SLC25A20 gene. The most prevalent missense variant in the SLC25A20 gene in Asia was c.199–10T > G. Due to the c.199–10T > G variant, CACT deficiency is a severe phenotype.
Materials and Methods: Herein, we present a neonatal case with c.199–10T > G variant in China and analyze the clinical, biochemical, and genetic aspects of 78 patients previously identified with CACT deficiency.
Results: The patient presented with a series of severe metabolic crises that rapidly deteriorated and eventually died 3 days after delivery. The sequencing of the patient's genome indicated that he was homozygous for the c.199–10T > G variant. 30 patients were found to have the c.199–10T > G mutation, of which 23 were Chinese and 22 were afflicted by the c.199–10T > G splicing variation. In China, c.199–10T > G allele frequency was 82.6%.
Conclusion: In CACT deficiency, prompt recognition and treatment are critical. Our data suggested that c.199–10T > G may be a potential hotspot SLC25A20 gene mutation in the Chinese population. Detection of single nucleotide polymorphism is possible for high-risk patients and parents in China.
Introduction
Carnitine-acylcarnitine translocase deficiency (CACT deficiency, OMIM # 212138) was first described by Stanley CA et al. in 1992 (1). It is a rare and life-threatening autosomal recessive disorder with an incidence of 1:60,000 in Hongkong and 1:1,017,593 in Zhejiang province China (2, 3). CACT deficiency, encoded by the SLC25A20 gene on chromosome 3p21.31, is the cause of this condition (4). To shuttle long-chain fatty acids through the inner mitochondrial membrane and into the mitochondrial matrix, where mitochondrial β-oxidation takes place, CACT is an essential part of the carnitine cycle (5). Mitochondrial β-oxidation serves as the primary energy source for cardiac and skeletal muscles, while ketogenesis in the liver fuels brains tissue during prolonged fasting and exercise (1, 6, 7). CACT deficiency is characterized by a wide spectrum of clinical manifestations including hypoketotic hypoglycemia, hyperammonemia, liver dysfunction, cardiomyopathy, severe neurologic impairment and progressive myopathy (8).
There are nine exons in the SLC25A20 gene, which produces a 301-amino-acid protein (9). At least 42 pathogenic variants have been detected in mutation databases like the HGMD around the world (Human Gene Mutation Database, www.hgmd.org). There are 20 missenses, 10 small deletions, 2 small insertions, 1 small indel, 4 large deletions, and 5 splicing mutations in the mutation spectrum (10). SLC25A20 gene missense variation c.199–10T > G was the most frequent in Asia. In our study, approximately 37.5% of pathogenic variants fall within this umbrella.
Though the spectrum of CACT deficiency is wide and continuous, there are two distinct clinical subtypes: a neonatal-onset severe form and an infancy-onset milder form (11). Severe classic presentation occurs at birth and has an extremely poor prognosis, with severe illness and debilitating symptoms. Moderate myopathy and hepatomegaly are seen in milder cases with more accessible residual transporter protein. Metabolic decompensation can be prevented and the prognosis improved with early detection and medicinal intervention (12).
In the present study, we described a patient with CACT deficiency which was failed to diagnosed and treated promptly and then leaded to rapid illness progression and eventual death. Furthermore, Patients previously diagnosed with CACT deficiency was reviewed systematically by describing the clinical, biochemical, and genetic characteristics and treatment to improve our understanding of this rare disorder.
Materials and methods
Case report
The patient was born at full term through a cesarean section. At 1 and 5 min, the baby's Apgar score was 10. The parents were healthy and had no history of consanguineous marriage. The mother's first child died at two days old from asphyxia, arrhythmia, and cardiac arrest. The baby seemed fine until he was 28 h old when he became very sleepy and showed no interest in breastfeeding. The patient was taken to the neonatal intensive care unit (NICU) for a checkup. During a physical exam, the baby's body temperature was 34.6°C, his blood pressure was 72/44 mmHg, and his SpO2 was 95%. Table 1 gives detailed information about the patient's health. After a test for sepsis was done, he was given antibiotics, and a 6.5 mg/ka/minute glucose infusion was started. The patient had recurrent ventricular tachycardia, bradycardia, and complete right bundle branch block between the ages of 47 and 51 h. Lidocaine, epinephrine, and milrinone were used to treat the patient for several days after the attack. Several attempts to save her life failed, and she died 3 days after giving birth. At 48 h, a small spot of dried blood was taken as a sample. Acylcarnitine profile by MS/MS analysis showed C14-acylcanitine, 1.13 µM (0.07–0.4); C16-acylcanitine, 14.37 µM (0.49–6); C16:1-acylcanitine, 1.40 µM (0.02–0.49); C18-acylcanitine, 3.73 µM (0.24–1.90); C18:1-acylcanitine, 5.83 µM (0.38–2.92), which were identified to have CPT2 (carnitine palmitoyl-transferases 2) deficiency or CACT deficiency. The patient's genomic DNA was taken out. High-throughput sequencing found that the patient's SLC25A20 gene had a homozygous c.199–10T > G splice site change (Figure 1). The child was diagnosed with CACT deficiency based on the signs and symptoms. Both parents were heterozygous carriers of the variation, but neither had any obvious symptoms.
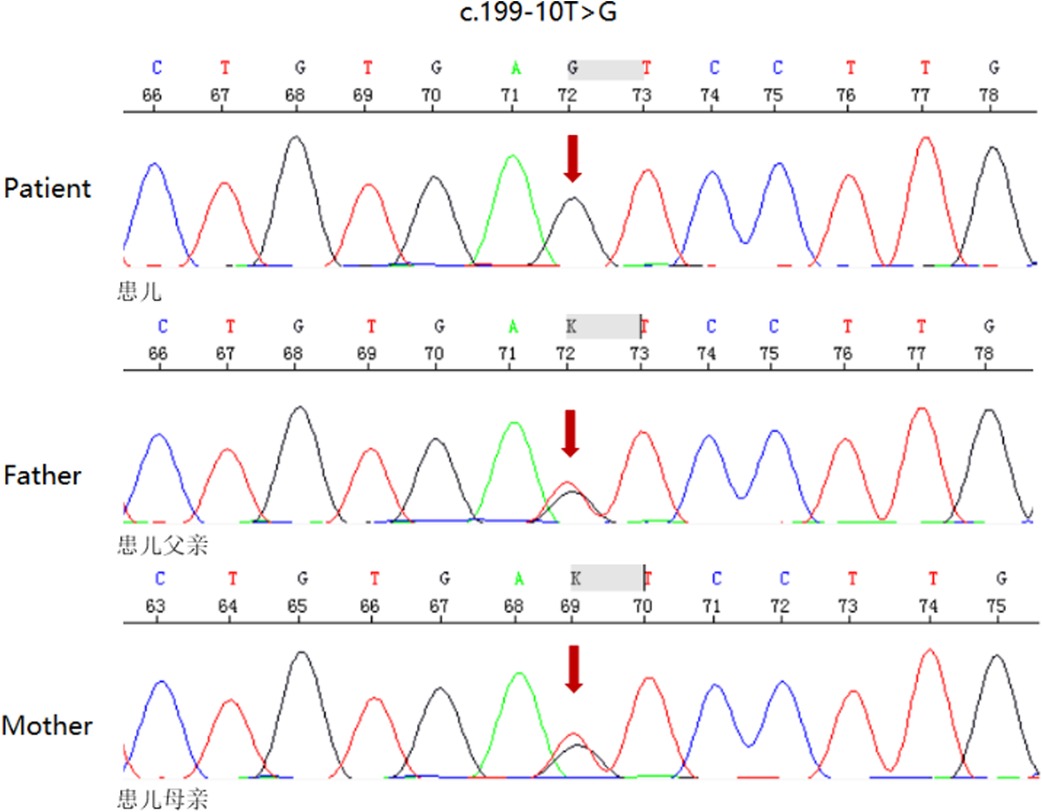
Figure 1. DNA sequence of SLC25A20 gene (a homozygous mutation of patient for maternally and paternally-inherited c.199–10 T > G variant. Parents were heterozygous carriers of the mutation. Mutation sites are indicated by red arrows)
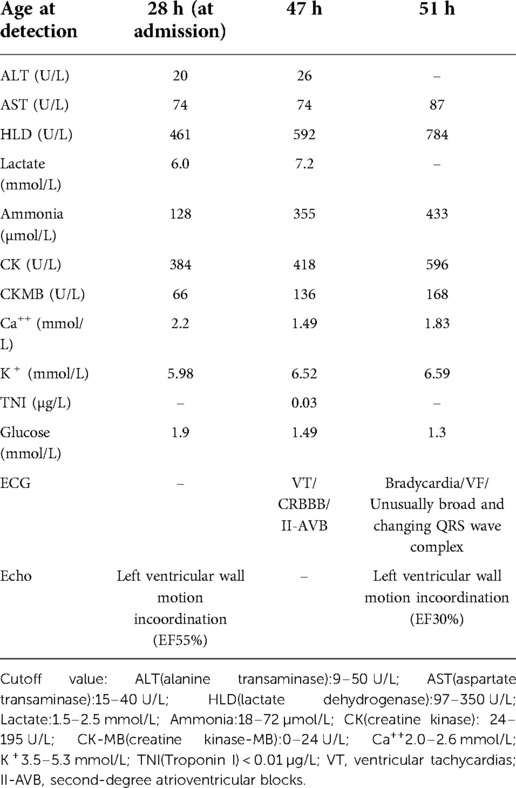
Table 1. Summary of the patient with CACT deficiency biochemistry, ECG and Echo
Literature search
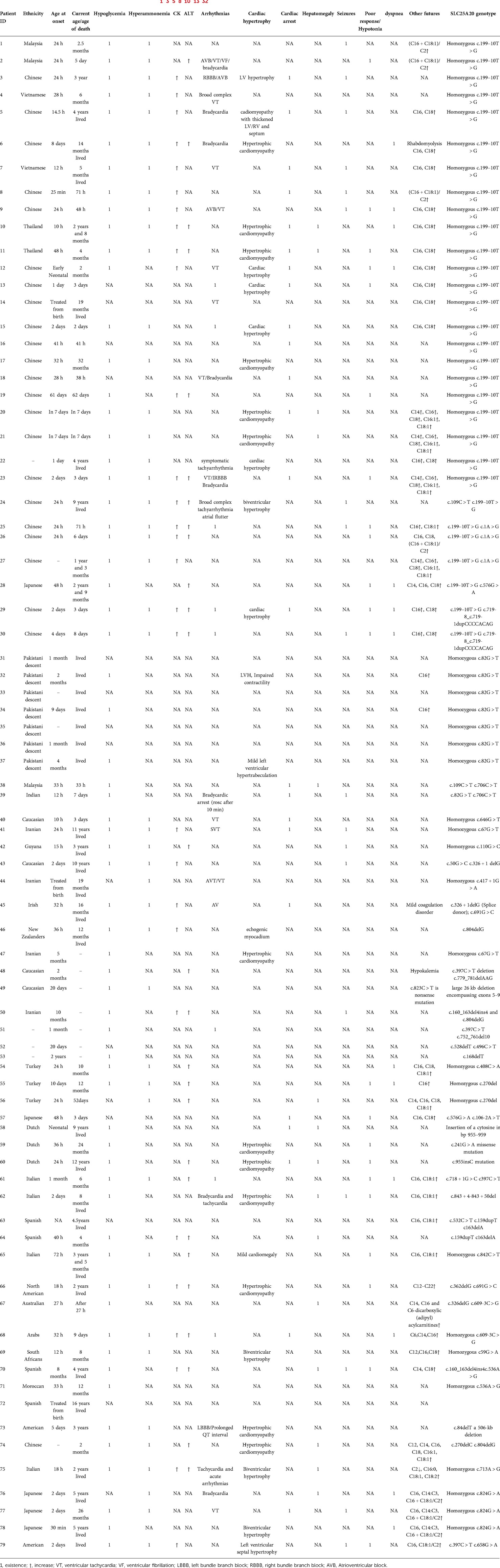
All patients previously genetically diagnosed with CACT deficiency were reviewed in the study. The literature search for “Carnitine-acylcarnitine translocase deficiency,” “Carnitine-acylcarnitine translocase,” “SLC25A20 gene,” “CACTD,” as keywords on PubMed, Elsevier and Medline from 1992 to June 2022. (Table 2).
Results
Individual case reports are available for all cases as online.
Case for CACT deficiency
Over 30 years, 81 children with CACT deficiency were identified, 3 of which lacked genetic testing to determine the mutation sites. Therefore, in addition to the newly identified patient with CACT deficiency, we included 78 previously diagnosed patients in our sample.
Genetic findings
Patients harbored either homozygous or heterozygous SLC25A20 mutations. 42 different variations have been discovered. The most prevalent splicing variant was homozygous c.199–10T > G (23/79). Heterozygous c.199–10T > G splicing variation (7/79) and homozygous c.82G > T splicing variation (7/79) were quite prevalent variations. Each of the forty remaining variants was detected one to three times. The c.199–10T > G variant was identified in thirty patients (38.0%), of whom 73.3% (22/30) were Chinese. 29.1% (23/79) of the patients were Chinese, and 95.7% (22/23) were affected by the c.199–10T > G splicing mutation. The remaining sufferers were scattered in different nations. The frequency of the c.199–10T > G allele was 33.5% in all cases, whereas it was 82.6% in China.
Biochemical and clinical specifications
Twenty of the thirty individuals with the c.199–10T > G variant exhibited clinical symptoms within 48 h (66.7%). There were arrhythmias in 18 patients (60%), cardiomyopathy in 13 patients (43.3%), hepatomegaly in 4 patients (13.3%), seizures in 9 patients (30%), hyperammonemia in 22 patients (73.3%), increase of CKMB and ALT in 17 patients (56.7%) and 12 patients (40%) respectively. 23 of 30 individuals (76.7%) perished due to the variation c.199–10T > G, which is linked to a severe phenotype.
7/79 cases were attenuated, and homozygosity for variation c.82G > T was confirmed. All of these patients of Pakistani heritage survived. Three patients were diagnosed with cardiomyopathy, although only one had seizures. There were no other clinical signs noted in these patients.
42/79 individuals harbored additional SLC25A20 mutations. 25 out of 42 participants exhibited clinical signs within 48 h (59.6%). These patients exhibited associated clinical manifestations: 14/42 (33.3%) had arrhythmias, 15/42 (35.7%) had bouts of cardiomyopathy, 11/42(26.2%) had hepatomegaly, and 7/42(16.7%) had seizures. Ammonia, CK and ALT levels were elevated in 22/42(52.3%), 11/42(26.2%) and 14/42(33.3%) patients, respectively.
Discussion
CACT deficiency appears to be very rare in the general population, except for a small number of ethnic subgroups. This study described the biochemical, clinical and genetic characteristics of patients with CACT deficiency, analyzed the distribution and ethnic specificity of the pathogenic genes, provided a theoretical basis of single nucleotide polymorphism detection, and thus contributed to the body of knowledge for early diagnosis and timely intervention in this rare disorder.
There is a wide variation in the prevalence of CACT deficiency among ethnic groups. About 30 cases of CTCT deficiency have been reported elsewhere, while there have been more than 50 reported in Asia. It is reported that the estimated incidence of CACT deficiency is 1/60,000 in Hongkong, 1/76,894 in Hunan, and at least 1/100,000 in Guangzhou China (2, 10, 15). Caucasian groups had a substantially lower incidence of CACT deficiency, which was reported to be 1:750,000–1:2,000,000. Indeed, the misleading clinical presentation, poor prognosis and the need to collect blood and urine specimens for metabolic investigation at an appropriate time in relation to the illness frequently limit the recognition of the disorder. For these reasons, the frequency of CACT deficient inborn defects is probably higher than recorded cases.
CACT deficiency is one of the most severe disorders of the carnitine transport system and mitochondrial fatty acid oxidation. The disorder results in deficient formation of energy-yielding substrates and toxicity of acylcarnitine accumulation which plays a pivotal role in the production of arrhythmias, and then presents a simultaneous dysfunction of the heart, liver, and skeletal muscle, associated with hypoketotic hypoglycemia (33). Severe classic manifestation, the most common, is accompanied by severe hypoketotic hypoglycemia, refractory hyperammonemia, elevated transaminase levels and CK, cardiomyopathy, and abrupt arrhythmias. The prognosis for those with the classic results is exceptionally dismal. Moderate myopathy and hepatomegaly are seen in milder cases, which are less common but do have more accessible residual transporter protein (11, 34, 35). Following an initial metabolic decompensation at birth, the neonate, in our case, developed hypoglycemia, hyperammonemia, and acute and severe arrhythmias before passing away. This may be related to the less residual enzyme activity and the increased accumulation of carnitine-acylcarnitine (33, 34). In our study, CACT deficiency due to the c.199–10T > G variation is a severe phenotype with a significantly higher mortality, arrhythmia, seizures, and hyperammonemia incidence than other variations, while CACT deficiency caused by the c.82G > T mutation is associated with milder phenotype (5). The most frequent mutation was a splicing site variation of c.199–10T > G.
Early recognition and timely treatment are crucial in CACT deficiency. NBS (Newborn screening) plays an important role in early detection of deficiency in enzymes of mitochondrial carnitine-acylcarnitinecycle. Most patients with CACT and CPT2 deficiency had a higher C12–C18:1 level than those without these (3). SLC25A20 gene mutational analysis is required to identify CACT deficiency, but this can be done without CACT activity assessment. Once a CACT deficiency has been diagnosed, the proper treatment must be implemented. To begin, sufficient glucose must be provided to prevent lipolysis from being broken down. Fasting prevention with frequent meals, a diet rich in carbohydrate, restricting long-chain fatty acids, supplementing with medium chain triglycerides (MCT) and essential polyunsaturated fatty acids are recommended as long-term treatments for CACT deficiency. Administration of carnitine is controversial, on the one hand it exerts to prevent arrhythmias, and on the other hand it causes an increase of acylcarnitines, responsible of arrhythmias (26, 33, 35). Triheptanoin can ameliorate acute cardiomyopathy and increase survival in patients with severe long-chain fatty acid oxidation disorders (26). CACT deficiency can also be treated with skimmed breast milk (30).
There are nine exons in the SLC25A20 gene, which spans more than 903 bp of genomic DNA on chromosome 3p21.31 (35). At least 42 pathogenic variants of SLC25A20 have been discovered so far. In contrast to the majority of pathogenic variations, c.199–10T > G and c.82G > T were shown to be shared in Asian and Pakistani origins, respectively. The founder mutation c.82G > T was detected in people of Pakistani ancestry. Our study's Chinese patients were found to have a wide range of homology, with seven distinct variants. The most common variant, c.199–10T > G, suggests that c.199–10T > G may be a hotspot of SLC25A20 gene mutation in the Chinese population. c.1A > G was detected only 3 times and was not yet found in other than Chinese populations, so it may be unique to the Chinese individuals. The remaining variant was detected only once or twice.
In conclusion, the biochemical, clinical, and genetic characteristics of Chinese patients with CACT deficiency identified in this investigation may aid early identification and intervention. In addition, it appeared from the data that the Chinese patients have a high degree of homozygosity. The c.199–10T > G variant, which is the most common one in this population, has the potential to be a hotspot SLC25A20 gene mutation. As a result, economical and rapid single nucleotide polymorphism and the genotyping assay can be performed for high-risk patients and their parents in China. In addition, prenatal or presymptomatic diagnosis can be performed in siblings.
Data availability statement
All data, models, and code generated or used during the study appear in the submitted article.
Ethics statement
Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
JS: contributed significantly to analysis and manuscript preparation. XL: performed the data analyses and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Construction Fund of Hangzhou Medical Key Discipline (2020-2024) (Grant No.: OO20200448).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Stanley CA, Hale DE, Berry GT, Deleeuw S, Boxer J, Bonnefont JP. Brief report: a deficiency of carnitine-acylcarnitine translocase in the inner mitochondrial membrane. N Engl J Med. (1992) 327(1):19–23. doi: 10.1056/NEJM199207023270104
2. Hui J, Tang NL, Li CK, Law LK, To KF, Yau P, et al. Inherited metabolic diseases in the Southern Chinese population: spectrum of diseases and estimated incidence from recurrent mutations. Pathology. (2014) 46(5):375–82. doi: 10.1097/PAT.0000000000000140
3. Zhou D, Cheng Y, Yin X, Miao H, Hu Z, Yang J, et al. Newborn screening for mitochondrial carnitine-acylcarnitine cycle disorders in Zhejiang Province, China. Front Genet. (2022) 13:823687. doi: 10.3389/fgene.2022.823687
4. Rubio-Gozalbo ME, Bakker JA, Waterham HR, Wanders RJ. Carnitine-acylcarnitine translocase deficiency, clinical, biochemical and genetic aspects. Mol Aspects Med. (2004) 25(5–6):521–32. doi: 10.1016/j.mam.2004.06.007
5. Ryder B, Inbar-Feigenberg M, Glamuzina E, Halligan R, Vara R, Elliot A, et al. New insights into carnitine-acylcarnitine translocase deficiency from 23 cases: management challenges and potential therapeutic approaches. J Inherit Metab Dis. (2021) 44(4):903–15. doi: 10.1002/jimd.12371
6. Loft A, Schmidt SF, Caratti G, Stifel U, Havelund J, Sekar R, et al. A macrophage-hepatocyte glucocorticoid receptor axis coordinates fasting ketogenesis. Cell Metab. (2022) 34(3):473–86.e9. doi: 10.1016/j.cmet.2022.01.004
7. Stanley CA. Carnitine deficiency disorders in children. Ann N Y Acad Sci. (2004) 1033:42–51. doi: 10.1196/annals.1320.004
8. Li X, Zhao F, Zhao Z, Zhao X, Meng H, Zhang D, et al. Neonatal sudden death caused by a novel heterozygous mutation in SLC25A20 gene: a case report and brief literature review. Leg Med. (2022) 54:101990. doi: 10.1016/j.legalmed.2021.101990
9. Vatanavicharn N, Yamada K, Aoyama Y, Fukao T, Densupsoontorn N, Jirapinyo P, et al. Carnitine-acylcarnitine translocase deficiency: two neonatal cases with common splicing mutation and in vitro bezafibrate response. Brain Dev. (2015) 37(7):698–703. doi: 10.1016/j.braindev.2014.10.005
10. Tang C, Liu S, Wu M, Lin S, Lin Y, Su L, et al. Clinical and molecular characteristics of carnitine-acylcarnitine translocase deficiency: experience with six patients in Guangdong China. Clin Chim Acta. (2019) 495:476–80. doi: 10.1016/j.cca.2019.05.018
11. Palmieri F, Scarcia P, Monné M. Diseases caused by mutations in mitochondrial carrier genes SLC25: a review. Biomolecules. (2020) 10(4):655. doi: 10.3390/biom10040655
12. Maguolo A, Rodella G, Dianin A, Nurti R, Monge I, Rigotti E, et al. Diagnosis, genetic characterization and clinical follow up of mitochondrial fatty acid oxidation disorders in the new era of expanded newborn screening: a single centre experience. Mol Genet Metab Rep. (2020) 24:100632. doi: 10.1016/j.ymgmr.2020.100632
13. Habib A, Azize NAA, Rahman SA, Yakob Y, Suberamaniam V, Nazri MIBA, et al. Novel mutations associated with carnitine-acylcarnitine translocase and carnitine palmitoyl transferase 2 deficiencies in Malaysia. Clin Biochem. (2021) 98:48–53. doi: 10.1016/j.clinbiochem.2021.10.002
14. Wang GL, Wang J, Douglas G, Browning M, Hahn S, Ganesh J, et al. Expanded molecular features of carnitine acyl-carnitine translocase (CACT) deficiency by comprehensive molecular analysis. Mol Genet Metab. (2011) 103(4):349–57. doi: 10.1016/j.ymgme.2011.05.001
15. Yan HM, Hu H, Ahmed A, Feng BB, Liu J, Jia ZJ, et al. Carnitine-acylcarnitine translocase deficiency with c.199–10 T > G and novel c.1A > G mutation: two case reports and brief literature review. Medicine. (2017) 96(45):e8549. doi: 10.1097/MD.0000000000008549
16. Gürbüz BB, Yılmaz DY, Özgül RK, Koşukcu C, Dursun A, Sivri HS, et al. Clinical and molecular characteristics of carnitineacylcarnitine translocase deficiency with c.270delC and a novel c.408C > A variant. Turk J Pediatr. (2021) 63(4):691–6. doi: 10.24953/turkjped.2021.04.017.
17. Fukushima T, Kaneoka H, Yasuno T, Sasaguri Y, Tokuyasu T, Tokoro K, et al. Three novel mutations in the carnitine-acylcarnitine translocase (CACT) gene in patients with CACT deficiency and in healthy individuals. J Hum Genet. (2013) 58(12):788–93. doi: 10.1038/jhg.2013.103
18. Pierre G, Macdonald A, Gray G, Hendriksz C, Preece MA, Chakrapani A. Prospective treatment in carnitine-acylcarnitine translocase deficiency. J Inherit Metab Dis. (2007) 30(5):815. doi: 10.1007/s10545-007-0518-x
19. Lee RS, Lam CW, Lai CK, Yuen YP, Chan KY, Shek CC, et al. Carnitine-acylcarnitine translocase deficiency in three neonates presenting with rapid deterioration and cardiac arrest. Hong Kong Med J. (2007) 13(1):66–817277394
20. Huizing M, Iacobazzi V, Ijlst L, Savelkoul P, Ruitenbeek W, van den Heuvel L, et al. Cloning of the human carnitine-acylcarnitine carrier cDNA and identification of the molecular defect in a patient. Am J Hum Genet. (1997) 61(6):1239–45. doi: 10.1086/301628
21. IJlst L, van Roermund CW, Iacobazzi V, Oostheim W, Ruiter JP, Williams JC, et al. Functional analysis of mutant human carnitine acylcarnitine translocases in yeast. Biochem Biophys Res Commun. (2001) 280(3):700–6. doi: 10.1006/bbrc.2000.4178
22. Iacobazzi V, Invernizzi F, Baratta S, Pons R, Chung W, Garavaglia B, et al. Molecular and functional analysis of SLC25A20 mutations causing carnitine-acylcarnitine translocase deficiency. Hum Mutat. (2004) 24(4):312–20. doi: 10.1002/humu.20085
23. Korman SH, Pitt JJ, Boneh A, Dweikat I, Zater M, Meiner V, et al. A novel SLC25A20 splicing mutation in patients of different ethnic origin with neonatally lethal carnitine-acylcarnitine translocase (CACT) deficiency. Mol Genet Metab. (2006) 89(4):332–8. doi: 10.1016/j.ymgme.2006.06.009
24. Chen M, Cai Y, Li S, Xiong H, Liu M, Ma F, et al. Late-onset carnitine-acylcarnitine translocase deficiency with SLC25A2° c.199–10T > G variation: case report and pathologic analysis of liver biopsy. Front Pediatr. (2020) 8:585646. doi: 10.3389/fped.2020.585646
25. Vitoria I, Martín-Hernández E, Peña-Quintana L, Bueno M, Quijada-Fraile P, Dalmau J, et al. Carnitine-acylcarnitine translocase deficiency: experience with four cases in Spain and review of the literature. JIMD Rep. (2015) 20:11–20. doi: 10.1007/8904_2014_382
26. Mahapatra S, Ananth A, Baugh N, Damian M, Enns GM. Triheptanoin: a rescue therapy for cardiogenic shock in carnitine-acylcarnitine translocase deficiency. JIMD Rep. (2018) 39:19–23. doi: 10.1007/8904_2017_36
27. Norris MK, Scott AI, Sullivan S, Chang IJ, Lam C, Sun A, et al. Tutorial: triheptanoin and nutrition management for treatment of long-chain fatty acid oxidation disorders. JPEN J Parenter Enteral Nutr. (2021) 45(2):230–8. doi: 10.1002/jpen.2034
28. Iacobazzi V, Pasquali M, Singh R, Matern D, Rinaldo P, Amat di San Filippo C, et al. Response to therapy in carnitine/acylcarnitine translocase (CACT) deficiency due to a novel missense mutation. Am J Med Genet A. (2004) 126A(2):150–5. doi: 10.1002/ajmg.a.20573
29. Chinen Y, Yanagi K, Nakamura S, Nakayama N, Kamiya M, Nakayashiro M, et al. A novel homozygous missense SLC25A20 mutation in three CACT-deficient patients: clinical and autopsy data. Hum Genome Var. (2020) 7:11. doi: 10.1038/s41439-020-0098-y
30. Kritzer A, Tarrant S, Sussman-Karten K, Barbas K. Use of skimmed breast milk for an infant with a long-chain fatty acid oxidation disorder: a novel therapeutic intervention. JIMD Rep. (2020) 55(1):44–50. doi: 10.1002/jmd2.12152
31. Chong SC, Law LK, Hui J, Lai CY, Leung TY, Yuen YP. Expanded newborn metabolic screening programme in Hong Kong: a three-year journey. Hong Kong Med J. (2017) 23(5):489–96. doi: 10.12809/hkmj176274
32. Fan X, Xie BB, Zhang Q, Yi S, Geng GX, Yang Q, et al. Analysis of four carnitine-acylcarnitine translocase deficiency cases caused by homozygous mutation of SLC25A2° c.199–10T > G. Zhonghua Er Ke Za Zhi. (2018) 56(7):545–9. doi: 10.3760/cma.j.issn.0578-1310.2018.07.014
33. Bonnet D, Martin D, De Lonlay P, Villain E, Jouvet P, Rabier D, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. (1999) 100(22):2248–53. doi: 10.1161/01.cir.100.22.2248
34. Lopriore E, Gemke RJ, Verhoeven NM, Jakobs C, Wanders RJ, Roeleveld-Versteeg AB, et al. Carnitine-acylcarnitine translocase deficiency: phenotype, residual enzyme activity and outcome. Eur J Pediatr. (2001) 160(2):101–4. doi: 10.1007/s004310000644
Keywords: CACT deficiency, SLC25A20 gene, c.199–10T > G, hotspot mutation, single nucleotide polymorphism detection
Citation: Li X and Shen J (2022) One potential hotspot SLC25A20 gene variants in Chinese patients with carnitine-acylcarnitine translocase deficiency. Front. Pediatr. 10:1029004. doi: 10.3389/fped.2022.1029004
Received: 26 August 2022; Accepted: 17 October 2022;
Published: 7 November 2022.
Edited by:
Mike Mikailov, United States Food and Drug Administration, United StatesReviewed by:
Siqi Hu, Seventh Medical Center of PLA General Hospital, ChinaFuad Al Mutairi, King Abdulaziz Medical City, Saudi Arabia
© 2022 Li and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Shen aHpzZnlianllYmtAMTYzLmNvbQ==
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics