 Dandan Yan1,2†Shaopei Chen3†Fengying Cai4†Jianbo Shu1,2Xiufang Zhi1,2Jie Zheng1,2Chunhua Zhang5Dong Li3*
Dandan Yan1,2†Shaopei Chen3†Fengying Cai4†Jianbo Shu1,2Xiufang Zhi1,2Jie Zheng1,2Chunhua Zhang5Dong Li3* Chunquan Cai1,2*
Chunquan Cai1,2*- 1Tianjin Pediatric Research Institute, Tianjin Children's Hospital (Tianjin University Children's Hospital), Tianjin, China
- 2Tianjin Key Laboratory of Birth Defects for Prevention and Treatment, Tianjin, China
- 3Department of Neurology, Tianjin Children's Hospital (Tianjin University Children's Hospital), Tianjin, China
- 4Department of Physiology, Tianjin Medical College, Tianjin, China
- 5Matsumoto Institute of Life Science (MILS) International, Yokohama, Japan
Background: The serine active site-containing protein 1 (SERAC1) biallelic variant usually causes MEGDEL syndrome, clinically characterized by increased excretion of 3-methylglutaconic in the urine, muscle hypotonia, sensorineural deafness, and Leigh-like lesions on brain MRI scans. In this study, we present a case from a Chinese family with disordered metabolism and dystonia owing to SERAC1 variants; the clinical phenotypes of the proband were different from those of MEGDEL syndrome but were similar to those juvenile-onset complicated hereditary spastic paraplegia. Thus, in this study, we aimed to confirm the relationship between SERAC1 variants and complicated hereditary spastic paraplegia.
Methods: MRI and laboratory tests, including gas chromatography/mass spectrometry (GC/MS), were carried out for the proband. Whole-exome sequencing was used to detect the candidate SERAC1 variants. Variants were verified using Sanger sequencing. Various software programs (PolyPhen-2, MutationTaster, PROVEAN, and SIFT) were used to predict the pathogenicity of novel variants.
Results: Brain MRI scans showed a symmetric flake abnormal signal shadow in the bilateral basal ganglia in T2-weighted image (T2WI) and fluid-attenuated inversion recovery (FLAIR) analyses. The excretion of 3-methylglutaconic acid was found to be increased in our GC/MS analysis. Whole-exome sequencing showed novel compound heterozygous variants, including a novel c.1495A>G (p.Met499Val) variant in exon 14 of SERAC1 inherited from the father and a novel c.721_722delAG (p.Leu242fs) variant in exon 8 inherited from the mother. The pathogenicity prediction results showed that these two variants were deleterious.
Conclusions: This study presented a patient with complicated hereditary spastic paraplegia caused by SERAC1 variants. These findings expand the number of known SERAC1 variants and the phenotypic spectrum associated with SERAC1 deficiency. This study may contribute to counseling and prevention of hereditary diseases through prenatal.
Introduction
Usually, an increase in 3-methylglutaconic acid (3-MGA) indicates 3-methylglutaconic aciduria (3-MGA-uria), which was first reported in 2006 by Wortmann and colleagues (1). 3-MGA-uria is a heterogeneous group of metabolic disorders, biochemically characterized by elevated 3-MGA and 3-methylglutaric acid excretion in urine. High uric 3-MGA is a major and rather common clinical characteristic of patients with suspected metabolic disorders, for which it is a phenotypic hallmark, and the key to diagnosis (2). In 2013, Wortmann and colleagues systematically classified 3-MGA-uria into five types (I–V) (3). According to this classification, type I refers to primary 3-MGA-uria, owing to the known pathological mechanism of 3-methylglutaconyl-CoA hydratase deficiency (4). Types unrelated to leucine metabolism are collectively referred to as secondary 3-MGA-uria (types II–V), among which types include type II, III, and V well-defined. However, type IV represents a confusing and ever-growing subgroup encompassing all “unclassified” symptoms (2, 5), which includes MEDGEL syndrome, characterized by 3-MGA-uria, dystonia-deafness, encephalopathy, and Leigh-like syndrome (MEGDEL, OMIM ID: 614739), known to be caused by serine active site containing 1 (SERAC1) variants.
MEGDEL syndrome is an autosomal-recessive disorder that presents clinically as 3-MGA-uria, feeding problems, liver failure, spasticity, dystonia, hearing loss, truncal hypotonia, and premature death, usually occurring between 10 and 20 years of age. Wortmann and colleagues reported several patients with 3-MGA-uria and MEGDEL syndrome in 2006 (1). To date, increasing evidence has confirmed the relationship between MEGDEL syndrome and SERAC1 variants (6–8). However, SERAC1 variants have been found to cause juvenile-onset complicated hereditary spastic paraplegia (cHSP) as well (9). Clinically, cHSP is a complicated condition, with predominant lower limb spasticity and additional neurological features; it can be inherited in autosomal dominant, autosomal recessive, or X-linked patterns (10). However, the mechanism has not yet been fully elucidated.
Here, we present a child with dyskinesia in both lower limbs, high uric 3-MGA, enhanced muscular tension and tendon reflexes, pyramidal tract injury, and abnormal MRI results, in whom we investigated SERAC1 variants by whole-exome sequencing (WES).
Materials and Methods
Study Proband
The individual included in this study was a 7-year-old female child admitted to Tianjin Children's Hospital in November 2020.
All procedures in this study were performed as per the ethical standards of the institutional review board and followed the Declaration of Helsinki. This study was approved by the Tianjin Children's Hospital Ethics Committee (Reference number 2016021). Written informed consent was obtained from the parents of the proband.
Genomic DNA Extraction
Genomic DNA was extracted from a whole peripheral blood sample using a Blood Genomic DNA Mini Kit (CWBio, Beijing, China) according to the manufacturer's instructions. The volume of DNA was approximately 100 μl, and the concentration of DNA was >10 ng/μl. The DNA was stored at −20°C until the analyses were performed.
Genomic Analysis
WES was performed using DNA samples from the proband and both of her parents. We used the Hg 19 human reference genome sequence, along with the Genome Analysis Tool Kit (GATK) software, Annovar, Thousand Human Genome Database, dbSNP, and OMIM, to obtain and notate the clinically relevant genomic variants data. To determine the variant sites, Sanger sequencing was performed on DNA samples from the proband and both of her parents. Chromas software was used to align the sequencing data using reference sequences from GenBank (NM_032861.3).
Bioinformatics Analysis
The online programs PolyPhen-2, MutationTaster, PROVEAN, and SIFT were used to predict the effects of genetic variants on the functions of SERAC1. Furthermore, FASTA-formatted amino acid sequences for SERAC1 were downloaded from the online program UniProt, and the conservation of the affected amino acids among ten species was analyzed using DNAMAN software.
Results
Clinical Analysis
The individual this case study was focused on was born at full term to a gravida 2, para 1, aborta 1, 27-year-old woman by cesarean section and was assigned female at birth. Both parents were healthy at the time of this study, were non-consanguineous, and did not have phenotypic disease characteristics. Physical examinations of the mother were found to be normal during pregnancy. The child had no postnatal asphyxia and achieved developmental milestones at the appropriate age. She had no history of convulsions, abnormal intelligence, or special genetic metabolic.
At the time of our study, the 7-year-old female child was hospitalized due to dyskinesia in the lower left limb for more than 3 years and motor disturbance in the lower right limb for longer than 1 month. Her weight was 16.8 kg (−3 SD to −2 SD), height 118 cm (−1 SD), and cranial circumference 49 cm. When a detailed medical history was taken, we found that the dyskinesia in the lower left limb fluctuated. When the dyskinesia was severe, the lower-left limb was limp when walking; she was able to stand, squat, and go up and down stairs independently, but she could not run or jump. When the dyskinesia was mild, there was no obvious limpness in the lower left limb, and she could run slowly and jump on both legs. One month before her admission, she had a severe limp in her lower right extremity and had myasthenia in both lower extremities while walking, but her upper limbs could move freely.
Clinical examination revealed that the muscle tone in the limbs was obviously increased. The myodynamia in both lower extremities was decreased, and the tendon reflexes were hyperactive. An ankle-clonus test was positive. All examination indicators of liver function that were examined were approximately normal. The levels of serum albumin (ALB), alanine aminotransferase (ALT), and aspartate transaminase (AST) were 45 g/L, 11 U/L, and 22 U/L, respectively, within the reference ranges of 38–54 g/L, 7–40 U/L, and 13–35 U/L, respectively. Laboratory tests in this period of time revealed mildly elevated levels of serum lactate (2.32 mmol/L, normal 0.5–2.2 mmol/L) and ammonia (92 μg/dl, normal 12–66 μg/dl). Cholesterol and triglycerides were normal. There were no abnormalities in electroneurogram or electromyography (EMG) results for both lower limbs. Eye fundus examinations were normal with no optic atrophy. Both pupils were equi-circular (diameter = 3 mm) and sensitive to light reflection; both eyeballs were able to move freely with no tremor. No hearing loss was noticed by the patient. Spinal and brain MRI scans of the patient revealed abnormalities in different sections. Spinal MRI scans showed a long T2 signal shadow located at the level of the 6–7th vertebrae. A T2-weighted image (T2WI) showed symmetric flake abnormal signal shadow in the bilateral basal ganglia and slightly widened ventricles (Figure 1A). Fluid-attenuated inversion recovery (FLAIR) analysis of the bilateral basal ganglia revealed a signal intensity that may indicate the involvement of white matter (Figure 1B). Organic acid analysis performed by gas chromatography/mass spectrometry (GC/MS) demonstrated increased excretion of 3-MGA in the urine (Figure 2).
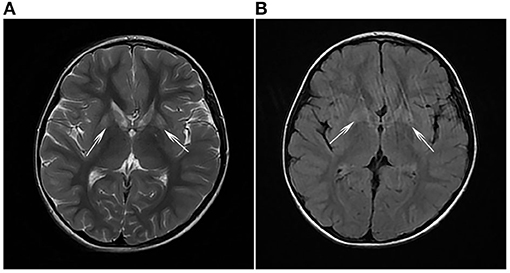
Figure 1. (A) T2-weighted imaging (T2WI) and (B) fluid-attenuated inversion recovery (FLAIR) analyses revealed symmetrical flake abnormal signal shadows in bilateral basal ganglia (white arrows).
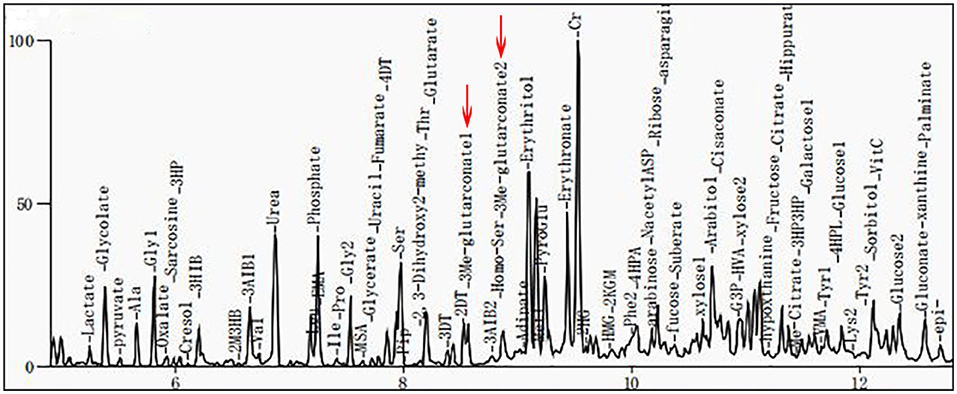
Figure 2. Gas chromatography/mass spectrometry (GC/MS) spectra for 3-methylglutaconic acid (3-MGA) and 3-methylglutaric acid. Results show that urinary excretion of 3-methylglutaric acid and 3-methylglutaric acid were increased (red arrows).
Genomic Analysis
WES revealed novel compound heterozygous variants in SERAC1 (NM_032861.3) in the proband, including a frameshift variant (c.721_722delAG resulting in Leu242fs) and a missense variant (c.1495A>G resulting in Met499Val). These variants were located on exons 8 and 14. Both variants were not found to be recorded in the Human Gene Mutation Database (HGMD), ESP6500siv2_ALL, 1000 Genomes project, and dbSNP147 database. Sanger sequencing confirmed that the variant c.721_722delAG was inherited from her healthy mother, and the variant c.1495A>G was inherited from her healthy father (Figure 3), both of whom were healthy at the time of this study.

Figure 3. Confirmation of the novel serine active site domain-containing protein (SERAC1) variants in the proband and her parents by Sanger sequencing. The proband had compound heterozygous variants in the SERAC1: c.1495A>G (p.Met499Val) in exon 14 inherited from her father and c.721_722del (p.Leu242fs) in exon 8 inherited from her mother, resulting in a premature stop codon and a truncated protein (red arrows).
Bioinformatics Analysis
The c.721_722del variant was found to be located in the upstream region of the serine-lipase domain, and the c.1495A>G variant was located within the serine-lipase domain (Figure 4). Prediction software analysis showed that the two variants were likely to be deleterious (Table 1). Amino acid sequence alignment revealed that the c.1495A>G variant was highly conserved among the ten species included in our study (Figure 5).
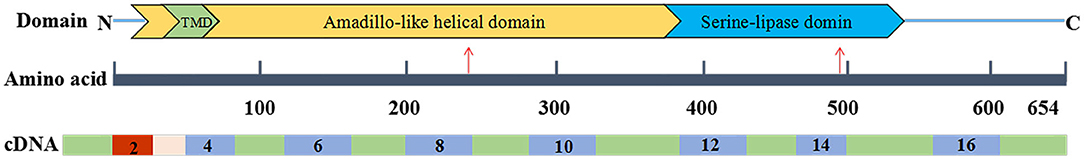
Figure 4. Serine active site domain-containing protein 1 (SERAC1) domain. The c.1495A>G variant (p.Met499Val) was located within the serine-lipase domain, and the c.721_722del variant (p.Leu242fs) was located upstream of the serine-lipase domain (indicated by the red arrows).
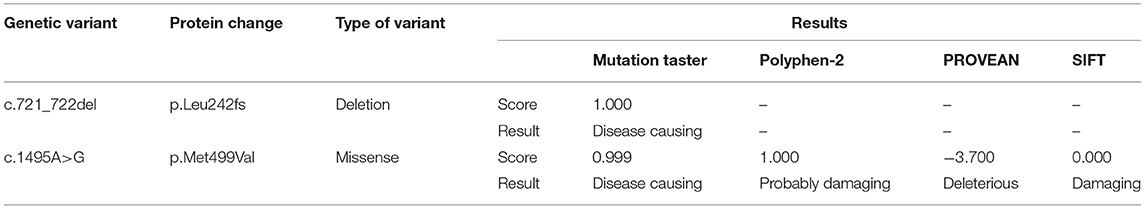
Table 1. Bioinformatics software prediction of the deleteriousness of the serine active site domain-containing protein (SERAC1) variants found in this case study.
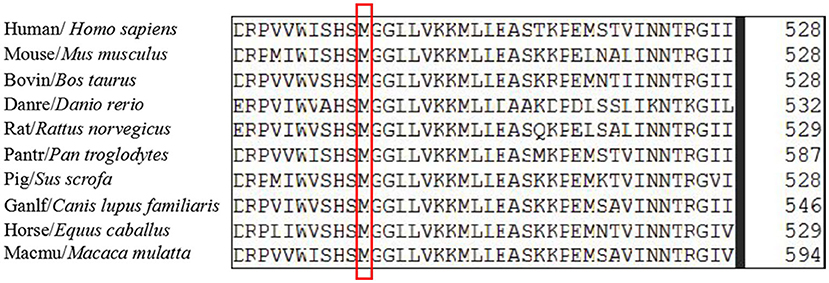
Figure 5. The amino acid sequences alignment of serine active site domain-containing protein (SERAC1) homologs among different species was highly conserved at position 499.
Discussion
Previous studies have identified a causal relationship between SERAC1 variants and MEGDEL syndrome (11–15). Approximately 70 cases of MEGDEL syndrome have been reported worldwide according to a report in 2020 (15). MEGDEL syndrome is serious, comprising an infantile-onset phenotype characterized by feeding problems, liver failure, spasticity, dystonia, hearing loss, truncal hypotonia, and premature death (16–18). The individual in this study only presented with one of the clinical characteristics of MEGDEL syndrome (3-MGA-uria), along with other symptoms not usually associated with this syndrome, including decreased myodynamia of both lower extremities, enhanced muscular tension and tendon reflexes, pyramidal tract injury, limp, and abnormal spinal and brain MRI; these symptoms were more similar to those of cHSP than MEGDEL syndrome. To the best of our knowledge, among more than 50 SERAC1 variants included in HGMD, only one has so far been reported to be related to mild cHSP in a large family (9). The author introduced the milder cHSP caused by a novel SERAC1 variant in a big family. Among the family members, none had a history of infantile feeding problems, liver failure, hearing loss, or truncal hypotonia. All were still able to walk several miles unaided aged between 10 and 20 years; the most severely affected of six siblings had signs of dystonia (9, 19). In brief, all these features were similar to those of the patient included in this study, which is only the second to report cHSP resulting from variants in SERAC1.
In our study, the proband was a 7-year-old female child with non-consanguineous, healthy Chinese parents. She had decreased myodynamia of both lower extremities, enhanced muscular tension and tendon reflexes, and pyramidal tract injury. Her brain MRI results showed bilateral basal ganglia alterations. These phenotypes were consistent with the clinical diagnosis of cHSP. A second MRI examination performed in June 2021 showed almost no change compared with the original MRI results. Clinical symptoms showed little change in 1 year since she was discharged from the hospital. The biochemical results showed 3-MGA-uria due to an elevated 3-MGA excretion in the urine. Our WES results, which covered more than 20,000 human nuclear genes and 37 human mitochondrial genes (including all 16,569 bases of mitochondrial DNA), found only SERAC1 variants, with variants in any other genes related to cHSP. Given that there was one previous report of SERAC1 variants resulting in cHSP, we concluded that the child in our study had juvenile-onset cHSP.
SERAC1, a novel cHSP gene located on chromosome 6q25.3, spans about 59 kb and contains 17 exons. The SERAC1 protein is 654 amino acids in length with a conserved serine-lipase domain (consensus lipase motif GxSxG); it is a member of the PGAP-like protein domain family (PFAM PF07819) (13, 19). SERAC1 is located within cells at the interface between the endoplasmic reticulum and the mitochondria and is involved in remodeling phosphatidylglycerol-34:1 (PG-34:1) to phosphatidylglycerol-36:1 (PG-36:1). This role is essential for both mitochondrial function and intracellular cholesterol trafficking. Previous researches have shown that SERAC1 variants can increase the PG 34:1/PG 36:1 ratio (6, 9), resulting in mitochondrial dysfunction. In our study, the c.721_722delAG variant (p.Leu242fs) led to a frameshift during translation and a premature termination codon. As a result, this variant was suspected to be deleterious according to the 2015 American College of Medical Genetics and Genomics variant classification guidance. The missense variant c.1495A>G (p.Met499Val) was also predicted to be deleterious. However, the clinical significance of this variant is not yet clear. The two variants were found to be located in different domains; the c.1495A>G variant was highly conserved among ten species. The superimposed effect of the two variants may have resulted in a diseased phenotype in the individual in this study, but no clear relationship between SERAC1 variants and phenotypes has been established (19), and the specific mechanism is also not clear. Therefore, further research is required to explore the potential mechanism.
The main finding of the study was that SERAC1 variants not only can lead to MEGDEL syndrome but also can cause cHSP. However, the specific relationship between SERAC1 variants and MEGDEL or cHSP is currently unclear. The mechanism by which SERAC1 variants may cause these diseases is also unclear. Although both of the diseases are associated with elevated 3-MGA in urine and SERAC1 variants, their clinical symptoms obviously differ. Therefore, functional validation experiments are required to explore the specific pathogenic mechanisms.
Conclusion
To date, few cases of SERAC1 variants associated with cHSP have been reported. Here, we reported a case of SERAC1 variants resulting in cHSP, which is only the second to demonstrate this phenomenon. This case therefore adds to a number of known SERAC1 variants and expands the phenotypic spectrum of SERAC1 deficiency. Thus, it will likely contribute to improved counseling and prevention of cHSP through a prenatal diagnosis of rare genetic metabolic diseases.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary files, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
XZ and JZ collected the clinical information. JS and CC designed the experiment and performed the genetic analysis. CZ performed part of the data analyses. DY and SC wrote the manuscript and analyzed the data. FC and DL edited the final manuscript. All authors agreed to be accountable for and ensure any questions relating to the accuracy, integrity of this work, and read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (grant number 81771589), the Tianjin Science and Technology Plan Program (grant number 18ZXDBSY00170), and the Public Health and Technology Project of Tianjin (grant number ZC20120).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the individual included in this study and her parents.
References
1. Wortmann S, Rodenburg RJ, Huizing M, Loupatty FJ, de Koning T, Kluijtmans LA, et al. Association of 3-methylglutaconic aciduria with sensori-neural deafness, encephalopathy, and leigh-like syndrome (MEGDEL association) in four patients with a disorder of the oxidative phosphorylation. Mol Genet Metab. (2006) 88:47–52. doi: 10.1016/j.ymgme.2006.01.013
2. Wortmann SB, Kluijtmans LAJ, Rodenburg RJ, Sass JO, Nouws J, van Kaauwen EP, et al. 3-3-Methylglutaconic aciduria-lessons from 50 genes and 977 patients. J Inherit Metab Dis. (2013) 36:913–21. doi: 10.1007/s10545-012-9579-6
3. Wortmann SB, Kluijtmans LA, Engelke UFH, Wevers RA, Morava E. The 3-methylglutaconic acidurias: what's new? J Inherit Metab Dis. (2012) 35:13–22. doi: 10.1007/s10545-010-9210-7
4. Tort F, García-Silva MT, Ferrer-Cortès X, Navarro-Sastre A, Garcia-Villoria J, Coll MJ, et al. Exome sequencing identifies a new mutation in SERAC1 in a patient with 3-methylglutaconic aciduria. Mol Genet Metab. (2013) 110:73–7. doi: 10.1016/j.ymgme.2013.04.021
5. Wortmann SB, Duran M, Anikster Y, Barth PG, Sperl W, Zschocke J, et al. Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature. J Inherit Metab Dis. (2013) 36:923–8. doi: 10.1007/s10545-012-9580-0
6. Wortmann SB, Vaz FM, Gardeitchik T, Vissers LELM, Renkema GH, Schuurs-Hoeijmakers JHM, et al. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat Genet. (2012) 44:797–802. doi: 10.1038/ng.2325
7. Sarig O, Goldsher D, Nousbeck J, Fuchs-Telem D, Cohen-Katsenelson K, Iancu TC, et al. Infantile mitochondrial hepatopathy is a cardinal feature of MEGDEL syndrome (3-Methylglutaconic aciduria type IV with sensorineural deafness, encephalopathy and leigh-Like Syndrome) caused by novel mutations inSERAC1. Am J Med Genet A. (2013) 161:2204–15. doi: 10.1002/ajmg.a.36059
8. Lumish HS, Yang Y, Xia F, Wilson A, Chung WK. The expanding MEGDEL phenotype: optic nerve atrophy, microcephaly, and myoclonic epilepsy in a child with SERAC1 mutations. JIMD Rep. (2014) 16:75–9. doi: 10.1007/8904_2014_322
9. Roeben B, Schüle R, Ruf S, Bender B, Alhaddad B, Benkert T, et al. SERAC1 deficiency causes complicated HSP: evidence from a novel splice mutation in a large family. J Med Genet. (2017) 55:39–47. doi: 10.1136/jmedgenet-2017-104622
10. Kara E, Tucci A, Manzoni C, Lynch DS, Elpidorou M, Bettencourt C, et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain. (2016) 139:1904–18. doi: 10.1093/brain/aww111
11. Dweikat IM, Abdelrazeq S, Ayesh S, Jundi T. MEGDEL syndrome in a child from palestine: report of a novel mutation in SERAC1 gene. J Child Neurol. (2015) 30:1053–6. doi: 10.1177/0883073814541474
12. Rodríguez-García ME, Martín-Hernández E, de Aragón AM, García-Silva MT, Quijada-Fraile P, Arenas J, et al. First missense mutation outside of SERAC1 lipase domain affecting intracellular cholesterol trafficking. Neurogenetics. (2016) 17:51–6. doi: 10.1007/s10048-015-0463-z
13. Radha Rama Devi A, Lingappa L. Novel mutations in SERAC1 gene in two Indian patients presenting with dystonia and intellectual disability. Eur J Med Genet. (2018) 61:100–3. doi: 10.1016/j.ejmg.2017.07.013
14. Snanoudj S, Mordel P, Dupas Q, Schanen C, Arion A, Gerard M, et al. Identification of a novel splice site mutation in the SERAC1 gene responsible for the MEGDHEL syndrome. Mol Genet Genomic Med. (2019) 7:e815. doi: 10.1002/mgg3.815
15. Alagoz M, Kherad N, Turkmen S, Bulut H, Yuksel A. A novel mutation in the SERAC1 gene correlates with the severe manifestation of the MEGDEL phenotype, as revealed by whole-exome sequencing. Exp Ther Med. (2020) 19:3505–12. doi: 10.3892/etm.2020.8658
16. Maas RR, Iwanicka Pronicka K, Kalkan Ucar S, Alhaddad B, AlSayed M, Al Owain MA, et al. Progressive deafness-dystonia due to SERAC1 mutations: a study of 67 cases. Ann Neurol. (2017) 82:1004–15. doi: 10.1002/ana.25110
17. Finsterer J, Scorza FA, Fiorini AC, Scorza CA. MEGDEL syndrome. Pediatr Neurol. (2020) 110:25–9. doi: 10.1016/j.pediatrneurol.2020.03.009
18. Giron C, Roze E, Degos B, Meneret A, Jardel C, Lannuzel A, et al. Adult-onset generalized dystonia as the main manifestation of MEGDEL syndrome. Tremor Other Hyperkinet Mov. (2018) 8:554. doi: 10.7916/D8VM5VBQ.eCollection_2018
Keywords: SERAC1, complicated hereditary spastic paraplegia, novel variant, MEGDEL syndrome, 3-methylglutaconic aciduria
Citation: Yan D, Chen S, Cai F, Shu J, Zhi X, Zheng J, Zhang C, Li D and Cai C (2022) Complicated Hereditary Spastic Paraplegia Caused by SERAC1 Variants in a Chinese Family. Front. Pediatr. 9:816265. doi: 10.3389/fped.2021.816265
Received: 16 November 2021; Accepted: 31 December 2021;
Published: 11 February 2022.
Edited by:
Andrea Lynne Gropman, Children's National Hospital, United StatesReviewed by:
Juan Dario Ortigoza-Escobar, Hospital Sant Joan de Déu Barcelona, SpainPatryk Lipiński, Children's Memorial Health Institute (IPCZD), Poland
Copyright © 2022 Yan, Chen, Cai, Shu, Zhi, Zheng, Zhang, Li and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Li, bGlkb25ndGpldHl5QDE2My5jb20=; Chunquan Cai, Y3FjbnM2QDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship