
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 05 October 2022
Sec. Pediatric Immunology
Volume 10 - 2022 | https://doi.org/10.3389/fped.2022.1017195
 Pedro Simão Coelho1*
Pedro Simão Coelho1* Catarina Gouveia2,3
Catarina Gouveia2,3 Marta Valente Pinto1Conceição Neves1
Marta Valente Pinto1Conceição Neves1 Ana Isabel Cordeiro1
Ana Isabel Cordeiro1 João Farela Neves1,3,4
João Farela Neves1,3,4
C3 is a crucial protein of the complement system. Congenital C3 deficiency is extremely rare and manifests through recurrent, severe infections and should always be considered as a differential diagnosis of recurrent pyogenic infections. We report a case of a patient with a novel C3 gene mutation, responsible for complete C3 deficiency with impaired complement system activation and recurrent infections.
Due to anatomical particularities and to the immaturity of the immune system, recurrent infections are very frequent in children. This can lead to a delay in the identification of the patients with congenital defects of immunity. Among children with recurrent infections, those presenting with pyogenic infections caused by encapsulated bacteria, such as Streptococcus pneumoniae, Neisseria meningitidis or Haemophilus influenzae, should always be screened for an underlying primary immunodeficiency (PID) (1). PIDs are a heterogeneous group of monogenic disorders that affect the development and/or function of both innate and adaptive immune system. PIDs can present as frequent, recurrent, or persistent infections, but also as early-onset and/or severe autoimmunity and inflammation (1).
The complement system is part of the immune system and participates not only in host defense against infections but also in the clearance of debris and in the maintenance of a homeostatic inflammatory status (2). In host defense, it is one of the first components engaged by pathogens and its two major functions are mediated by opsonization and membrane perturbation (3). C3 cleavage is the converging point of all complement pathways and, therefore, it is a central step of the complement system (4). Immune complexes are a potent activator of the complement system, and in normal circumstances, are maintained in a soluble state by C3, preventing them to act as a stimulus for inflammation. When C3 is compromised, immune complexes may deposit on endothelial cells and trigger an inflammatory response (3).
Inherited C3 deficiency is an extremely rare primary immunodeficiency, with, until recently, approximately 40 cases described. Its main clinical features are severe recurrent infections mainly caused by encapsulated bacteria, as well as autoimmunity and diseases caused by immune complexes deposition, such as membranous glomerulonephritis (5, 6).
We report a case of a patient with a novel C3 gene mutation responsible for complete C3 deficiency presenting with severe and recurrent bacterial infections.
A 4-year-old boy was admitted to the hospital with serogroup B Neisseria meningitidis septic shock and meningitis requiring fluid resuscitation and ceftriaxone. His family history was remarkable for parental consanguinity (parents were second degree cousins, Figure 1A) and he had a personal history of recurrent pyogenic infections since birth, with previous hospital admissions caused by orbital cellulitis and Streptococcus pneumoniae lobar pneumonia. He also presented recurrent acute otitis media and tonsillitis.
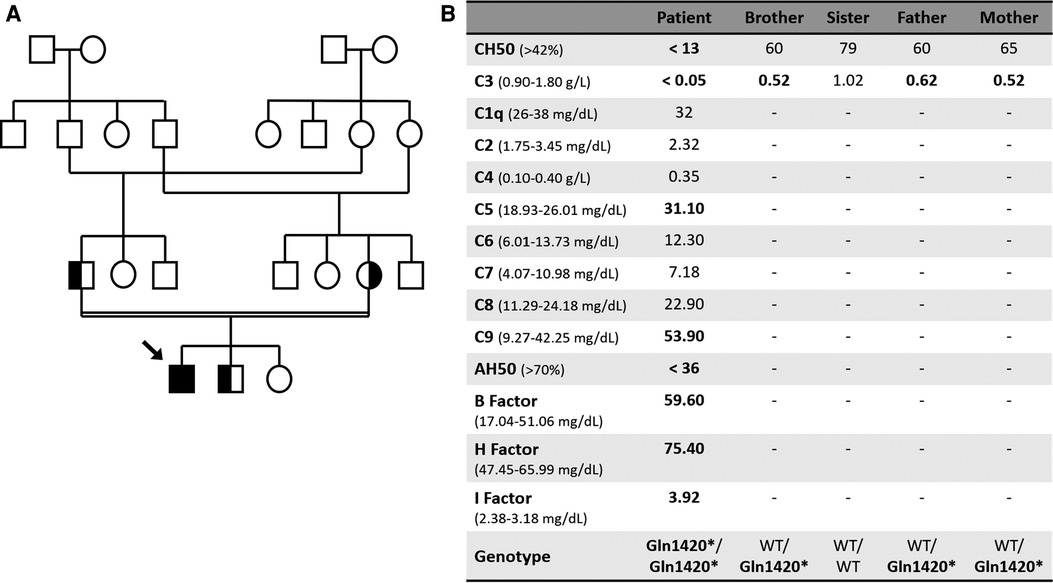
Figure 1. (A) Family tree. (B) Complement laboratory evaluation. WT, Wild type.
Diagnostic workup revealed undetectable classical complement pathway activity (CH50) and C3 levels (Figure 1B). Further study demonstrated a reduced alternative pathway activity with increased complement B, H and I factors, and raised C5 and C9 levels, probably related to an underlying inflammatory status (Figure 1B). Immunoglobulin G, A and M were within normal range. There was no clinical or laboratory evidence of organ damage after resolution of the infectious disease. The patient was suspected to have a C3 deficiency and was promptly vaccinated against meningococcus and pneumococcus and, because he had frequent and severe infections, antibiotic prophylaxis with amoxicillin was commenced with good adherence and no more hospital admissions. Genetic testing was performed using Illumina next-generation sequencing for a panel of genes involved in complement deficiencies, and revealed a novel homozygotic nonsense mutation in the C3 gene (p.Gln1420*)) that leads to a stop codon, thus being predicted to be deleterious and pathogenic. The mutation was confirmed by Sanger sequencing. The patient's family was screened and both parents and his 3-year-old brother were shown to be healthy carriers of the mutation (Figures 1A,B). The patient is being monitored quarterly and has been presenting good evolution, free from severe infections. Genetic counselling was offered to the family.
C3 is a major component of the complement system. It is the converging point of all three pathways and is responsible for full activation of the complement system. It is also involved in a variety of homeostatic processes, such as tissue regeneration and tumor cell progression control (6, 7). It derives from a precursor molecule with 1663 amino acids encompassed by 41 exons located on chromosome 19. C3 mRNA is translated into pro-C3 and cleaved into β chain (exons 1–16) and α chain (exons 16–41), which are subsequently linked by disulfide bonds to make the mature C3 molecule (8).
The C3 is cleaved by both classical (C4bC2a) and alternative (C3bBb) pathway convertases, resulting in C3a and C3b. C3a is an anaphylatoxin responsible for degranulation of mast cells and basophils, leading to inflammatory reactions (3, 9). C3b is the major opsonin of the complement system and promotes adherence between opsonized microbes and immune cells. Through iC3b, a product of C3b degradation, enhances complement mediated phagocytosis and acts as a natural adjuvant for antibody production by reducing B cell receptor stimulation threshold (7, 10).
A dysregulation of the complement system may be involved in a decrease in functional activity that leads to increased susceptibility to bacterial infections, particularly pneumococcal for early and meningococcal for late pathway, autoimmunity and undesirable tissue damage (3). Complement deficiencies are considered rare, but they are estimated to be responsible for between 1% and 10% of all PIDs (9). Most congenital complement deficiencies display an autosomal recessive inheritance and heterozygous carriers usually remain asymptomatic, with defects being identified through medical history and family analysis (9). In most cases, the onset of infections begins in childhood but the undervaluation of this type of disease may reflects a lack of awareness among practitioners (6). Lack of C3, besides recurrent encapsulated pyogenic bacteria infections, also leads to an impaired inflammatory response and decreases the patient's ability to clear immune complexes, resulting in renal, pulmonary and vascular damage and might also promote the development of antibodies to self-antigens (3, 9), which did not happen in our patient.
The European Society of Immunodeficiencies has issued recommendations regarding management of complement deficiencies, with emphasis on vaccination, namely on conjugated vaccines against pneumococcus, Haemophilus influenzae and Neisseria meningitidis, as one of the most important tools to prevent severe infections (5, 11).
In the case of C3 deficiency, due to the increased susceptibility to invasive pneumococcal infections and recurrent pyogenic infections, some authors recommend that in addition to vaccination it may be important to evaluate pneumococcal antibody levels, since patients with low level responses should receive booster vaccinations (5).
Regarding antibiotics, there is no formal recommendation and should therefore be evaluated for every patient, based on an individualized risk stratification. Most patients with complement deficiencies may be managed with an as-needed plan, with prompt use within the first signs of infection. Antibiotic prophylaxis is controversial, as the benefits should be balanced against the risks, particularly the development of resistance to antibiotics. When used, it may be penicillin or macrolide-based, as it should target encapsulated bacteria (5, 12). The authors propose that it should only be used in patients with recurrent infections despite appropriate vaccination, namely in those with difficult access to healthcare services, as this may delay medical assistance and worsen their prognosis.
In summary, we report the first Portuguese patient with complete C3 deficiency caused by a novel C3 mutation that was diagnosed after a severe meningococcal infection on a patient with recurrent pyogenic infections and with a consanguineous family.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
PC collected data and composed the manuscript. CG and JN are the physicians responsible for the patient and have given permission to PC to report this case. Senior co-authors CG, MP, CN, AC and JN contributed to the elaboration of the manuscript and provided critical revision for important intellectual content. All authors contributed to the article and approved the submitted version.
The present publication was funded by Fundação Ciência e Tecnologia, IP national support through CHRC (UIDP/04923/2020).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Devonshire AL, Makhija M. Approach to primary immunodeficiency. Allergy Asthma Proc. (2019 Nov 1) 40(6):465–9. doi: 10.2500/aap.2019.40.4273
2. Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol. (2007) 188(2):183–94. doi: 10.1111/cei.12952
3. Atkinson JP. Complement system in disease. In: Goldman L, Shaffer A, editors. Goldman-cecil medicine, 26th ed. Philadelphia: Elsevier (2020). p. 220–226.e1.
4. Angioi A, Fervenza FC, Sethi S, Zhang Y, Smith RJ, Murray D, et al. Diagnosis of complement alternative pathway disorders. Kidney Int. (2016) 89:278–88. doi: 10.1016/j.kint.2015.12.003
5. Brodszki N, Frazer-Abel A, Grumach AS, Kirschfink M, Litzman J, Perez E, et al. European society for immunodeficiencies (ESID) and European reference network on rare primary immunodeficiency, autoinflammatory and autoimmune diseases (ERN RITA) complement guideline: deficiencies, diagnosis, and management. J Clin Immunol. (2020) 40(4):576–91. doi: 10.1007/s10875-020-00754-1
6. Reis ES, Falcão DA, Isaac L. Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H. Scand J Immunol. (2006) 63(3):155–68. doi: 10.1111/j.1365-3083.2006.01729.x
7. Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 - The “Swiss Army Knife” of innate immunity and host defense. Immunol Rev. (2016) 274(1):33–58. doi: 10.1111/imr.12500
8. Kida M, Fujioka H, Kosaka Y, Hayashi K, Sakiyama Y, Ariga T. The first confirmed case with C3 deficiency caused by compound heterozygous mutations in the C3 gene; a new aspect of pathogenesis for C3 deficiency. Blood Cells Mol Dis. (2008) 40(3):410–3. doi: 10.1016/j.bcmd.2007.11.002
9. Grumach AS, Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol Immunol. (2014) 61(2):110–7. doi: 10.1016/j.molimm.2014.06.030
10. West EE, Kolev M, Kemper C. Complement and the regulation of T cell responses. Annu Rev Immunol. (2018) 36:309–38. doi: 10.1146/annurev-immunol-042617-053245
11. Rubin LG, Levin MJ, Ljungman P, Davies EG, Avery R, Tomblyn M, et al. Infectious diseases society of America. 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clin Infect Dis. (2014) 58(3):309–18. doi: 10.1093/cid/cit816
Keywords: primary immunodeficiency, complement deficiency, C3 deficiency, C3 gene mutation, recurrent infections
Citation: Coelho PS, Gouveia C, Pinto MV, Neves C, Cordeiro AI and Neves JF (2022) Recurrent pyogenic infections caused by a novel Gln1420* mutation in the C3 gene. Front. Pediatr. 10:1017195. doi: 10.3389/fped.2022.1017195
Received: 11 August 2022; Accepted: 20 September 2022;
Published: 5 October 2022.
Edited by:
Luis Ignacio Gonzalez-Granado, University Hospital, SpainReviewed by:
Margarita López-Trascasa, Universidad Autónoma de Madrid, Spain© 2022 Coelho, Gouveia, Pinto, Neves, Cordeiro and Neves. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pedro Simão Coelho cGVkcm8uY29lbGhvM0BjaGxjLm1pbi1zYXVkZS5wdA==
Specialty Section: This article was submitted to Pediatric Immunology, a section of the journal Frontiers in Pediatrics
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.