 Hui Wang
Hui Wang Dian Wu
Dian Wu De-Hua Wu3
De-Hua Wu3 Hai-Feng Li
Hai-Feng Li Ke-Wen Jiang
Ke-Wen Jiang Chao-Chun Zou
Chao-Chun Zou- 1Department of Rehabilitation, The Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou, China
- 2Department of Psychological, The Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou, China
- 3Department of Urology, The Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou, China
- 4Department of Endocrinology, The Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou, China
The myelin regulatory factor (MYRF; MIM# 608329) gene was first identified as a critical transcription factor involved in oligodendrocyte differentiation and central nervous system myelination. With the recent development of exome sequencing, pathogenic variants of MYRF had been considered as the cause of cardiac-urogenital syndrome (CUGS), 46,XY and 46,XX disorders/differences of sex development (DSDs), and nanophthalmos. Herein, we described a 4-year-7-month-old “girl” with ventricular septal defect, atrial septal defect, patent ductus arteriosus, severe pulmonary hypertension, moderate-to-severe tricuspid regurgitation, enlarged coronary sinus, left superior vena cava, and right lung hypoplasia at birth. Later, the patient developed short stature and amblyopia. Further examination revealed a karyotype 46,XY and visible uterus, whereas the presence of gonads were not explored. Laparoscopy revealed dysplasia of testicular gonad. Whole-exome sequencing (WES) was performed and a de novo heterozygous mutation in MYRF was identified, known as c.2817G > A/p. W939* (NM_001127392.3). Therefore, this case report presented multiple clinical manifestations with syndromic symptoms of CUGS, 46,XY DSD, and ocular symptoms. These new data expanded the phenotype of the MYRF variant and may benefit to characterize the phenotypes caused by the variants of this gene.
Introduction
Myelin regulatory factor (MYRF)-related disorders, including cardiac-urogenital syndrome (CUGS, MIM#618280) and wild encephalitis/encephalopathy with reversible myelin vacuolization (MMERV, MIM#618113), are caused by heterozygous mutations in the MYRF (MIM#608329) gene on chromosome 11q12. The MYRF gene was first identified as an essential transcription factor for oligodendrocyte differentiation and central nervous system myelination (1–4). Kurahashi et al. (5) identified a heterozygous c.1208A > G transition (NM_001127392) in the MYRF gene, resulting in a p.Q403R substitution at a highly conserved residue in the DNA-binding domain, in 9 individuals from 2 different families who had mild encephalitis/encephalopathy and reversible myelin vacuolization in Japan.
In addition to the central nervous system, MYRF is also expressed in the heart, lungs, diaphragm, and genitourinary tract. A series of de novo heterozygous mutations in MYRF were identified in patients with CUGS (6–10). The symptoms of CUGS included partial anomalous pulmonary venous return associated with tracheal anomalies, pulmonary hypoplasia, congenital diaphragmatic hernia (CDH), thyroid fibrosis, thymic involution, cleft spleen, penoscrotal hypospadias, and cryptorchidism (6). A few patients also suffered from short stature, speech delay, intellectual disability and motor delay, and intestinal malrotation. Hamanaka et al. (11) found MYRF playing a key role in gonadal cells and tissues development such as Müllerian derivatives, ovaries, Sertoli cells, and Leydig cells. They demonstrated that MYRF haploinsufficiency caused a type of disorders/differences of sex development (DSDs) (11). In addition, other studies established MYRF as a nanophthalmos gene (12) and the correlation between MYRF truncation mutation and high hyperopia (13). These reports proved that MYRF played a role in the development of many other organs in addition to regulating cardiac and urogenital development.
Herein, we first reported the case of a male patient with a deleterious MYRF variant causing a loss of MYRF activity.
Case presentation
Case description
A 4-year-7-month-old “girl” was referred to our unit in December 2021 because of the short stature. The current height was 2 standard deviations behind peers; and her height was more than one standard deviation below average in each physical examination. Both eyes were amblyopic. She was her family's second living child (G4P2). The “girl” was born at 36+5 weeks, with a birth weight of 2,430 g (z-score: −1.91), length of 49 cm (z-score: −0.08), and a head circumference of 34.2 cm (z-score: 0.27). She underwent a premature cesarean section because of maternal uterine scarring, gestational diabetes, and fetal heart malformation. The Apgar scores were 9 at 1 and 5 min, 5 at 7 min, and 8 at 10 min. After birth, she presented breathing difficulty due to ventricular septal defect (VSD), atrial septal defect (ASD), patent ductus arteriosus (PDA), persistent superior vena cava, and pulmonary hypertension. Besides, the patient also suffered from right-sided pulmonary hypoplasia. She was sent to NICU immediately for respiratory support and other rescue treatments. After 1 month, she underwent surgery to repair ASD, VSD and PDA. She recovered well postoperatively. As the “girl” grows up, the motor and language development was delayed compared to other children of the same age, raising head, climbing, walking and speaking at 6 months, 8 months, 16 months and 18 months respectively. So far, she had been living as a girl since birth.
In her family, 52-year-old father and 17-year-old brother are medium build, her 47-year-old mother has a height of 147 cm and a weight of 48 kg. In addition, her mother had two early pregnancy loss, one due to fetal heart rate disappearance and the other no specific reasons.
Our physical examination showed a weight of 13.5 kg (−1.86 SD), height of 97 cm (−2.21 SD), well-proportioned figure. There was a surgical scar approximately 8 cm long in the median line of the chest. Cardiopulmonary auscultation, nervous system was unremarkable. Both breasts were not enlarged (stage B1). The abdomen was flat and soft, the liver and spleen were not touched under the ribs. Genitourinary physical examination showed female external genitalia with a slightly enlarged clitoris, urethral opening, and vaginal opening in the perineum. No gonadoid masses were found in the inguinal region. No pubic hair was noted. No coffee spot was found all over the body.
The serum levels of sex hormones showed basal luteinizing hormone (LH) of 0.85 IU/L (normal range: <0.32 IU/L), follicle-stimulating hormone (FSH) of 33.15 IU/L (normal range: 0.25–5.89 IU/L), prolactin (PRL) of 242.5 mIU/L (normal range: 108.8–557.0 mIU/L), estradiol (E2) of <36.7 pmol/L (normal range: <115.6 pmol/L), testosterone (T) of 0.46 nmol/L (normal range: <1.31 nmol/L), and human chorionic gonadotropin (HCG) of <1.2 mIU/ml (normal range: <5.0 mIU/ml). Adrenocorticotropic hormone (ACTH) and cortisol at 8 AM were <5 pg/ml (normal range: 0–46 pg/ml) and 92.4 μg/L (normal range: 50–250 μg/L). The blood fasting glucose and insulin level was 4.7 mmol/L (normal range: 3.6–6.11 mmol/L) and 7.5 pmol/L (normal range: 13.0–161.0 pmol/L), respectively. The peak growth hormone (GH) was 14.2 ng/ml (normal range: >10.0 ng/ml) with an insulin-like growth factor 1 (IGF-1) of 41.5 ng/ml (normal range: 35.0–232.0 ng/ml). The SRY gene (14) was positive in peripheral blood examination. Renal function, liver function, electrolytes, thyroid function, alpha fetal protein, carcinoembryonic antigen, and genetic metabolic disease maps were all in the normal range. Interpreting these results in an integrated manner, growth hormone therapy and endocrinology outpatient follow-up were recommended.
The heart and large vessels CT angiography showed postoperative changes of CHD, with an enlarged coronary sinus, residual left superior vena cava, and an enlarged right atrium and right ventricle. There was no obvious patchy shadow in the two lungs. Electrocardiogram examination suggested incomplete right bundle branch block with right ventricular hypertrophy, and ST-segment changes. Ultrasonography of the uterus and ovaries demonstrated that the bilateral adnexal area was slightly thickened, with a thickness of 0.27 cm on the left and 0.23 cm on the right. The gonads were not explored, and the uterus was visible and may have cord-like gonads in thickened bilateral adnexal areas. Ultrasonography of the perineum, pelvic cavity, and groin revealed no testicular echoes. Pituitary MRI suggested a suspected pituitary Rathke fissure while the radiographs of carpal bone showed a bone age of approximately 4.5 years. The ultrasonography of the abdominal and kidneys were normal during our research period, as well as the electroencephalogram.
Cystoscopy showed the vagina was sized 4.8 cm, and the cervical opening was visible, no obvious abnormalities in bladder and bilateral ureteral orifices. Laparoscopy showed muscular tissue in the space between the bladder and rectum, from bottom to top, along the left pelvic cavity to the lower pole of the spleen, accompanied by vasculiform tissue with a slightly enlarged end. There was no vas deferens in the right pelvic cavity, a testicular gonad was found between the lower pole of the right kidney and the opening of the inner ring, not well developed, approximately 1.2 × 0.3 cm (Figures 1A,B). That testicular gonad was removed with the consent of the parents. Pathological examination revealed that the left abdominal mass was an accessory middle renal duct cyst, and the right gonadal tissue was testicular (Figures 1C,D). Regular follow-up was done in the urology department.
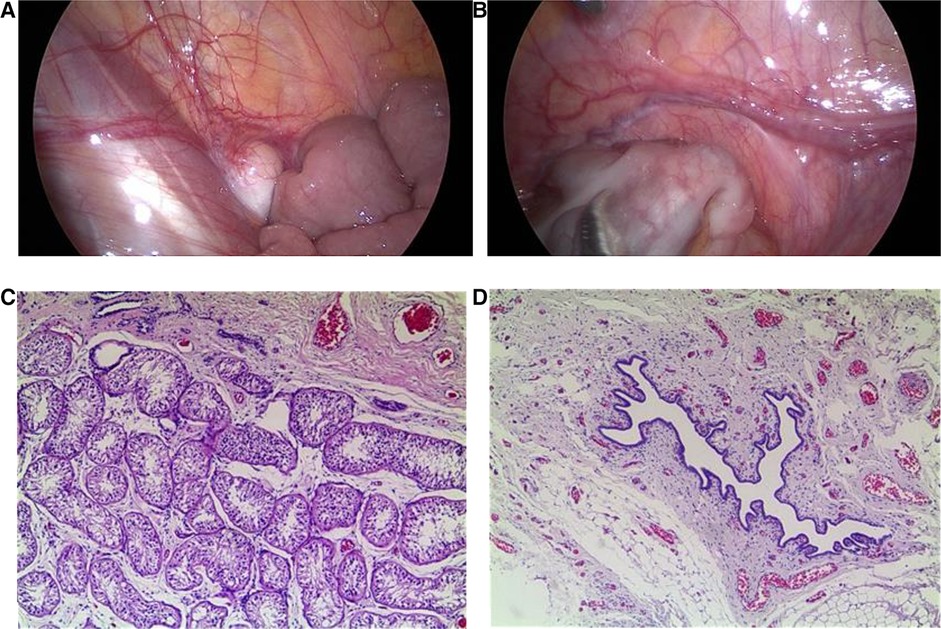
Figure 1. Laparoscopic findings and pathological examination. (A) Laparoscopy showed testicular gonads between the lower pole of the right kidney and the opening of the inner ring in the right pelvic cavity. (B) Laparoscopy showed muscular tissue in the space between the bladder and rectum in the left pelvic cavity, accompanied by vascular tissue with a slightly enlarged end. (C) Pathological examination revealed that the right gonadal tissue was consistent with testicular tissue (×50). (D) Pathological examination revealed that the left abdominal mass was consistent with an accessory middle renal duct cyst (×50).
Genetic testing
To further clarify the diagnosis, WES was performed on the DNA of the patient and her unaffected parents after informed consent. DNA was extracted from the peripheral blood of the patient and the parents with normal phenotypes for WES. Copy number variation analysis confirmed the karyotype of 46,XY, carrying SRY (Figures 2A,B). One de novo heterozygous variant in MYRF was identified, namely c.2817G > A (NM_001127392.3) (Figure 2C), which resulted in early termination (p. W939*). This variation had not been reported or registered in several variant databases, including 1,000 Genomes, GnomAD, LOVD, and HGVD. The ClinVar database annotated it as “pathogenic”, but further literature search revealed no relevant reports. According to the ACMG Classification Standards and Guidelines for Genetic Variations (15), the variant showed strong evidence of pathogenicity because it was a nonsense variant (PVS1), and a de novo variant (PS2), and was not included in 1,000 Genomes, GnomAD, LOVD, and HGVD (PM2). Analysis of conserved sequences suggested that this variant was located in highly conserved sequences across several species (PP3) (Figure 3). Finally, we regarded the variant identified in our patient as a pathogenic variant (PVS1 + PS2 + PM2 + PP3) associated with CUGS.
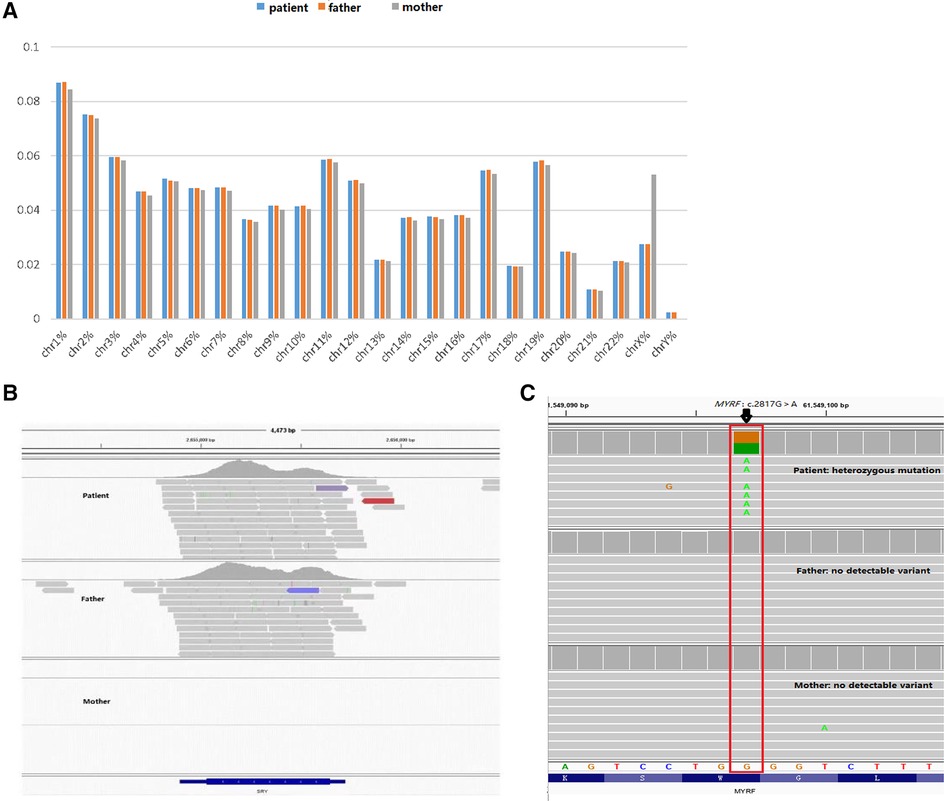
Figure 2. Genetic test results. (A,B) Suggests that the patient has a male karyotype (46,XY, carrying SRY). (C) c.2817G > A (p.W939*) de novo heterozygous mutation in the MYRF gene by WES.
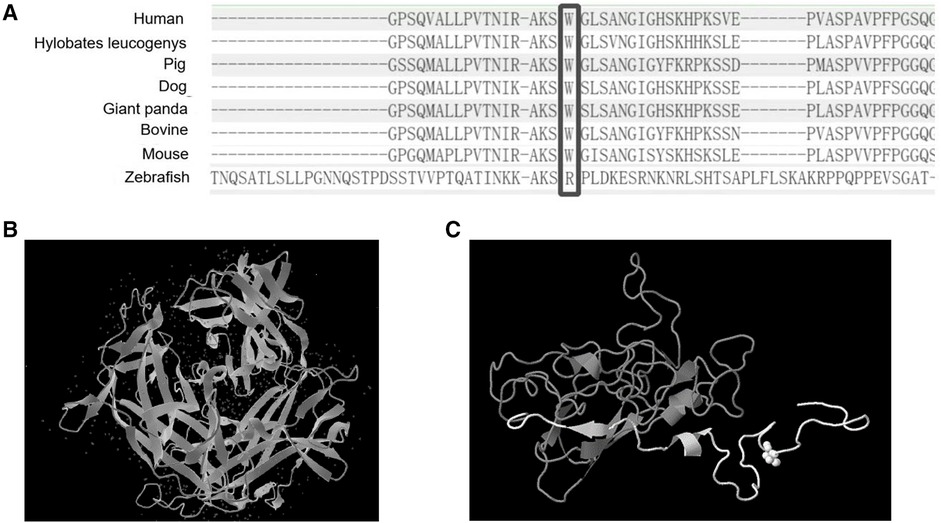
Figure 3. The protein sequence from several organisms and the predicted 3D structure of the variant. (A) Comparison of MYRF protein sequences from several organisms. (B) Normal 3D structure of MYRF protein. (C) The predicted 3D structure of MYRF protein residues 890–1,138 was modeled by Genome3D (residues 890–939 are colored white, and residues 940–1,138 are colored gray). As we could see from the predicted 3D structure, the variant caused a premature termination codon, resulting in protein truncation (approximately 15% of protein length is missing), and damaging the protein structure (region colored in gray).
Literature review
We searched the PubMed database, OMIM, HGMD, and China National Knowledge Infrastructure (CNKI) using “cardiac-urogenital syndrome (CUGS)” “disorders/differences of sex development (DSDs)” and “MYRF” as keywords. The search period was from the database's inception to April 30, 2022. Five documents reporting on CUGS and MYRF were retrieved (6–10). The clinical characteristics and MYRF gene variants of twenty patients (male: 16, female: 4) with CUGS who carried MYRF gene variants, as documented in these studies, are summarized in Table 1. The most common clinical presentation of syndromic MYRF is as follows: CHD (18/20, 90.0%), genitourinary anomalies (14/17, 82.4%), diaphragmatic anomalies (13/20, 65.0%), and pulmonary hypoplasia (9/19, 47.4%). The rare clinical presentations of CUGS are nanophthalmos (n = 1, c.789dupC/p.S264Qfs*74) (10) and short stature (n = 1, c.1435C > G/p. L479V) (8).
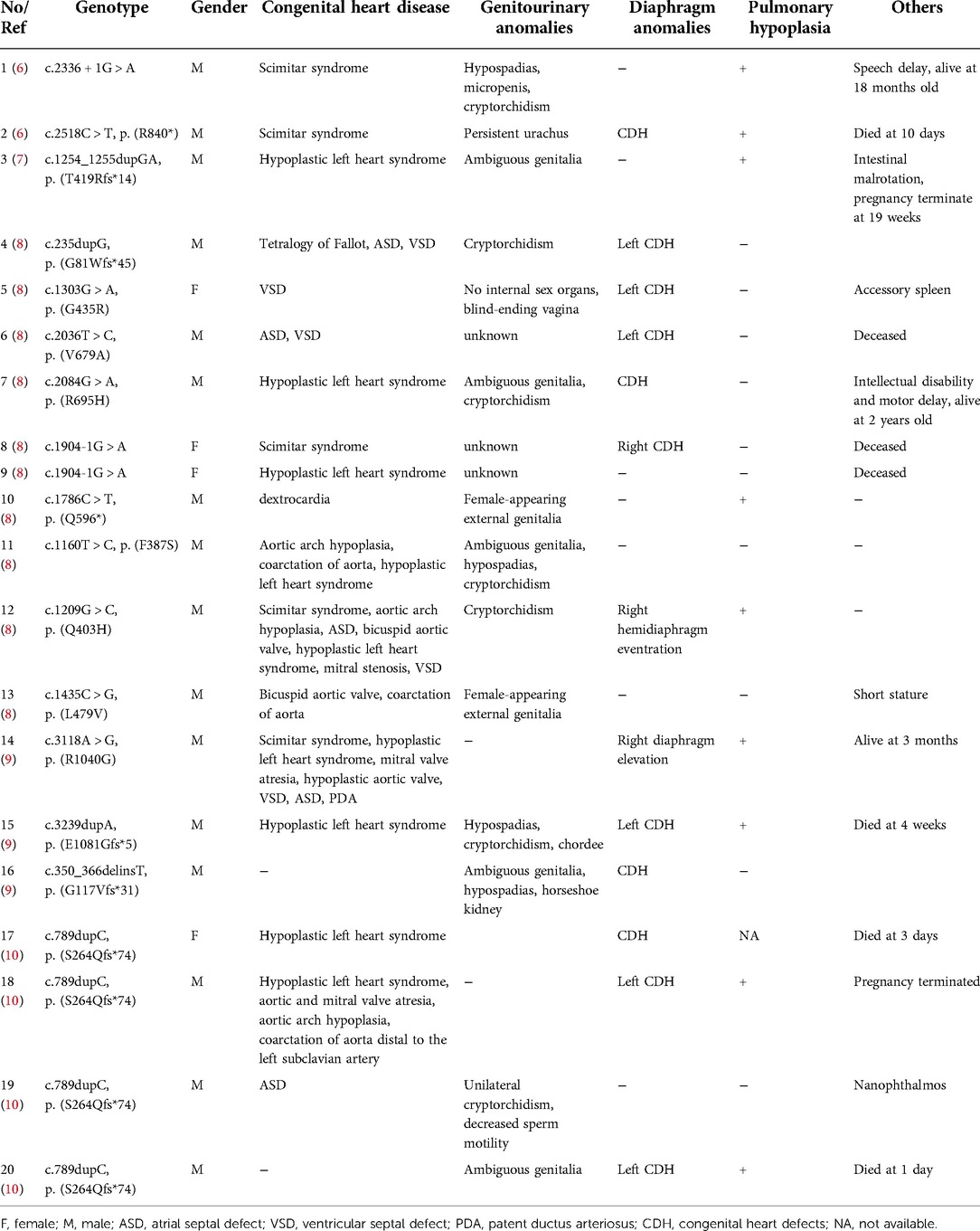
Table 1. The clinical and genetic findings in CUGS cases with MYRF variants.
Two documents regarding DSDs and MYRF were retrieved (11, 16). Seven patients (karyotype 46,XY: 5, karyotype 46,XX: 2) with DSDs carried MYRF gene variants; DSD with nanophthalmos (n = 1, c.2572 + 1G > A) (16), their clinical characteristics and MYRF gene variants are summarized in Table 2.

Table 2. Summary of clinical and genetic findings in DSD cases with MYRF variants.
Previous studies reported a nonsyndromic form of isolated nanophthalmos (n = 10, c.789delC/p.S264Afs*8, c.789dupC/p.S264Qfs*74, c.1433G > C/p.R478P, c.2956C > T/p.R986X, c.3377delG/p.G1126Vfs*31, c.3194 + 2T > C, and c.3274_3275delAG/p.L1093Pfs*22) (10, 13, 17), nanophthalmos with mitral valve prolapse, unilateral cryptorchidism and micropenis (n = 1, c.789dupC/p.S264Qfs*74) (12), nanophthalmos with unilateral cryptorchidism and ASD (n = 1, c.789dupC/p.S264Qfs*74) (10), nanophthalmos with posterior fossa cyst (n = 1, c.1553C > T/p.T518M) (18), and 2 large nanophthalmos families with or without dextrocardia or CDH (c.789dupC/p.S264Qfs*74, c.3376-1G > A, and c.3361delC/p.R1121Gfs*36) (12, 19), their clinical characteristics and MYRF gene variants are summarized in Table 3.
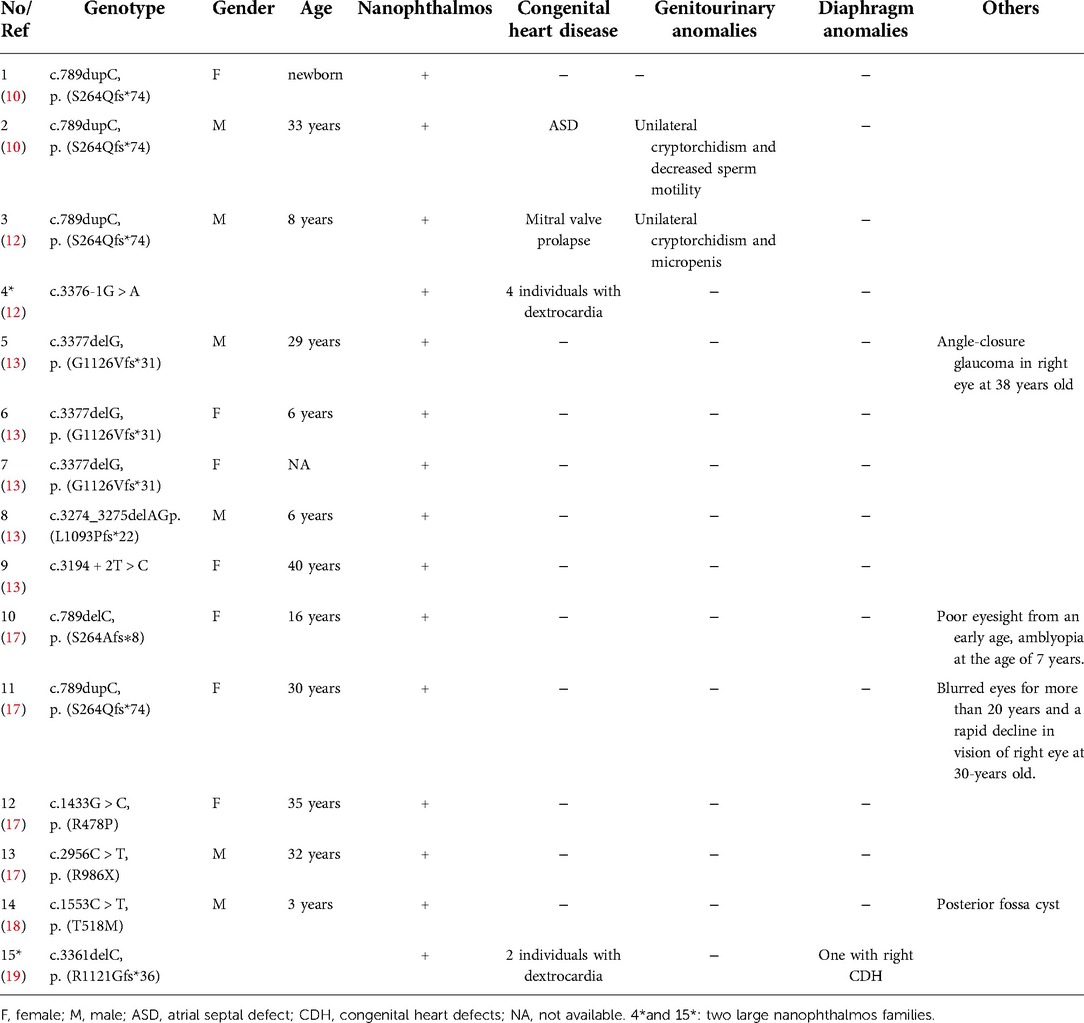
Table 3. Summary of clinical and genetic findings in nanophthalmos cases with MYRF variants.
Discussion
MYRF consists of the N-terminal proline-rich domain, DNA binding domain (DBD), intramolecular chaperone autoprocessing (ICA), or peptidase S74 domain in the cytoplasm, transmembrane helix, and luminal domain in the lumen of the endoplasmic reticulum. In these conserved domains, the DBD directly binds to the promoters of target genes, and the ICA domain triggers the homotrimerization of MYRF protein and subsequently results in autoproteolytic cleavage to release an N-terminal transcriptional activator translocating to the nucleus, which is important for transcriptional activation (20, 21). Mutations could be present in any domain of MYRF and result in haploinsufficiency or single amino acid changes, and single amino acid changes were mostly located in the DNA binding domain and peptidase S74 domain. Both syndromic presentations and nanophthalmos or high hyperopia were associated with missense, nonsense, frameshift, and essential splice variants in MYRF (6–9, 11, 19) (Figure 4).
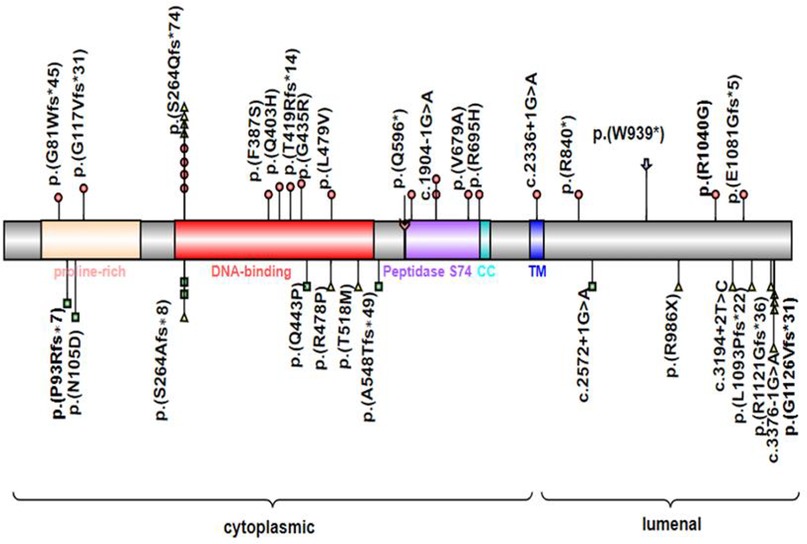
Figure 4. MYRF protein schematic, showing the location of individual domains, and relative positions of reported disease-associated variants associated with CUGS (colored circles) or DSDs (colored squares) or nanophthalmos or high hyperopia (colored triangles). Symbol () indicates the variant in this study. CC, coiled-coil domain; TM, transmembrane domain; Symbol () indicates the autolytic cleavage site.
Here, we reported the case of a 4-year-7-month-old “girl” who presented with multiple clinical manifestations, including VSD, ASD, PDA, persistent superior vena cava, pulmonary hypertension, female external genitalia with a slightly enlarged clitoris, uterus and vagina with no ovary, 46,XY karyotype, vanishing testis with testicular gonadal tissue in the right pelvis, vasculiform tissue in the left pelvis, Tanner stage I (breast) and I (pubis), short stature, and amblyopia. Syndromic symptoms of CUGS and 46,XY DSD were overlapping. Exome sequencing revealed a heterozygous de novo c.2817G > A (p.W939*) nonsense mutation of MYRF in exon 22. Analysis of conserved sequences suggested that this variant was located in highly conserved sequences across several species. A DECIPHER database search revealed that the variant caused haploinsufficiency (%HI = 40.26) and loss-of-function (pLI = 1.00). The mutation generated a premature termination codon, leading to protein truncation (approximately 15% of the protein length was missing) and damage to the protein structure (22) (Figure 3).
Previously, 30 pathogenic variants of MYRF had been described in patients with CUGS, DSDs, and nanophthalmos. The syndromic CUGS and DSDs MYRF variants were de novo, whereas variants with nanophthalmos or high hyperopia showed kindred transmission. Twenty cases of CUGS with structural congenital disabilities affecting the heart, lungs, diaphragm, and genitourinary system had been reported to carry variants of MYRF, including frameshift (n = 8), missense (n = 7), splice site (n = 3), and nonsense (n = 2) variants. Seven patients with DSDs showed MYRF variants, including 4 frameshifts, 2 missense mutations, and 1 splice site. Most of these variants (17/22) affected the 5′ end of the transmembrane domain (TM) in MYRF, except the variant p.(E1081Gfs*5) closest to the C-terminal reported in a syndromic case, which lies in exon 25. This suggested that loss of MYRF function, triggering nonsense-mediated mRNA decay was the likely mode of action of these syndromic variants. We also observed male predominance in the published cases of CUGS (16/20) and DSDs (5/7), with genitourinary anomalies present in most of the cases. This also suggested that loss of function of the MYRF transcription factor leads to the transcriptional dysregulation of genes related to sex development (11). Further studies are warranted to confirm this hypothesis. Compared with the variants presenting severe syndromic forms, the variants that manifested nanophthalmos occurred in the final six exons of MYRF or their associated splice sites (after exon 22), except for the variants c.789delC (p.S264Afs*8) and c.789dupC (p.S264Qfs*74). The c.789dupC (p.S264Qfs*74) had been observed in variable phenotypic manifestations (10, 12, 17), while c.789delC (p.S264Afs*8) had only been described in the milder clinical presentation of DSDs and isolated nanophthalmos in previous reports (11, 17), although they had the same position of de novo mutations. Therefore, we agree that severe syndromic manifestations are relevant to MYRF haploinsufficiency, while isolated nanophthalmos may sometimes affect MYRF homotrimerization, autoproteolysis, or transcriptional activity (19).
Previous study also considered that introduction of a premature termination codon in the terminal exon may cause the variant at the 3′ end of the penultimate exon of MYRF avoiding nonsense-mediated decay (23). Therefore, the isolated nanophthalmos and incompletely penetrant dextrocardia (12) or high hyperopia were possibly associated with further downstream variants. However, in our report, the variant at the 3′ end of exon 22 caused a genetic syndrome of CUGS and DSD, similar to the variants c.3118A > G (p.R1040G) in exon 23 and c.3239dupA (p. E1081Gfs*5) in exon 25 that Rossetti et al. (9) reported, which also are likely to trigger nonsense-mediated mRNA decay. This suggests that loss of MYRF function, even in the last C-terminal exons, is still the most likely mode of action of these variants. Besides, Garnai et al. (12) considered MYRF as a dosage-sensitive transcription factor, because the ocular tissues were more sensitive than the cardiac and urogenital tissues, especially to changes in the C-terminus of the protein. In our case amblyopia was also observed. Since strabismus and amblyopia in early childhood may develop to uncorrected high hyperopia (17), further follow-up is required for subsequent visual loss. Further, it is possible that ocular symptoms have not yet been observed in individuals with MYRF variants causing severe syndromic symptoms, because they did not survive infancy and owing to plausible lack of detailed ocular examination. Despite these assumptions, the detailed action mode of the C-terminal region in MYRF remains unknown. Deeper research is necessary to determine the mechanisms that explain these individuals' ocular and systemic phenotypes.
In summary, we reported a 4-year-7-month-old patient presenting multiple clinical manifestations with syndromic symptoms of CUGS and 46,XY DSD, ocular symptoms, and short stature. WES identified a novel truncation mutation in the 3′ end of MYRF, located in exon 22. Our findings suggest further studies to illustrate the function of the critical MYRF as well as how different variants affect the function of MYRF in different cell types in human disorders are necessary.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
CCZ: conceptualized and designed the study, and reviewed and revised the manuscript. HW: drafted the initial manuscript. DHW, HJT and DW: collected the patient data. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation (81371215, 81670786) and Key R & D Projects of Zhejiang Provincial Department of Science and Technology (2021C03094).
Acknowledgments
We extend our gratitude to the patient and her parents for their enthusiasm to participate in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, et al. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell. (2009) 138:172–85. doi: 10.1016/j.cell.2009.04.031
2. Hornig J, Fröb F, Vogl MR, Hermans-Borgmeyer I, Tamm ER, Wegner M. The transcription factors Sox10 and mYRF define an essential regulatory network module in differentiating oligodendrocytes. PLoS Genet. (2013) 9:e1003907. doi: 10.1371/journal.pgen.1003907
3. Kim D, Choi J, Fan C, Shearer RS, Sharif M, Busch P, et al. Homo-trimerization is essential for the transcription factor function of mYRF for oligodendrocyte differentiation. Nucleic Acids Res. (2017) 45:5112–25. doi: 10.1093/nar/gkx080
4. Koenning M, Jackson S, Hay CM, Faux C, Kilpatrick TJ, Willingham M, et al. Myelin gene regulatory factor is required for maintenance of myelin and mature oligodendrocyte identity in the adult CNS. J Neurosci. (2012) 32:12528–42. doi: 10.1523/JNEUROSCI.1069-12.2012
5. Kurahashi H, Azuma Y, Masuda A, Okuno T, Nakahara E, Imamura T, et al. MYRF Is associated with encephalopathy with reversible myelin vacuolization. Ann Neurol. (2018) 83:98–106. doi: 10.1002/ana.25125
6. Pinz H, Pyle LC, Li D, Izumi K, Skraban C, Tarpinian J, et al. De novo variants in myelin regulatory factor (MYRF) as candidates of a new syndrome of cardiac and urogenital anomalies. Am J Med Genet A. (2018) 176:969–72. doi: 10.1002/ajmg.a.38620
7. Chitayat D, Shannon P, Uster T, Nezarati MM, Schnur RE, Bhoj EJ. An additional individual with a de novo variant in myelin regulatory factor (MYRF) with cardiac and urogenital anomalies: further proof of causality: comments on the article by Pinz et al. Am J Med Genet A. (2018) 176:2041–3. doi: 10.1002/ajmg.a.40360
8. Qi H, Yu L, Zhou X, Wynn J, Zhao H, Guo Y, et al. De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PLoS Genet. (2018) 14:e1007822. doi: 10.1371/journal.pgen.1007822
9. Rossetti LZ, Glinton K, Yuan B, Liu P, Pillai N, Mizerik E, et al. Review of the phenotypic spectrum associated with haploinsufficiency of MYRF. Am J Med Genet A. (2019) 179:1376–82. doi: 10.1002/ajmg.a.61182
10. Gupta N, Endrakanti M, Gupta N, Dadhwal V, Naini K, Manchanda S, et al. Diverse clinical manifestations and intrafamilial variability due to an inherited recurrent MYRF variant. Am J Med Genet A. (2022) 188:1–5. doi: 10.1002/ajmg.a.62265
11. Hamanaka K, Takata A, Uchiyama Y, Miyatake S, Miyake N, Mitsuhashi S, et al. MYRF haploinsufficiency causes 46,XY and 46,XX disorders of sex development: bioinformatics consideration. Hum Mol Genet. (2019) 28:2319–29. doi: 10.1093/hmg/ddz066
12. Garnai SJ, Brinkmeier M, Emery B, Aleman TS, Pyle LC, Veleva-Rotse B, et al. Variants in myelin regulatory factor (MYRF) cause autosomal dominant and syndromic nanophthalmos in humans and retinal degeneration in mice. PLoS Genet. (2019) 15:e1008130. doi: 10.1371/journal.pgen.1008130
13. Xiao X, Sun W, Ouyang J, Li S, Jia X, Tan Z, et al. Novel truncation mutations in MYRF cause autosomal dominant high hyperopia mapped to 11p12-q13.3. Hum Genet. (2019) 138:1077–90. doi: 10.1007/s00439-019-02039-z
14. She ZY, Yang WX. Sry and SoxE genes: how they participate in mammalian sex determination and gonadal development? Semin Cell Dev Biol. (2017) 63:13–22. doi: 10.1016/j.semcdb.2016.07.032
15. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
16. Globa E, Zelinska N, Shcherbak Y, Bignon-Topalovic J, Bashamboo A, MсElreavey K. Disorders of sex development in a Large Ukrainian cohort: clinical diversity and genetic findings. Front Endocrinol. (2022) 13:810782. doi: 10.3389/fendo.2022.810782
17. Guo C, Zhao Z, Chen D, He S, Sun N, Li Z, et al. Detection of clinically relevant genetic variants in Chinese patients with nanophthalmos by trio-based whole-genome sequencing study. Invest Ophthalmol Vis Sci. (2019) 60:2904–13. doi: 10.1167/iovs.18-26275
18. Hagedorn J, Avdic A, Schnieders MJ, Roos BR, Kwon YH, Drack AV, et al. Nanophthalmos patient with a THR518MET mutation in MYRF, a case report. BMC Ophthalmol. (2020) 20:388. doi: 10.1186/s12886-020-01659-8
19. Siggs OM, Souzeau E, Breen J, Qassim A, Zhou T, Dubowsky A, et al. Autosomal dominant nanophthalmos and high hyperopia associated with a C-terminal frameshift variant in MYRF. Mol Vis. (2019) 25:527–34.31700225
20. Bujalka H, Koenning M, Jackson S, Perreau VM, Pope B, Hay CM, et al. MYRF is a membrane-associated transcription factor that autoproteolytically cleaves to directly activate myelin genes. PLoS Biol. (2013) 11:e1001625. doi: 10.1371/journal.pbio.1001625
21. Li Z, Park Y, Marcotte EM. A bacteriophage tailspike domain promotes self-cleavage of a human membrane-bound transcription factor, the myelin regulatory factor MYRF. PLoS Biol. (2013) 11:e1001624. doi: 10.1371/journal.pbio.1001624
22. Lewis TE, Sillitoe I, Andreeva A, Blundell TL, Buchan DW, Chothia C, et al. Genome3D: exploiting structure to help users understand their sequences. Nucleic Acids Res. (2015) 43:D382–6. doi: 10.1093/nar/gku973
Keywords: MYRF gene, cardiac-urogenital syndrome, disorders/differences of sex development, nanophthalmos, short stature
Citation: Wang H, Wu D, Wu D, Tian H, Li H, Jiang K and Zou C (2022) Case Report: De novo variant in myelin regulatory factor in a Chinese child with 46,XY disorder/difference of sex development, cardiac and urogenital anomalies, and short stature. Front. Pediatr. 10:1027832. doi: 10.3389/fped.2022.1027832
Received: 25 August 2022; Accepted: 24 October 2022;
Published: 18 November 2022.
Edited by:
Anatoly Tiulpakov, Federal State Budgetary Scientific Institution Research Centre for Medical Genetics (RCMG), RussiaReviewed by:
Gabriel C. Dworschak, University of Bonn, GermanyLaura Audí, Vall d’Hebron University Hospital, Spain
© 2022 Wang, Wu, Wu, Tian, Li, Jiang and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chao-Chun Zou emNjMTRAemp1LmVkdS5jbg==
†These authors have contributed equally to this work
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics