 Haiyan Luo1,†
Haiyan Luo1,† Yan Yang1,†Xinrong Wang1Fangping Xu2Cheng Huang1Danping Liu1Liuyang Zhang1Ting Huang1Pengpeng Ma1Qing Lu1Shuhui Huang1Bicheng Yang1
Yan Yang1,†Xinrong Wang1Fangping Xu2Cheng Huang1Danping Liu1Liuyang Zhang1Ting Huang1Pengpeng Ma1Qing Lu1Shuhui Huang1Bicheng Yang1 Yongyi Zou1*Yanqiu Liu1*
Yongyi Zou1*Yanqiu Liu1*
- 1Department of Medical Genetics, Jiangxi Key Laboratory of Birth Defect Prevention and Control, Jiangxi Maternal and Child Health Hospital, Nanchang, China
- 2Department of Obstetrics, Jiangxi Provincial Maternal and Child Health Hospital, Nanchang, China
Background and aims: Concurrent hearing and genetic screening of newborns have been widely adopted as an effective strategy in early diagnosis and intervention for hearing loss in many cities in China. Here, we aimed to firstly explore the efficacy of combining conventional hearing screening with genetic screening among the large-scale newborns in Jiangxi Province.
Methods: A total of 24,349 newborns from Jiangxi Maternal and Child Health Hospital were enrolled in our study from April 2021 to June 2022. Newborn hearing screening was conducted using otoacoustic emission (OAE) and automated auditory brainstem response (AABR). Meanwhile, newborn dried blood spots were collected and twenty common variants in four genes, including GJB2, SLC26A4, MT-RNR1(12SrRNA), and GJB3, were screened using a BGISEQ-500 next generation sequencing platform. Whole coding regions sequencing of GJB2 and SLC26A4 were performed by Sanger sequencing and NGS, respectively. Following up of hearing for the newborns was undertaken by phone interviews.
Results: Among the 24,349 newborns, 7.00% (1,704/24,349) were bilaterally or unilaterally referred in their initial hearing screening, whereas 1.30% (316/24,349) exhibited bilateral or unilateral hearing loss in the repeated screening. Genetic screening revealed that 4.813% (1,172/24,349) of the screened newborns were positive for at least one mutant allele (heterozygote, homozygote, or compound heterozygote in one gene, mtDNA homoplasmy or heteroplasmy and combined variants in different genes). A total of 1,146 individuals were identified with mutant allele in one gene, including 525 of GJB2, 371 of SLC26A4, 189 as homoplasmic or heteroplasmic of MT-RNR1, and 61 of GJB3, indicating that GJB2 and SLC26A4 are the most common endemic deafness-associated genes among newborns in Jiangxi Province. Nineteen newborns were detected with combined heterozygous variants in different genes, with “c.235delC heterozygous and c.919-2A > G heterozygous” as the most prevalent genotype. Additionally, seven newborns were screened as homozygotes or compound heterozygotes responsible for congenital or late-onset prelingual hearing loss, including three cases with GJB2 c.235delC homozygous and one with SLC26A4 c.919-2A > G homozygous variant, one case with compound heterozygous variants for GJB2 and two with compound heterozygous variants for SLC26A4. Coding regions sequencing of GJB2 or SLC26A4 for overall 265 infants revealed that 14 individuals were identified as compound heterozygote with a second pathogenic variant not screened by our genetic panel.
Conclusions: Herein our study firstly investigated the efficacy of concurrent hearing screening and genetic screening of common hearing impairment variants among large-scale newborns in Jiangxi Province. Concurrent screening provides a more comprehensive approach for management of congenital or delayed onset prelingual hearing loss and prevention of drug-induced hearing impairment for newborns at risk as well as their maternal relatives. An insight into the molecular epidemiology for hearing loss genes among Jiangxi population will also be beneficial to the genetic counseling and birth defect prevention.
Introduction
Hearing loss (HL) is one of the most common human disorders, affecting nearly 1 to 3 newborns per 1,000 live births (1). Based on a correlated report released by the WHO in 2018, over 466 million people worldwide are suffering from moderate to profound hearing loss in unilateral or bilateral ears, while most cases with hearing impairment occur in developing countries (2). In China, there are approximately 21 million people affected with hearing impairment and approximately 0.8 million are children younger than 7 years old (3). Studies have revealed that this number has continued to increase and about 30,000 neonates with congenital hearing loss are born annually in China (4). Congenital or late-onset prelingual hearing impairment in children can lead to detrimental effects on language acquisition, behavior and psychosocial interaction, educational and cognitive outcomes (5).
Both genetic causes and environmental factors are related to etiologies of this sensory disorder (6). It is estimated that an approximately estimated 50% of hearing impairment are caused by genetic factors, which can also be defined as hereditary hearing loss (7). Hereditary HL is extremely phenotypic heterogeneous and can be grouped into non-syndromic hearing loss (NSHL) and syndromic hearing loss (SHL) based on the distinctive clinical accompanying symptoms. NSHL makes up a substantial portion of nearly 70% in genetic HL, while the remaining 30% of genetic HL is attributed to SHL (8). Hereditary HL is also a complex disorder with diverse genetic heterogeneities and there have been over 223 genes reported to be associated with hearing loss according to the Hereditary Hearing Loss Homepage database (https://deafnessvariationdatabase.org) up to now. Molecular mutation spectrum of hereditary HL can be distinct in different geographic areas and ethnic groups. According to previous epidemiological studies on molecular etiology analysis among Chinese population, GJB2, GJB3, SLC26A4, and MT-RNR1 have been proved to be highly prevalent and causative genes in NSHL patients (9).
Conventional universal newborn hearing screening (UNHS) has been implemented worldwide as one of the important strategies for early identification, diagnosis and management of hearing impairment due to its feasibility and cost-efficiency (10, 11). However, evaluation of benefits and effectiveness of UNHS still demonstrates its inherent limitations. Firstly, UNHS only detects existing hearing impairment at the time of screening, while neonates with late-onset or progressive hearing impairment or drug-induced hearing loss usually show normal hearing at birth and cannot be detected and predicted timely (12, 13). Moreover, UNHS may not elucidate the common etiology for most cases with hereditary hearing impairment while supplementary genetic testing may assist in determining the genetic causes (3). Therefore, concurrent hearing screening and genetic screening of common deafness genes in neonates have been investigated in many cities to work as a scientific approach for early diagnosis and intervention of hearing defects including delayed-onset and aminoglycoside-antibiotic induced ototoxicity hearing loss. In recent decades, many specific molecular etiology reports are available in multiple geographic areas among Chinese population, especially for Northern and Eastern China, which is beneficial to genetic counseling for hearing loss in different regional background (14, 15). However, there has been no large-scale study on combined neonatal screening of hearing and deafness genetic screening in Jiangxi Province. Herein we firstly explored the benefits of concurrent hearing screening and genetic screening, and meanwhile reported the first analysis of mutation spectrum of the 20 common deafness gene variants in a large cohort of infants in Jiangxi Province, which would facilitate timely detection, prevention and management of hearing loss in newborns efficiently.
Materials and methods
Research subjects
A total of 24,349 newborns from Jiangxi Maternal and Child Health Hospital were enrolled in our study between April 2021 and July 2022, including 13,413 males and 10,936 females. Inclusion criteria of the enrolled newborns were as follows: (1) the infants were born between April 2021 and July 2022 in our hospital; (2) the infants were healthy enough to tolerate the screening procedures; (3) the parents or guardians agreed with participating in the concurrent newborn genetic and hearing screening program. Exclusion criteria were as follows: (1) the infants' samples were unqualified for the genetic tests or (2) the infants were lost to hearing or genetic follow-up; (3) the infants underwent prenatal diagnosis with definite deafness mutations in utero. A flow diagram of the recruited participants in our research was shown as in Figure 1. All the general newborn information, such as delivery modes, birth weight, length, and head circumference for gestational age was collected into a newborn screening information database. At least two telephone numbers from the guardians of each newborn were recorded for long-time telephone interview. Written informed consent was obtained from the infants' parents or guardians involved in the study. This study was approved by the Ethics Committee of Jiangxi Maternal and Child Health Hospital.
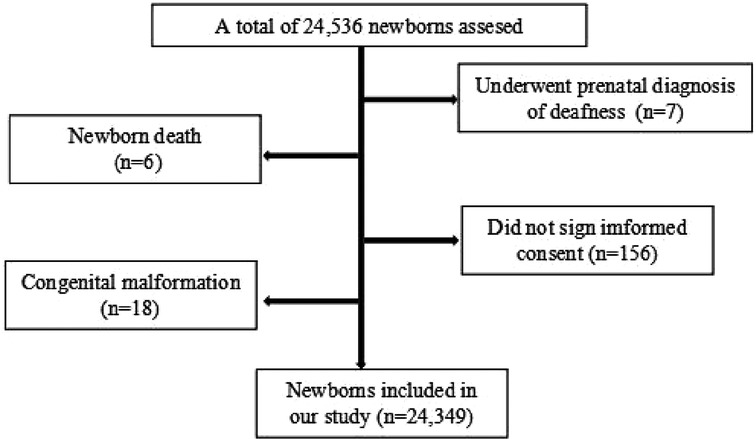
Figure 1. The flow diagram of the participants inclusion and exclusion in our research.
Universal newborn hearing screening
All the enrolled newborns underwent a two-step screening approach. Each newborn was initially tested on hearing using OAE within 72 h after delivery in a state of natural sleep in the maternity ward by an otolaryngologist. If the newborn failed the initial test, either unilaterally or bilaterally, which was also defined as “refer” in our study, a repeated OAE test combined with automatic auditory brainstem repose (AABR) assessment were performed by the age of 42 days. Newborns who failed the 42-day re-screening should be further evaluated with comprehensive audiological assessment by audiologists, preferably at the age of 3 months.
Molecular genetic screening and variant validation by Sanger sequencing
Three appropriate-sized heel peripheral blood spots were obtained using a customized dried blood collection card for genetic screening within 72 h of birth from all newborns. Nucleic acid was extracted from dried blood spots according to the manufacturers' instructions (BGI, Shenzhen, China). Library preparation and targeted NGS were performed according to the standard operation procedure by Combined Probe-Anchored Synthesis Kit using the BGISEQ-500 platform. The genetic screening panel included 20 NGS targeted common sites in GJB2, SLC26A4, MT-RNR1, and GJB3 (Table 1). Genetic screening results were categorized as three types: “Pass” (no screened variants), “Carrier” (one variant or combined variants), “Refer” (homozygous or compound heterozygous for GJB2 or SLC26A4; homoplasmic or heteroplasmic for MT-RNR1). All the positive samples screened were further verified by Sanger sequencing.
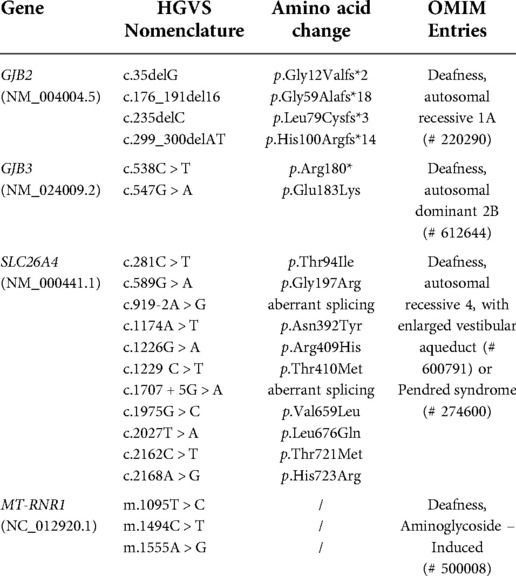
Table 1. Twenty common hearing-loss-associated variants targeted in newborns in our study.
DNA sequence analysis of the entire coding exons and flanking introns in GJB2 or SLC26A4
Newborns tested positive in the genetic screening were subsequently recommended to the Department of Medical Genetics for genetic counseling. For newborns screened as carriers of GJB2 and/or SLC26A4, the presence of a second pathogenic allele in the same gene not screened by our panel was taken into consideration. Based on the principle of voluntary participation, the entire coding regions sequencing and approximately 20 bp of exon–intron boundaries of GJB2 and SLC26A4 were further conducted by respective Sanger sequencing and NGS (BGI, Shenzhen, China). Primers used for direct sequencing in GJB2 were as listed in Table 2. Four dried heel peripheral blood spots were obtained from the infants and the DNA extraction was undertaken for detection according to the manufactures' instructions.

Table 2. Primers used for the entire coding regions analysis of GJB2 by Sanger sequencing.
Parental validation for newborns with suspected compound heterozygous variants
Two variants inherited from both parents were regarded as compound heterozygous. Since the detection of two heterozygous variants in one gene is insufficient for a genetic diagnosis, newborns with suspected compound heterozygous variants by genetic screening or the entire coding regions sequencing were further investigated on parental validation to determine whether the two variants were inherited from both parents, which was also defined as “in trans”. Two mL of peripheral venous blood sample from both parents were obtained and subject to parental validation.
Statistical analysis
The statistical analysis was performed using SPSS 20.0 software. The referrals rate of hearing and genetic screening, as well as the frequency of genes and variants were represented by n%. Chi-squared tests were performed to determine the statistical significance of differences in mutation carrier rates between the current and previous studies.
Results
Outcomes of hearing screening
A total of 24,349 newborns were screened with hearing in our study. The overall hearing screening results was summarized in Table 3. In the first step, OAE test for infants within72 h after delivery revealed that 22,645 (93.00%, 22,645/24,349) newborns passed the initial screening in all. Among those who were referred during the first hearing screening, 715 cases failed the bilateral hearing test while 989 cases exhibited the unilateral hearing screening failure. Overall, a total of 1,704 newborns (7.00%, 1,704/24,349) failed the initial screening. For those who were referred after the initial test, repeated tests of OAE plus AABR were administrated at 42 days after birth. The second test showed that a total of 316 referred infants did not pass the re-screening, including 128 cases with bilateral referral (0.53%, 128/24,349) and 188 cases with unilateral referral (0.77%, 188/24,349), with a failure rate of 1.30% in the re-screening test.
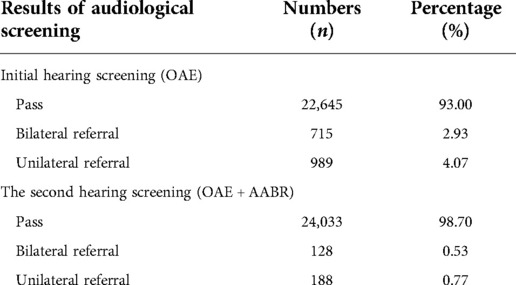
Table 3. Hearing screening results of 24,349 newborns in our study.
Analysis of genetic screening for 20 common deafness-related variants and Sanger sequencing confirmation
The genetic screening data of the 20 NSHL-related variants in this study among 24,349 neonates was shown in Table 4. A total of 1,172 infants were identified with at least one variant in the screening panel, with the overall carrier rate counting up to 4.813% (1,172/24,349) of all the neonates. Among the neonates screened, 957 newborns (3.93%, 957/24,349) exhibited heterozygous variant in one gene (GJB2, GJB3 and SLC26A4), together with 189 infants (0.78%, 189/24,349) showing simple heteroplasmic or homoplasmic variant in MT-RNR1. Furthermore, 19 cases (0.08%, 19/24,349) exhibited combined variants in two genes and seven newborns (0.03%, 7/24,349) harboured compound heterozygous variants or homozygous variants in GJB2 or SLC26A4. The highest prevalence rate was found for GJB2 at a rate of 2.24% (545/24,349), followed by the SLC26A4 gene at 1.57% (383/24,349). The GJB2 c.235delC and SLC26A4 IVS7-2A > G were the most frequently detected variant in our study, with the respective carrier rate of 1.83% (445/24,349) and 0.98% (238/24,349). A genetic carrier rate of 0.25% (62/24,349) for GJB3 heterozygous variant was identified, with c.538C > T and c.547G > A making up almost a similar proportion. MT-RNR1 was identified with a carrier rate of 0.81% (198/24,349) among the newborns, with homoplasmic m.1095T > C being the most endemic variant at a rate of 0.59% (137/24,349). All the positive samples by genetic screening showed concordant results as verified by Sanger sequencing.

Table 4. Frequency of the genetic variants in common deafness genes in newborns.
Integrated and comprehensive analysis of concurrent genetic and hearing screening
A comprehensive analysis and the associations between hearing and genetic screening of the tested newborns were shown in Figure 2. In our study, among the 205 (205/24,349, 0.84%) infants genetically referred, 177 (177/205, 86.34%) of them passed the hearing re-screening. Twenty-eight (28/205, 13.66%) of these referred from genetic screening also failed the second hearing screening. A total of 967 infants (967/24,349, 3.97%) were simple or combined variants carriers of GJB2, GJB3 and SLC26A4, while 75 (75/967, 7.76%) of them were referred on the second hearing screening, and the remaining 892 (892/967, 92.24%) infants passed. Notably, united hearing and genetic screening informed us of the risk of delayed-onset or drug-induced hearing loss even though these genetically referred infants had passed their two-step hearing screening. Accordingly, all these infants referred from the hearing screening should be further scheduled to receive diagnostic audiological tests for confirmation of hearing.

Figure 2. Comprehensive analysis and associations between the newborn hearing screening and genetic screening in this study. AABR, automatic auditory brainstem repose; OAE, otoacoustic emission.
Results of the entire coding regions sequencing of GJB2 and SLC26A4
A total of 265 infants were subject to the entire coding regions sequencing after voluntary parental decision, including 167 for GJB2 and 98 for SLC26A4. Results revealed that 13 infants were compound heterozygous for GJB2, with one case as GJB2 c.176_191del16/c.164C > A, two cases as GJB2 c.299_300delAT/c.109G > A, and ten cases as c.235delC/c.109G > A. GJB2 c.109G > A (p.V37I) was detected in 12 of these infants as a high frequent mutation allele. One infant was identified as c.919-2A > G/c.1615-1G > T compound heterozygote for SLC26A4. Detailed genotypes were listed as in Table 5. Chromatograms of Sanger sequencing for the three mutations GJB2 c.109G > A, GJB2 c.164C > A and SLC26A4 c.1615-1G > T were shown as in Figure 3.
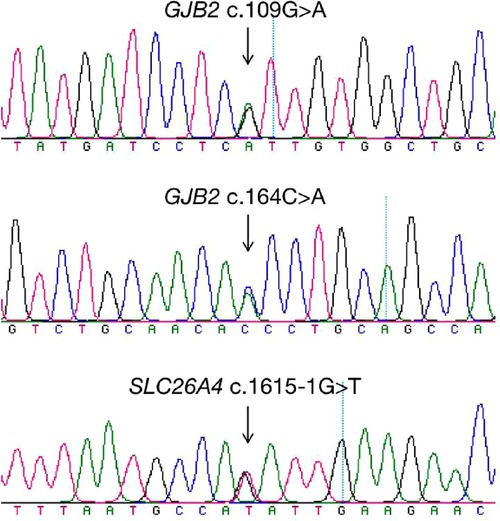
Figure 3. Chromatograms of three mutations identified by whole coding regions sequencing.

Table 5. Results of the entire coding regions sequencing of GJB2 and SLC26A4.
Discussion
Hearing impairment is one of the most common birth defects in human beings and affects approximately 60,000 children in China every year (16, 17). In recent decades, NGS has been widely used in genetic testing for hearing loss and has been shown to increase the diagnostic rate greatly (18). Although NGS could expand the number of HL-related genes evaluated, it is still challenging to apply expanded NGS panel to large-scale newborn screening due to its high cost, long turnaround time, and increased interpretation burden (14). In spite of the high genetic heterogeneity involved in numerous genes related with deafness, studies have revealed that there are specific common hereditary deafness causative genes in Chinese population (19, 20). Nearly 70% of the deafness-causative gene variation can be attributed to the four common deafness-associated genes GJB2, SLC26A4, 12SrRNA and GJB3 (9, 21). Targeted newborn genetic screening panel may cover varied numbers of hotspot variants in these four genes, usually varying from 9 to 20 sites in distinct screening kits. A meta-analysis study including 46 studies on subjects from 19 provinces in China has provided us the spot numbers and detection method most widely used in China, with most of which focus on 20 variants using microarray chip or matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) (22). In our study, we targeted the limited 20 common variants in GJB2, SLC26A4, 12SrRNA and GJB3 by NGS on universal hearing loss genetic screening for newborns in Jiangxi Province, which showed the advantages of cost efficiency, high throughput and easy interpretation.
Genetic mutation spectrum of hearing loss genes varies among different regions and ethnic populations caused by diverse genetic background (23). Several previous typical studies of common genetic screening of hearing loss among neonates in different areas of China were reviewed in Table 6, including the screening method and tested spot numbers. Herein we demonstrated a total carrier rate of the 20 common variants screened among newborns in Jiangxi Province at 4.813% (1,172/24,349). Compared with five previous studies entailing genotyping 20 common variants among newborns in different geographic areas, 18 identical mutation sites were commonly contained except for GJB2 c.167delT and MT-RNR1 m.1095T > C (24–28). Our findings revealed that Jiangxi Province has a significantly higher carrier rate than that in southeastern China such as Dongguan (3.74%) in Guangdong Province (28) and Liuzhou (2.3%) in Guangxi Province (29), while a slightly lower rate than that in Tianjin (5.52%) of northern China (26). In our study, GJB2 was the most prevalent deafness-associated gene, with the carrier rate of the four screened variants accounting for nearly 45.14% (529/1,172) of all the positive samples (p < 0.05). Of the four variants, GJB2 c.235delC was the most prevalent mutant allele with a frequency rate of 1.822%, which is also the most common variant in Asian population (30). The GJB2 c.35delG mutation constitutes about 70% of the pathologic alleles in European and American Caucasian deaf populations (4), but we detected this mutation in only one infant (1/24,349, 0.004%) in Jiangxi Province. SLC26A4 was the second most prevalent deafness-associated gene in Jiangxi Province, with the carrier rate of the screened variants accounting for nearly 32.94% (386/1,172) of all the positive samples. Among the eleven variants screened, c.919-2A > G was the most common mutant allele in Jiangxi Province, while c.2168A > G mutation exhibits the highest prevalence in the Japanese (31) and Korean population (32). In our study, SLC26A4 c.281C > T was identified in none of the infants, whereas a report on newborn hearing loss genetic screening in Tianjin showed the carrier frequency of 0.03% for SLC26A4 c.281C > T (26), which might be attributed to the differences between the ethnic compositions of southern and northern China or the differences resulting from the sample size. Notably, we found that the mitochondrial variant m.1095T > C had a higher rate than the m.1494 C > T and m.1555A > G in our study, with the respective incidence rate of 0.59% (144/24,349), 0.02% (5/24,349), and 0.20% (49/24,349), which showed the indispensability of adding this mutation into traditional screening panel for an increased detection rate of mtDNA variants. Sixty-two infants (0.25%, 62/24,349) were screened with GJB3 heterozygous variant, with a slightly lower frequency rate than that in nationwide China (0.34%) (24).
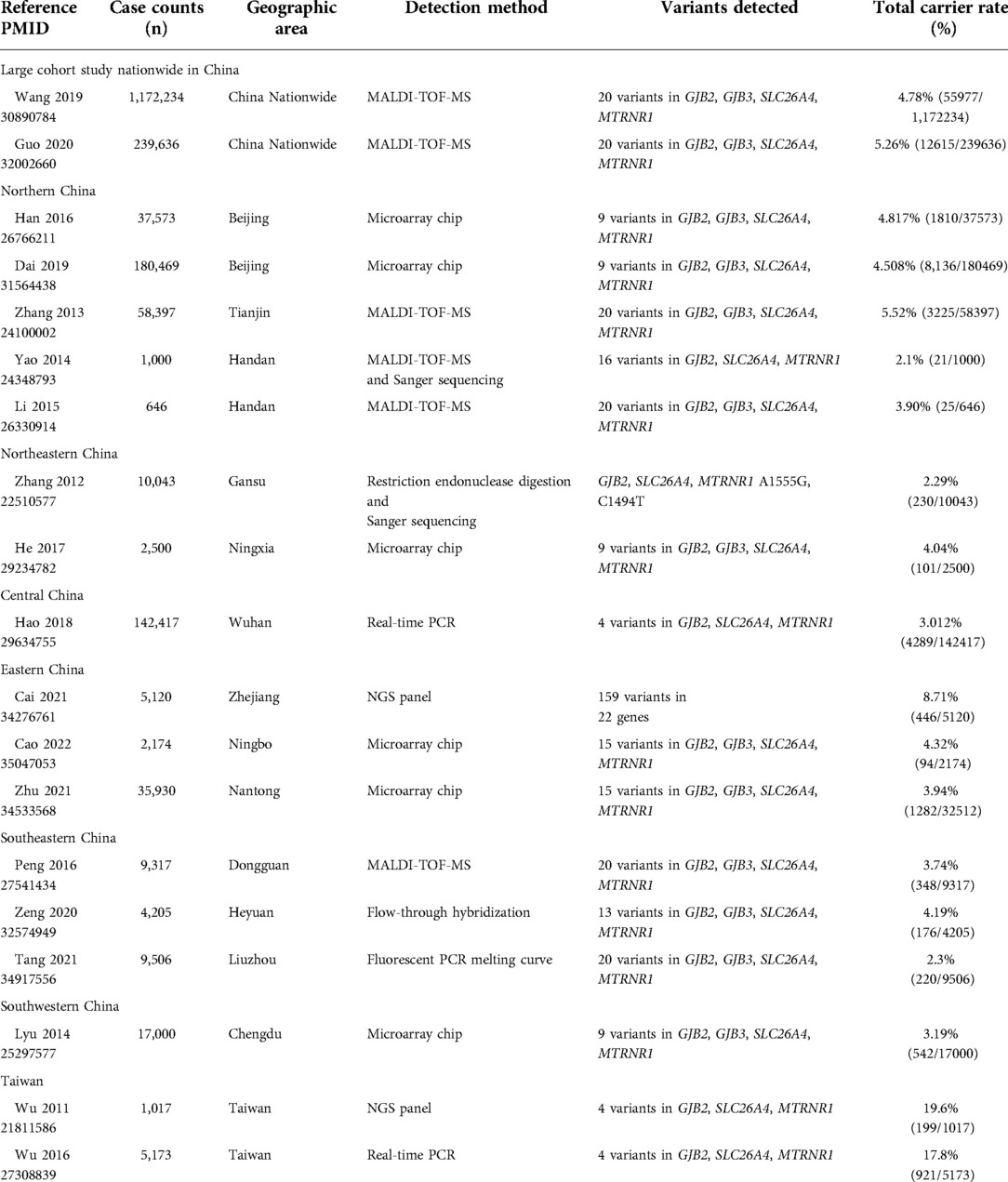
Table 6. Previous studies on genetic screening of neonates in different areas of China.
Based on a survey by the Chinese National Bureau of Statistics, the number of births in 2021 in Jiangxi Province was 377,000, which suggests that an estimated number of 18,145 individuals may be positive for the 20 common hearing loss variants screened. In our study, we identified newborns with congenital or prelingual HL at the rate of 0.03%, indicating that almost 113 individuals may be diagnosed as congenital or prelingual HL at much earlier time by genetic screening. Besides, we detected mitochondria mutations at a rate of 0.081% in our study, which indicates that nearly 305 infants and their maternal family members can be protected from aminoglycoside-antibiotic induced HL. Assume a deafness treatment needs 500,000 Yuan, this would save a total of 20.9 million Yuan each year in Jiangxi Province. What's more, a predicted number of 8,444 carriers for GJB2 (2.240%*377,000) and 5,967 carriers for SLC26A4 (1.583%*377,000) can be instructed on their future pre-pregnancy or prenatal genetic counseling by genetic screening. Therefore, genetic screening in all the newborns are more cost effective than only in the patients who are at higher risk or those who failed the hearing screening in identifying late-onset or progressive or drug-induced hearing loss, as well as identifying the carriers of hearing loss mutations for the guidance of future pre-pregnancy or prenatal genetic counseling.
As previously reported, the heterozygous c.538C > T variant in GJB3 has been reported to be associated with late-onset high-frequency sensorineural HL (21). Nevertheless, a recent study showed no evidence of the pathogenicity of GJB3 variants in autosomal recessive, dominant, or digenic hearing loss (33). Further investigations on the association between hearing loss and GJB3 variants are essential in our future work. Individuals with an MT-RNR1 mutation are predisposed to ototoxicity, and drug-susceptible hearing loss can be eliminated without inadvertent aminoglycoside exposure. Therefore, the vast majority of newborns without the use of aminoglycoside-antibiotics just display normal hearing in the hearing screening. Notably, one infant with deafness-causative GJB2 c.235delC homozygous variant and one with SLC26A4 c.1229 C > T/c.1975G > C passed the two-step hearing screening in our study, while the remaining five individuals with homozygous or compound heterozygous variants failed both the hearing screenings. Findings in our study were likely to the data in the previous report. A large-scale study revealed that almost 25% of the infants with two pathogenic combinations of GJB2 or SLC26A4 variants passed initial or second-tier hearing screening, while most of them would develop into HL before the age of five (14). Importantly, longitudinal auditory outcomes of all these cases with causative variants are crucial in our future work, especially for their diagnostic testing. In our work, the vast majority of newborns identified as carriers by genetic screening passed the initial hearing screening with a portion of approximately 92.2% and the hearing re-screening with the portion of 86.34%. However, additional testing of the entire coding regions sequencing for a second pathogenic mutant allele should be taken into consideration for carriers of GJB2 or SLC26A4, especially those who failed the hearing screening. The entire exons sequencing of GJB2 and SLC26A4 revealed 14 infants as causative compound heterozygous variants, including thirteen as GJB2 compound heterozygotes and one as SLC26A4 compound heterozygote. Since GJB2 c.109G > A is a highly frequent mutant allele, with an allele frequency of almost 6.7% in the Chinese population (19), it also demonstrated a relatively high detection rate of 7.19% (12/167) in Jiangxi Province in our study. However, studies have shown that compound heterozygous or homozygous mutation allele of GJB2 c.109G > A has significantly variable penetrance and expressivity of deafness, associated with normal hearing to moderate progressive SNHL in humans (34). In our study, all the 12 infants with GJB2 c.109G > A compound heterozygous variants passed the two-step hearing screening with a limited sample size, but longitudinal and periodic audiological evaluation should also be scheduled for them. Two infants failing both the hearing screening were identified with a second rare pathogenic mutant allele, including c.164C > A in GJB2 (c.176_191del16/c.164C > A) and c.1615-1G > T in SLC26A4 (c.919-2A > G/c.1615-1G > T) by the entire coding regions sequencing, which otherwise could be neglected only by conventional UNHS and genetic screening. The additional testing with entire coding regions sequencing assisted in elucidating the molecular etiologic of hearing loss for these two infants, facilitating early detection and intervention of congenital hearing infects.
In spite of the strength above, several limitations were present in our study. Firstly, not all the genetic causes of hearing loss could be determined. We only genotyped the 20 frequent variants in four common genes in the present study. Further efforts are needed to analyze other frequent deafness-predisposing genes, such as TMC1, USH2A, CDH23 and MYO15A in an expanded panel in future studies. Secondly, an increased hearing loss risk in the “carrier” genotype group with only one heterozygous pathogenic variant identified in GJB2 or SLC26A4 may be attributed to the inclusion of affected individuals whose second pathogenic allele was undetectable by the screening, but only a limited number of individuals underwent the entire coding regions sequencing in our study. Thirdly, congenital HCMV infection, one of the leading causes of congenital hearing loss, was not included in this study. In addition, we haven't recorded the diagnostic audiological results of these infants referred from the second hearing screening so far. Therefore, long-term follow-up investigations of this cohort are, warranted.
Conclusion
In summary, this is the first study providing an insight into the effective strategy for prevention of common hereditary HL and early identification of late-onset prelingual HL by concurrent newborn hearing and genetic screening in a large cohort newborns in Jiangxi Province. The results of our study highlighted the importance of newborn genetic screening in elucidating common etiologies, identifying infants with late-onset or progressive hearing impairment, and recognizing newborns and their maternal relatives at-risk of increased susceptibility to ototoxicity undetectable by newborn hearing screening. Our study may also provide as a reference in promoting the implementation of universal concurrent newborn hearing and genetic screening in the whole area in Jiangxi Province in the future.
Data availability statement
The authors acknowledge that the data presented in this study must be deposited and made publicly available in an acceptable repository, prior to publication. Frontiers cannot accept a manuscript that does not adhere to our open data policies.
Ethics statement
The studies involving human participants were reviewed and approved by EC-KT-202216. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
HL and YY were in charge of the genetic analysis, conceptualized the study, and wrote the manuscript original draft preparation. XW, DL and LZ were responsible for the clinical evaluation. FX, CH, TH, PM, QL did the investigation and data analysis. SH, BY, YZ and YL wrote, reviewed, and edited the manuscript. All authors have read and agreed to the published version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Provincial Health Commission Program of Jiangxi (Grant No. SKJP220211247 to Haiyan Luo and No. 20201095 to Yongyi Zou). Jiangxi Provincial Key Laboratory of Birth Defect for Prevention and Control (No. 20202BCD42017 to Yanqiu Liu). National Natural Science Foundation of China (Grant No. 82160318 to Yongyi Zou) and Postgraduate Innovation Special Fund Project of Jiangxi Province in 2022 of Cheng Huang.
Acknowledgments
We sincerely express our thanks to all the newborns enrolled and their parents or guardians for their participation in our study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tucci D, Merson MH, Wilson BS. A summary of the literature on global hearing impairment: current status and priorities for action. Otol Neurotol. (2010) 31(1):31–41. doi: 10.1097/MAO.0b013e3181c0eaec
2. Cai L, Liu Y, Xu Y, Yang H, Lv L, Li Y, et al. Multi-center in-depth screening of neonatal deafness genes: Zhejiang, China. Front Genet. (2021) 12:637096. doi: 10.3389/fgene.2021.637096
3. Wang QJ, Zhao YL, Rao SQ, Guo YF, He Y, Lan L, et al. Newborn hearing concurrent gene screening can improve care for hearing loss: a study on 14,913 Chinese newborns. Int J Pediatr Otorhinolaryngol. (2011) 75(4):535–42. doi: 10.1016/j.ijporl.2011.01.016
4. Han B, Zong L, Li Q, Zhang Z, Wang D, Lan L, et al. Newborn genetic screening for high risk deafness-associated mutations with a new Tetra-primer ARMS PCR kit. Int J Pediatr Otorhinolaryngol. (2013) 77(9):1440–5. doi: 10.1016/j.ijporl.2013.05.040
5. Fink D. Review of hearing loss in children. JAMA. (2021) 325(12):1223–4. doi: 10.1001/jama.2021.0387
6. Nieman CL, Oh ES. Hearing loss. Ann Intern Med. (2020) 173(11):ITC81–96. doi: 10.7326/AITC202012010
7. Marazita ML, Ploughman LM, Rawlings B, Remington E, Arnos KS, Nance WE. Genetic epidemiological studies of early-onset deafness in the U.S. School-age population. Am J Med Genet. (1993) 46(5):486–91. doi: 10.1002/ajmg.1320460504
8. Najmabadi H, Kahrizi K. Genetics of non-syndromic hearing loss in the Middle East. Int J Pediatr Otorhinolaryngol. (2014) 78(12):2026–36. doi: 10.1016/j.ijporl.2014.08.036
9. Yuan Y, You Y, Huang D, Cui J, Wang Y, Wang Q, et al. Comprehensive molecular etiology analysis of nonsyndromic hearing impairment from typical areas in China. J Transl Med. (2009) 7:79. doi: 10.1186/1479-5876-7-79
10. Clemens CJ, Davis SA. Minimizing false-positives in universal newborn hearing screening: a simple solution. Pediatrics. (2001) 107(3):E29. doi: 10.1542/peds.107.3.e29
11. Watkin PM, Baldwin M. Identifying deafness in early childhood: requirements after the newborn hearing screen. Arch Dis Child. (2011) 96(1):62–6. doi: 10.1136/adc.2010.185819
12. Morton CC, Nance WE. Newborn hearing screening–a silent revolution. N Engl J Med. (2006) 354(20):2151–64. doi: 10.1056/NEJMra050700
13. Fortnum HM, Summerfield AQ, Marshall DH, Davis AC, Bamford JM. Prevalence of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: questionnaire based ascertainment study. Br Med J. (2001) 323(7312):536–40. doi: 10.1136/bmj.323.7312.536
14. Dai P, Huang LH, Wang GJ, Gao X, Qu CY, Chen XW, et al. Concurrent hearing and genetic screening of 180,469 neonates with follow-up in Beijing, China. Am J Hum Genet. (2019) 105(4):803–12. doi: 10.1016/j.ajhg.2019.09.003
15. Zhu QW, Li MT, Zhuang X, Chen K, Xu WQ, Jiang YH, et al. Assessment of hearing screening combined with limited and expanded genetic screening for newborns in nantong, China. JAMA Netw Open. (2021) 4(9):e2125544. doi: 10.1001/jamanetworkopen.2021.25544
16. Kennedy CR, McCann DC, Campbell MJ, Law CM, Mullee M, Petrou S, et al. Language ability after early detection of permanent childhood hearing impairment. N Engl J Med. (2006) 354(20):2131–41. doi: 10.1056/NEJMoa054915
17. Liu M, Liu C. Advances in research on the prevalence and prevention of disabilities in China. Zhonghua Liu Xing Bing Xue Za Zhi. (2011) 32(6):533–8.21781466
18. García-García G, Berzal-Serrano A, García-Díaz P, Villanova-Aparisi R, Juárez-Rodríguez S, de Paula-Vernetta C, et al. Improving the management of patients with hearing loss by the implementation of an NGS panel in clinical practice. Genes (Basel). (2020) 11(12):1467. doi: 10.3390/genes11121467
19. Dai P, Yu F, Han B, Liu X, Wang G, Li Q, et al. GJB2 Mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J Transl Med. (2009) 7:26. doi: 10.1186/1479-5876-7-26
20. Dai P, Yuan Y, Huang D, Zhu X, Yu F, Kang D, et al. Molecular etiology of hearing impairment in Inner Mongolia: mutations in SLC26A4 gene and relevant phenotype analysis. J Transl Med. (2008) 6:74. doi: 10.1186/1479-5876-6-74
21. Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP, et al. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat Genet. (1998) 20(4):370–3. doi: 10.1038/3845
22. Chen S, Liang Z, Chen B, Yao F, Huang G, Zhu J, et al. The prevalence of deafness-associated mutations in neonates: a meta-analysis of clinical trials. Int J Pediatr Otorhinolaryngol. (2019) 121:99–108. doi: 10.1016/j.ijporl.2019.01.012
23. Guomei C, Luyan Z, Lingling D, Chunhong H, Shan C. Concurrent hearing and genetic screening among newborns in Ningbo, China. Comput Math Methods Med. (2022) 2022:1713337. doi: 10.1155/2022/1713337
24. Wang Q, Xiang J, Sun J, Yang Y, Guan J, Wang D, et al. Nationwide population genetic screening improves outcomes of newborn screening for hearing loss in China. Genet Med. (2019) 21(10):2231–8. doi: 10.1038/s41436-019-0481-6
25. Guo L, Xiang J, Sun L, Yan X, Yang J, Wu H, et al. Concurrent hearing and genetic screening in a general newborn population. Hum Genet. (2020) 139(4):521–30. doi: 10.1007/s00439-020-02118-6
26. Zhang J, Wang P, Han B, Ding Y, Pan L, Zou J, et al. Newborn hearing concurrent genetic screening for hearing impairment-a clinical practice in 58,397 neonates in Tianjin, China. Int J Pediatr Otorhinolaryngol. (2013) 77(12):1929–35. doi: 10.1016/j.ijporl.2013.08.038
27. Li SX, Chen DL, Zhao SB, Guo LL, Feng HQ, Zhang XF, et al. Cordblood-based high-throughput screening for deafness gene of 646 newborns in Jinan area of China. Clin Exp Otorhinolaryngol. (2015) 8(3):211–7. doi: 10.3342/ceo.2015.8.3.211
28. Peng Q, Huang S, Liang Y, Ma K, Li S, Yang L, et al. Concurrent genetic and standard screening for hearing impairment in 9317 Southern Chinese newborns. Genet Test Mol Biomarkers. (2016) 20(10):603–8. doi: 10.1089/gtmb.2016.0055
29. Tang X, Liu L, Liang S, Liang M, Liao T, Luo S, et al. Concurrent newborn hearing and genetic screening in a multi-ethnic population in South China. Front Pediatr. (2021) 9:734300. doi: 10.3389/fped.2021.734300
30. Kenneson A, Van Naarden Braun K, Boyle C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genet Med. (2002) 4(4):258–74. doi: 10.1097/00125817-200207000-00004
31. Miyagawa M, Nishio SY, Usami S. Mutation spectrum and genotype-phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: a large cohort study. J Hum Genet. (2014) 59(5):262–8. doi: 10.1038/jhg.2014.12
32. Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M, et al. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet. (2003) 40(4):242–8. doi: 10.1136/jmg.40.4.242
33. Huang S, Huang B, Wang G, Kang DY, Zhang X, Meng X, et al. The relationship between the GJB3 c.538C > T variant and hearing phenotype in the Chinese population. Int J Pediatr Otorhinolaryngol. (2017) 102:67–70. doi: 10.1016/j.ijporl.2017.09.001
Keywords: newborn hearing screening, genetic screening, hearing loss, targeted next generation sequencing (NGS), genetic counseling
Citation: Luo H, Yang Y, Wang X, Xu F, Huang C, Liu D, Zhang L, Huang T, Ma P, Lu Q, Huang S, Yang B, Zou Y and Liu Y (2022) Concurrent newborn hearing and genetic screening of common hearing loss variants with bloodspot-based targeted next generation sequencing in Jiangxi province. Front. Pediatr. 10:1020519. doi: 10.3389/fped.2022.1020519
Received: 17 August 2022; Accepted: 6 October 2022;
Published: 31 October 2022.
Edited by:
Bassam R Ali, United Arab Emirates University, United Arab EmiratesReviewed by:
Hongyang Wang, People's Liberation Army General Hospital, ChinaFiras Alzoubi, Jordan University of Science and Technology, Jordan
© 2022 Luo, Yang, Wang, Xu, Huang, Liu, Zhang, Huang, Ma, Lu, Huang, Yang, Zou and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanqiu Liu bHlxMDkxNEAxMjYuY29t Yongyi Zou em91eW9uZ3lpQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics