 Meng Dong
Meng Dong Tianyu Zhang
Tianyu Zhang Ruimei Hu
Ruimei Hu Meng Li
Meng Li Xinjie Liu
Xinjie Liu- Department of Pediatrics, Qilu Hospital, Shandong University, Jinan, China
Objective: STXBP1 mutations are associated with early onset epileptic encephalopathy (EOEE). Our aim was to explore the phenotype spectrum, clinical treatment and prognosis of STXBP1-related encephalopathy (STXBP1-E).
Methods: Clinical and genetic data were collected from 10 patients with STXBP1 mutations. These patients were examined and diagnosed from 2015 to 2021 at the Pediatric Department of Qilu Hospital. Blood samples were collected and sequenced by next generation sequencing and Candidate pathogenic variants were identified using Sanger sequencing in all family members.
Results: All of the patients showed severe epilepsy, varying degrees of intellectual disability and delayed motor. The patients developed multiple seizure types and abnormal electroencephalography (EEG) results at onset, and focal seizures were the most frequent seizure type. Among the patients, 2 were diagnosed with Ohtahara syndrome, 2 patient was diagnosed with West syndrome. The other 6 patients could not be diagnosed with any specifically recognized epilepsy syndrome. Five of the 10 patients had a history of fever with seizures, 4 of whom had eliminated intracranial infection according to the results of cerebrospinal fluid (CSF) examinations, and the other patient was diagnosed with anti-myelin oligodendrocyte glycoprotein (MOG) -associated encephalitis. We identified one patient with a complete deletion of STXBP1 and 9 patients with de novo heterozygous mutations of STXBP1. Among those mutations, 4 were novel (c.56°C > T, c.1315A > T, c.751G > C, and c.554_559del), and 5 had been previously reported [c.364C > T, c.569G > A (2 cases), c.748C > T, and c.1651C > T]. For 8 of our patients, different combinations of anti-seizure medications (ASMs) led to seizure freedom. One patient with MOG antibodies in his serum obtained a poor therapeutic effect from the traditional ASMs treatment, so he had to achieve seizure-free status through vagus nerve stimulation (VNS), which had little effect on his psychomotor ability. Fortunately, in one case, patient psychomotor ability was improved through VNS.
Conclusion: Our study shows that STXBP1 screening should be considered in patients with neonatal seizures with intellectual disability, and frequent seizures with fever should also be considered with the STXBP1 mutation when intracranial infection is eliminated. VNS has expanded outcome measures to include behavioral and developmental function as well as seizure control.
Introduction
Early-onset epileptic encephalopathy (EOEE) embodies the notion that continuous and repeated epileptic activity itself may contribute to severe neurological and cognitive impairment and prominent interictal epileptiform discharges during the neonatal or early infant period (1). After the initial identification of mutations for cryptogenic West syndrome and Ohtahara syndrome (2), in the genes ARX, CDKL5, STXBP1, SLC25A22, SPTAN1, PLCb1, MAGI2, PNKP, SCN1A, numerous other mutated genes for West syndrome, Dravet syndrome and various other types of childhood-onset epilepsy have been found, revealing that a significant proportion of cryptogenic EOEEs are single-gene disorders (3–5). West syndrome, also known as epileptic spasms or infantile spasms which belong to infantile epileptic spasms syndrome (IESS) (6). In 2008, the first mutation in the syntaxin binding protein 1 (STXBP1) gene was confirmed to be associated with EOEE (7). Since then, mutations in STXBP1 have been described in different patient cohorts, the phenotypic spectrum of patients with STXBP1 mutations has expanded, and an increasing number of studies have indicated that epilepsy and intellectual disease (ID) are 2 major features of STXBP1 encephalopathy (STXBP1-E) (7, 8). However, Hamdan et al. reported a STXBP1-positive patient with mild nonsyndromic ID without epilepsy (9). Presently, STXBP1-E is considered a complex neurodevelopmental disorder rather than a primary epileptic encephalopathy (10). Previous studies have reported that vigabatrin, valproic acid (VPA), levetiracetam (LEV), adrenocorticotropic hormone and a ketogenic diet (KD) are effective at seizure control (11–15). However, the prognosis of the child is not good, so it is necessary to deeply understand this disease and improve the prognosis. Our study not only expands the phenotypic spectrum associated with STXBP1-E but also describes the role of VNS in controlling epilepsy and improving cognition.
Materials and methods
Clinical data and phenotypes groups
All the patients diagnosed EOEE were detected by next-generation sequencing from Jan 2015 to Jul 2021 at the Pediatric Department in Qilu Hospital and only ten previously unreported patients with a STXBP1 mutation were enrolled in this study. Clinical data [classification in epileptic syndrome, epileptic spasms, ambulatory or video EEG monitoring, magnetic resonance imaging (MRI), history of status epilepticus, treatments, and follow-up until July 2021] were collected from their clinical cases. Approximately 2 ml of peripheral blood (plus EDTA anticoagulant) was obtained from the patient and their parents after receiving written informed consent.
We refer to the study from Wolking et al. (16), they discerned 4 different phenotypic groups and divided into four groups [developmental and epileptic encephalopathies, DEE; genetic epilepsies with febrile seizures (FS) plus, GEFS+; genetic generalized epilepsy, GGE; focal epilepsy] according to their clinical characters.
The investigation was performed with the approval of the ethics committee of Qilu Hospital.
Genetic analysis
Genomic DNA was extracted using a QIAamp Blood Midi Kit (QIAGEN, Valencia, CA). To identify disease-causing gene variants, a GenCap panel with 175 genes (including STXBP1, Table 1) associated with epilepsy was customized, and a capture strategy was performed using the GenCap custom enrichment kit (MyGenostics Inc, Beijing, China). An Illumina NextSeq 500 sequencer (Illumina, San Diego, CA, United States) was used with 150 bp paired-end reads. An ABI3730xl sequencer (Applied Biosystems, United States) was used to apply the Sanger sequencing method, and the results were compared to the capture sequencing results.

Table 1. Detected gene list.
After sequencing, the raw data were saved in FASTQ format. Quality control (QC) filters were applied to remove reads with low quality. Then, the clean reads were assembled and spliced using the second-generation sequencing analysis platform provided by MyGenostics and the coverage and sequencing quality of the target region were evaluated. Finally, flash analysis platform was used to analyze the pathogenicity of variation, and the possible variation loci were determined. The pathogenicity of variation loci was also analyzed according to ACMG (American College of Medical Genetics and Genomics) genetic variation classification criteria and guidelines.
We performed a conservative analysis of the eight mutant amino acid sequences using Clustal Omega. Domains of STXBP1 were identified based on the National Center for Biotechnology Information (NCBI) Conserved Domain Database. Multiple sequence alignment of STXBP1 was performed using the ClustalW program. Three-dimensional structural models of STXBP1 were predicted with the Swiss-model web tool. Protein structure images were generated using the PDB file and PyMOL. Hydrogen bonds in the proteins were demonstrated using PyMOL to predict changes in mutant stability.
Results
STXBP1 molecular analysis
In total, we identified 9 de novo STXBP1 mutations in 10 patients by next generation sequencing to sequence all the STXBP1 exons and splice junction boundaries from the genomic DNA. Upon searching in the HGMD (The Human Gene Mutation Database), 6 of them had been reported previously [c.364C > T, c.569G > A (2 cases), c.748C > T, c.1651C > T, c.56°C > T, one complete deletion], and the other 3 were novel mutations (c.1315A > T, c.751G > C, c.554_559del). Two unrelated patients (patient No. 2 and patient No. 8) had the same STXBP1 nucleotide alteration (c.569G > A). The details of the STXBP1 mutations are summarized in Table 2 and Figure 1. According to ACMG (American College of Medical Genetics and Genomics) criteria, we have determined that the variants are Pathogenic variants (Supplementary Figures S1, S2).
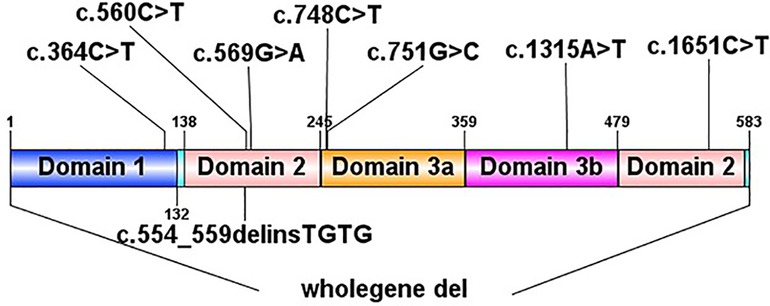
Figure 1. Pathogenic STXBP1 mutations.

Table 2. Summary of the variants in the STXBP-1 mutations.
Clinical characterization of STXBP1-positive of the 10 patients
The clinical features of this study with EOEE having STXBP1 defects are summarized in Table 3. The median age of ascertainment was 14.5 months (n = 10, range = 1–35 months), with a median age of seizure onset of 8.5 months (range = 0–22 months), and most patients had intellectual disability prior to seizure onset (60%). At the last follow-up, all patients had development delay. Patient 1 and patient 2 were diagnosed with Ohtahara syndrome, and patient 1 developed into West syndrome at the age of 5 months. Two patients had West syndrome (patient 4, 9), and 6 patients were diagnosed with unclassified EOEEs (patients 2, 3, 5, 6, 7, and 8). Various seizure types presented during the course of the disease, including epileptic spasms, partial seizures, atonic seizures, absence seizures, myoclonic seizures, and tonic seizures. The initial seizure types of the patients were partial seizures in 4 cases (patients 3, 5, 6, and 7), tonic seizures in 1 case (patient 2), tonic-clonic seizures in 2 cases (patients 1 and 10) and spasms in 3 cases (patients 4, 8, and 9). One case (patient 3) had an average number of seizures a few times a month, and all were associated with high fever. Three cases (patients 2, 6, and 8) had an average number of seizures of ≤5 times per day, and 6 cases (patients 1, 4, 5, 7, 9, and 10) had an average number of 10–20 seizures per day. Five patients (patients 2, 3, 6, 7, and 8) had a history of seizure with fever, and intracranial infection was eliminated in 4 patients (patients 2, 3, 7, and 8). In patient 6, cerebrospinal fluid examination showed positive MOG antibody. MRI showed abnormal signal near white matter in lateral ventricle triangle. There was still epileptic seizure after glucocorticoid and gamma globulin treatment, and the genetic examination showed STXBP1 gene mutation.
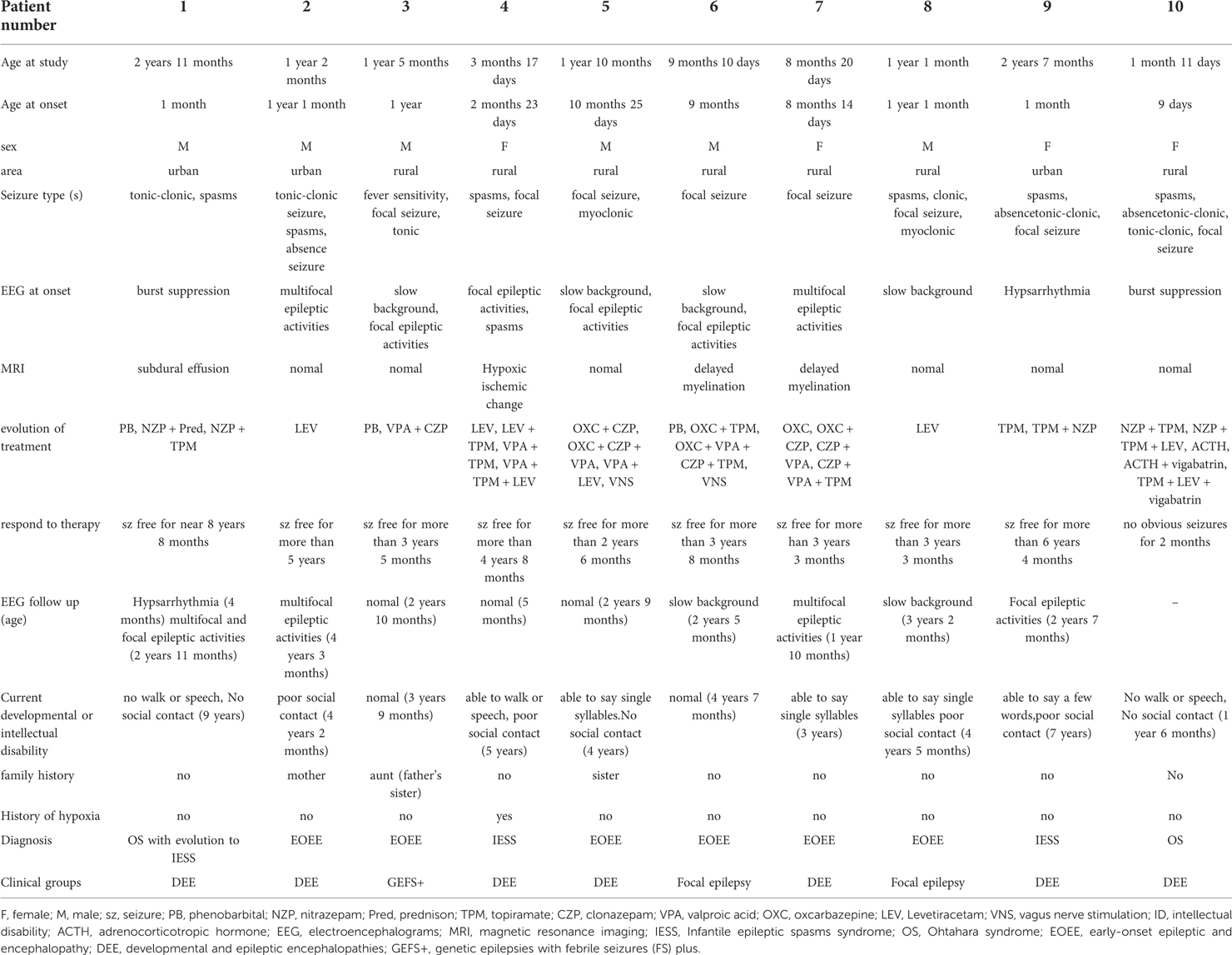
Table 3. Clinical features of patients with STXBP-1 mutations.
Reviewing the above clinical characteristics of all patients, we could distinguish 3 different phenotypic groups. (1) Seven patients (patient 1, 2, 4, 5, 7, 9, 10) had intractable seizures, occurrence of developmental stagnation or regression after seizure onset, and additional neuropsychiatric deficits compatible with developmental and epileptic encephalopathy (DEE). (2) Patient 3 had an average number of seizures a few times a month, and all were associated with high fever. All the times, two seizure types presented during the course of the disease, focal seizures and tonic seizures, and had a relatively benign course, generally good drug response, normal development, and mild neuropsychiatric symptoms, corresponding to genetic epilepsies with febrile seizures plus (GEFS+). (3) The last 2 patients (patient 6, 8) with some form of focal epilepsy.
EEG abnormalities were observed in all patients, including burst suppression (patients 1 and 10), hypsarrhythmia (patient 9), multifocal epileptic activities (patients 2, 3, 7), focal epileptic activities (patients 4, 5, and 6), and slow background (patients 3, 5, 6, 8). The initial EEG of 7 patients with STXBP1 mutations is shown in Figure 2. The other 3 patients had no initial EEG. At the last follow-up, EEG showed normalization after treatment in 3 cases (patients 3, 4, and 5). The initial brain MRI did not present any apparent structural abnormality. Of the nine patients included here, one patient had subdural effusion (patient 1), one had hypoxic-ischemic change (patient 4), and two had delayed myelination (patients 6 and 7). Blood biochemical examination showed no apparent abnormalities.
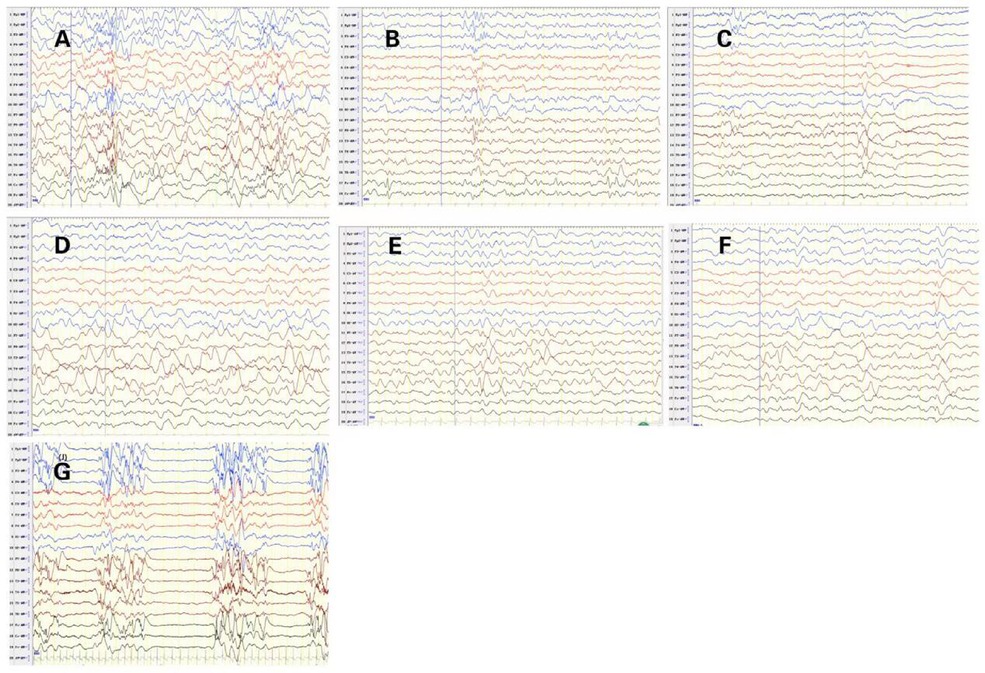
Figure 2. Initial electroencephalograms of patients with syntaxin-binding protein 1 (STXBP1) mutations. (A) Patient2 1year and 1months, Multifocal spikes and slow waves during sleep status. (B) Patient3 1year and 5months, slow background, multifocal slow waves. (C) Patient4 2 months and 29 days, spasms and focal epileptic activities originating from the left Anterior and middle temporal area. (D) Patient5 1 year and 2 months, slow background and Slow wave activity originating from the left middle and posterior temporal regions. (E) Patient6 9 months and 10days, sharp-slow waves originating from the right Posterior temporal area during sleep status on interictal EEG. (F) Patient7 8 months and 20 days, slow and spikes-slow waves originating from bilateral Parietal, Occipital, mid-posterior temporal areas during sleep status. (G) Patient10 1 months, burst suppression (BS).
Treatment and prognosis
With regard to the treatment and prognosis, including the use of VNS, huge improvements in seizure activity were found in all of our cohorts. With ongoing brain maturation, patient 1, who did not become seizure-free during the first few months of life, developed epileptic spasms. Together with the observation that none of the patients with STXBP1 mutations evolved to West syndrome later in life after controlling seizure. Four cases (patients 2, 4, 5, and 8) were free of seizures after treatment with LEV. Two patients received LEV monotherapy, they got seizures free when the dose of LEV got 40 mg/kg/d. Others received LEV combination therapy. Initially, patient 4 was treated with bolus doses of topiramate (TPM), nitrazepam (NZP) and VPA, but there was no significant improvement. Next, NZP was replaced with LEV, and the seizures were completely controlled. Patient 5 had failed with bolus doses of oxcarbazepine (OXC), clonazepam (CZP) and VPA. Next, CZP and OXC were replaced with LEV, and the seizures were completely controlled after treatment with LEV and VPA. To make things even better, he tried VNS after being seizure-free and improved his psychomotor ability. Patients 2 and 8 were free of seizures after treatment with LEV alone. Patient 6 had frequent convulsions after a two-day history of fever. The CSF analysis showed an elevated white count of 14 per cubic millimeter. Serum MOG antibodies were requested, and he was started on intravenous immunoglobulin, OXC, TPM, and glucocorticoid (GC). Two weeks later, although his repeat lumbar puncture (LP) was negative and the serum titer of MOG antibodies disappeared, his seizure status had not changed. Next, CZP and VPA were added successively; however, the seizures failed to respond to any medical treatment. Fortunately, he tried VNS, and his seizures promptly ceased after 20 days of VNS therapy. Although the effects of treatment on seizure frequency might be quantifiable, the effect on intellectual disability is not so easily assessed. The good news is that one case (patient 5) experienced improved psychomotor ability after VNS.
Discussion
STXBP1 is a member of the evolutionarily conserved Sec1/Munc-18 gene family that plays a central role in vesicular docking and fusion. Splicing mutations, a frameshift mutation, and nonsense mutations can lead to loss-of-function, which is a common mechanism underlying STXBP1-E (22). In this study, two novel recurrent missense mutations in STXBP1 (c.1315A > T, c.751G > C) were detected in three patients with unclassified epileptic encephalopathy (patients 5 and 7). We found that all the patients with novel mutations had similar clinical symptoms: early onset seizures and intellectual disability.
Barcia et al. (15) and Wolking et al. (16) demonstrated that the clinical features in STXBP1-E are mostly shared with early onset seizures, a poor prognosis with severe intellectual disability and a high mortality rate and a frequent evolution to infantile spasms (IS). However, in our study, 10 patients eventually became seizure-free, three of the 10 patients were West syndrome and they were reducing the development of West syndrome. We consider this result to be related to prompt and early seizure control. If the notion is that seizures must be controlled as soon as possible to optimize prognosis, then optimal targeted therapies are needed to shorten exposure to repetitive seizures.
EOEE are characterized by recurrent clinical seizures and prominent interictal epileptiform discharges seen during the early infantile period. The underlying genetic cause often results in developmental delay in its own right, with the epileptic encephalopathy further adversely affecting development. Frequent epileptiform activity that impacts adversely on development, typically causing slowing or regression of developmental skills. Similar to the study from Wolking et al. (16), all the patients were also divided into four groups (DEE, GEFS+, GTCS and focal epilepsy) according to their clinical characters. In our study, we noted that developmental delay and epilepsy are characteristic features of STXBP1-E, in keeping with previous reports (7, 8). Although developmental delay can be seen at seizure onset, some degree of developmental delay is present prior to the onset of seizures in many patients (patients 1, 3, 4, 5, 6, and 10). Furthermore, in 2009, Hamdan et al. (8) reported de novo mutations, p. R388X and c.169 + 1G > A in 2 cases, with intellectual disability and nonsyndromic epilepsy. In 2011, Hamdan et al. (9) reported 1 case with a novel de novo truncating mutation, c.1206delT/p.402X, presenting with nonsyndromic intellectual disability deficit and no history of epilepsy. These findings suggest no correlation between seizures and intellectual disability, as described previously (23). STXBP1 plays an important role in many aspects of neurodevelopment.
The initial EEG was abnormal in all patients. The main EEG finding was (multi)focal abnormality, while burst suppression or hypsarrhythmia was observed in three patients. This type of alteration is much less specific. At the last follow-up, the repeat EEG recordings of 3 cases (patients 3, 4, and 5) were normal. Structural imaging with MRI was normal in most cases, although delayed myelination was present in two patients, subdural effusion was observed in one patient, and hypoxic-ischemic change was observed in one patient.
Treating STXBP1-E involves a multidisciplinary approach, including epilepsy control and neurological rehabilitation. Currently, there is no treatment for intellectual, motor or behavioral disturbances, which exert a significant impact on the quality of life of patients. Several studies have reported antiepileptic effects of LEV through modulating the synaptic vesicle release affected by STXBP1 mutations (11, 24). LEV had a specific effect on the children with STXBP1-E in our study, and four patients presented a dramatic response to LEV with full epilepsy control. Among the various antiepileptic drugs, valproic acid and topiramate were partially effective in some patients. Serum MOG antibodies were found in patient 6, and he was given immunomodulation treatment. Although the study from Lopez et al. (25) have identified STXBP1 as an important player in cytotoxic lymphocytes function, the immunomodulation treatment did not reduce seizure status in patient 6. Further studies are needed to clarify the correlation between STXBP1-E and autoimmune mechanism.
VNS has been used in the treatment of epilepsy in clinic, but the mechanism of its application is not completely clear, its mechanism may be: 1. VNS can increase the number of neurotransmitters, such as norepinephrine, 5-hydroxytryptamine and γ-aminobutyric acid (γ-aminobutyricacid) which can produce antiepileptic effect (26, 27). VNS treatment can cause the desynchronization of cortical electrical activity, and the number of spike waves and electrodes decreased significantly during the opening phase (28). The mechanism of VNS in the treatment of epilepsy might be the asynchrony of neural circuits in the hippocampus and thalamic cortex (29). At present, it is believed that the indications for the treatment of epilepsy with VNS include drug refractory epilepsy, unable to take other surgical treatment or poorly controlled epilepsy after other types of surgery. VNS is a relatively safe surgical method, which can be tolerated by most patients, it has low incidence of complications. Because the mechanism of this operation is not clear, ASMs is still the first choice for epileptic control. Fortunately, 1 patient showed improvement in cognitive function after VNS. This improvement may be a direct effect of VNS on behavior, concentration, and affect (30) and may be related to seizure reduction, reduced ASM load in association with successful antiepileptic treatment or putative effects of VNS on mood (26). The most favorable treatment regimens should note the improvement of the prognosis on both seizures and psychomotor ability. VNS may represent the patient's option for optimal seizure and cognitive outcomes. A recent study identified miR-218 and miR-424 as regulators of STXBP1 expression. Inhibiting their interaction with STXBP1 resulted in an increase in STXBP1 protein levels (27). Guiberson et al. identified three chemical chaperones, trehalose, sorbitol, and 4-phenylbutyrate, which are able to restore STXBP1 protein levels (28) and rescue synaptic deficits. A clinical pilot trial of 4-phenylbutyrate in a small group of STXBP1 patients began in 2020, and it will be the first trial of a disease-modifying therapy in this patient population (27). Further studies are required to screen for and identify molecules that are effective for both wild-type and mutant STXBP1.
In conclusion, STXBP1 analysis should be considered for infants with seizures and severe ID, and we have shown that the major clinical features of STXBP1 mutations are frequent seizures from epilepsy, abnormal initial EEG, and intellectual disability. Frequent seizures with fever should also be considered with the STXBP1 mutation when intracranial infection is eliminated. This study demonstrates better response to LEV in STXBP1 disorder. Thus, we would suggest early consideration of the use of LEV in this population. VNS may be worthy of consideration as an option for treating STXBP1-E. Further studies are needed to determine the adequacy and ideal duration of VNS for optimal management.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by The ethics committee in Qilu Hospital of Shandong University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
XL and RH: conceived and designed the study, and critical revision of manuscript for intellectual content. ML and GW: collected the patients’ clinical data. MD and TZ: wrote the paper. All authors contributed to the article and approved the submitted version.
Acknowledgments
This work is supported by grants from the National Natural Science Foundation of China (82171352). We greatly thank the technical support from MyGenostics Co. Ltd. (Beijing) in the interpretation of gene results. We also thank the parents of the patient for signing the written consent for the publication of the cases.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2022.1010886/full#supplementary-material.
References
1. Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. (2010) 51(4):676–85. doi: 10.1111/j.1528-1167.2010.02522.x
2. Yamatogi Y, Ohtahara S. Age-dependent epileptic encephalopathy: a longitudinal study. Folia Psychiatr Neurol Jpn. (1981) 35(3):321–32. doi: 10.1111/j.1440-1819.1981.tb00233.x
3. Mastrangelo M, Leuzzi V. Genes of early-onset epileptic encephalopathies: from genotype to phenotype. Pediatr Neurol. (2012) 46(1):24–31. doi: 10.1016/j.pediatrneurol.2011.11.003
4. Carvill GL, Weckhuysen S, McMahon JM, Hartmann C, Møller RS, Hjalgrim H, et al. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology. (2014) 82(14):1245–53. doi: 10.1212/WNL.0000000000000291
5. Deprez L, Weckhuysen S, Holmgren P, Suls A, Dyck TV, Goossens D, et al. Clinical spectrum of early-onset epileptic encephalopathies associated with STXBP1 mutations. Neurology. (2010) 75(13):1159–65. doi: 10.1212/WNL.0b013e3181f4d7bf
6. Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, et al. Ilae classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the Ilae task force on nosology and definitions. Epilepsia. (2022) 63:1349–97. doi: 10.1111/epi.17239
7. Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. (2008) 40(6):782–8. doi: 10.1038/ng.150
8. Hamdan FF, Piton A, Gauthier J, Lortie A, Dubeau F, Dobrzeniecka S, et al. De novo STXBP1 mutations in mental retardation and nonsyndromic epilepsy. Ann Neurol. (2009) 65(6):748–53. doi: 10.1002/ana.21625
9. Hamdan FF, Gauthier J, Dobrzeniecka S, Lortie A, Mottron L, Vanasse M, et al. Intellectual disability without epilepsy associated with STXBP1 disruption. Eur J Hum Genet. (2011) 19(5):607–9. doi: 10.1038/ejhg.2010.183
10. Gupta A. STXBP1-related EOEE - early onset epilepsy and encephalopathy, or is it early onset epileptic encephalopathy? Epilepsy Curr. (2016) 16(5):302–4. doi: 10.5698/1535-7511-16.5.302
11. Liu S, Wang L, Cai XT, Zhou H, Yu D, Wang Z. Therapeutic benefits of ACTH and levetiracetam in STXBP1 encephalopathy with a de novo mutation: a case report and literature review. Medicine. (2018) 97(18):e0663. doi: 10.1097/MD.0000000000010663
12. Li T, Cheng M, Wang J, Hong S, Li M, Liao S, et al. De novo mutations of STXBP1 in Chinese children with early onset epileptic encephalopathy. Genes Brain Behav. (2018) 17(8):e12492. doi: 10.1111/gbb.12492
13. Mignot C, Moutard ML, Trouillard O, Gourfinkel-An I, Jacquette A, Arveiler B, et al. STXBP1-related Encephalopathy presenting as infantile spasms and generalized tremor in three patients. Epilepsia. (2011) 52(10):1820–7. doi: 10.1111/j.1528-1167.2011.03163.x
14. Milh M, Villeneuve N, Chouchane M, Kaminska A, Laroche C, Barthez MA, et al. Epileptic and nonepileptic features in patients with early onset epileptic encephalopathy and STXBP1 mutations. Epilepsia. (2011) 52(10):1828–34. doi: 10.1111/j.1528-1167.2011.03181.x
15. Barcia G, Chemaly N, Gobin S, Milh M, Bogaert PV, Barnerias C, et al. Early epileptic encephalopathies associated with STXBP1 mutations: could we better delineate the phenotype? Eur J Med Genet. (2014) 57(1):15–20. doi: 10.1016/j.ejmg.2013.10.006
16. Wolking S, May P, Mei D, Møller RS, Balestrini S, Helbig KL, et al. Clinical spectrum of STX1B-related epileptic disorders. Neurology. (2019) 92(11):e1238–49. doi: 10.1212/WNL.0000000000007089
17. Liu J, Tong L, Song S, Niu Y, Li J, Wu X, et al. Novel and de novo mutations in pediatric refractory epilepsy. Mol Brain. (2018) 11(1):48. doi: 10.1186/s13041-018-0392-5
18. Butler KM, da Silva C, Alexander JJ, Hegde M, Escayg A. Diagnostic yield from 339 epilepsy patients screened on a clinical gene panel. Pediatr Neurol. (2017) 77:61–6. doi: 10.1016/j.pediatrneurol.2017.09.003
19. Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. (2016) 18(7):696–704. doi: 10.1038/gim.2015.148
20. Weckhuysen S, Holmgren P, Hendrickx R, Jansen AC, Hasaerts D, Dielman C, et al. Reduction of seizure frequency after epilepsy surgery in a patient with STXBP1 encephalopathy and clinical description of six novel mutation carriers. Epilepsia. (2013) 54(5):e74–80. doi: 10.1111/epi.12124
21. Yamamoto T, Shimojima K, Yano T, Ueda Y, Takayama R, Ikeda H, et al. Loss-of-function mutations of STXBP1 in patients with epileptic encephalopathy. Brain Dev. (2016) 38(3):280–4. doi: 10.1016/j.braindev.2015.09.004
22. Stamberger H, Nikanorova M, Willemsen MH, Accorsi P, Angriman M, Baier H, et al. STXBP1 Encephalopathy: a neurodevelopmental disorder including epilepsy. Neurology. (2016) 86(10):954–62. doi: 10.1212/WNL.0000000000002457
23. Dilena R, Striano P, Traverso M, Viri M, Cristofori G, Tadini L, et al. Dramatic effect of levetiracetam in early-onset epileptic encephalopathy due to STXBP1 mutation. Brain Dev. (2016) 38(1):128–31. doi: 10.1016/j.braindev.2015.07.002
24. Hallböök T, Lundgren J, Stjernqvist K, Blennow G, Strömblad LG, Rosén I. Vagus nerve stimulation in 15 children with therapy resistant epilepsy; its impact on cognition, quality of life, behaviour and mood. Seizure. (2005) 14(7):504–13. doi: 10.1016/j.seizure.2005.08.007
25. Lopez J, Noori T, Minson M, Jovanoska LL, Thia K, Hildebrand MS, et al. Bi-Allelic mutations in STXBP2 reveal a complementary role for STXBP1 in cytotoxic lymphocyte killing. Front Immunol. (2018) 9:529. doi: 10.3389/fimmu.2018.00529
26. Henry TR. Therapeutic mechanisms of Vagus nerve stimulation. Neurology. (2002) 59(6 Suppl 4):S3–14. doi: 10.1212/wnl.59.6_suppl_4.s3
27. DeGiorgio CM, Schachter SC, Handforth A, Salinsky M, Thompson J, Uthman B, et al. Prospective long-term study of Vagus nerve stimulation for the treatment of refractory seizures. Epilepsia. (2000) 41(9):1195–200. doi: 10.1111/j.1528-1157.2000.tb00325.x
28. Yokoyama R, Akiyama Y, Enatsu R, Suzuki H, Suzuki Y, Kanno A, et al. The immediate effects of Vagus nerve stimulation in intractable epilepsy: an intra-operative electrocorticographic analysis. Neurol Med Chir. (2020) 60(5):244–51. doi: 10.2176/nmc.oa.2019-0221
29. Ilyas A, Toth E, Pizarro D, Riley KO, Pati S. Modulation of neural oscillations by Vagus nerve stimulation in posttraumatic multifocal epilepsy: case report. J Neurosurg. (2018):1–7. doi: 10.3171/2018.6.JNS18735
Keywords: STXBP1 mutations, early infantile epileptic encephalopathy, intellectual disability, phenotype spectrum treatment, prognosis
Citation: Dong M, Zhang T, Hu R, Li M, Wang G and Liu X (2022) Genotype and phenotype spectrum of 10 children with STXBP1 gene-related encephalopathy and epilepsy. Front. Pediatr. 10:1010886. doi: 10.3389/fped.2022.1010886
Received: 3 August 2022; Accepted: 26 October 2022;
Published: 11 November 2022.
Edited by:
Weida Tong, National Center for Toxicological Research (FDA), United StatesReviewed by:
Makoto Nabetani, Yodogawa Christian Hospital, JapanJianxiang Liao, Shenzhen Children’s Hospital, China
Wei-De Lin, China Medical University, Taiwan
© 2022 Dong, Zhang, Hu, Li, Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinjie Liu bGl1eGluamllLWVydEAxNjMuY29t
†These authors have contributed equally to this work
Specialty Section: This article was submitted to Genetics of Common and Rare Diseases, a section of the journal Frontiers in Pediatrics