
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 20 January 2022
Sec. Pediatric Rheumatology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.805919
This article is part of the Research Topic Hereditary Periodic Fevers and Autoinflammatory Diseases View all 7 articles
 Kübra Öztürk1*
Kübra Öztürk1* Taner Coşkuner2
Taner Coşkuner2 Esra Baglan3
Esra Baglan3 Hafize Emine Sönmez4
Hafize Emine Sönmez4 Gülçin Otar Yener5
Gülçin Otar Yener5 Figen Çakmak6
Figen Çakmak6 Fatma Gül Demirkan6
Fatma Gül Demirkan6 Ayşe Tanatar6
Ayşe Tanatar6 Serife Gül Karadag7
Serife Gül Karadag7 Semanur Ozdel3
Semanur Ozdel3 Ferhat Demir2
Ferhat Demir2 Mustafa Çakan8
Mustafa Çakan8 Nuray Aktay Ayaz6
Nuray Aktay Ayaz6 Betül Sözeri2
Betül Sözeri2Familial Mediterranean fever (FMF) is the most common monogenic autoinflammatory disease manifesting phenotypic heterogeneity. It is a clinically diagnosed disease supported by MEditerranean FeVer (MEFV) gene mutation analysis. However, the phenotype-genotype correlation is not yet established clearly. We aimed to determine the clinical findings, phenotype-genotype correlation, and treatment outcomes within a large pediatric FMF cohort. The medical charts of children with FMF who were diagnosed and followed up at the eight pediatric rheumatology units were reviewed retrospectively. All patients in the cohort were analyzed for sequence variants in exon 2,3,5 and 10 of the MEFV gene. Patients without any mutations or with polymorphisms including R202Q were excluded. A total of 3,454 children were involved in the study. The mean ± standard deviation of current age, age at symptom onset, and age at diagnosis were 12.1 ± 5.2, 5.1 ± 3.8, and 7.3 ± 4.0 years, respectively. Of 3,454 patients, 88.2% had abdominal pain, 86.7% had fever, 27.7% had arthritis, 20.2% had chest pain, 23% had myalgia, and 13.1% had erysipelas-like erythema. The most common MEFV mutation patterns were homozygous (32.5%) and heterozygous (29.9%) mutations of exon 10. Homozygous M694V was present in 969 patients (28.1%). Allele frequencies of common mutations were M694V (55.3%), M680I (11.3%), V726A (7.6%), and E148Q (7.2%). Children carrying homozygous or compound heterozygous exon 10 mutations had an earlier age of disease onset (4.6 vs. 5.6 years, p = 0.000) and a higher number of attacks per year (11.1 vs. 9.6, p = 0.001). Although 8% of the patients had a family history of amyloidosis, 0.3% (n = 11) had the presence of amyloidosis. M694V homozygosity was detected in nine patients who developed amyloidosis. Colchicine resistance was present in 4.2% of our patients. In this largest pediatric cohort reviewed and presented to date, patients with exon 10 mutations, particularly the M694V homozygous mutation, have been demonstrated earlier disease onset, annual attack count, and more frequent colchicine-resistant cases. Although E148Q is considered as a polymorphism in some populations, it was identified as a disease-causing mutation in our cohort. Secondary amyloidosis is still happening in adults however, it is extremely rare among children, presumably due to increased awareness, tight control, and the availability of anti-IL1 agents in colchicine-resistant cases.
Familial Mediterranean fever (FMF) is the most common inherited monogenic autoinflammatory disease manifesting with phenotypic heterogeneity (1). The disease results from the gain-of-function mutations located on the MEFV (MEditerraneanFeVer) gene. The MEFV gene encodes the protein pyrin which acts a part in the activation of the caspase-1 molecule and the production of interleukin (IL)-1-β (2, 3). To date, more than 350 sequence variations have been identified in the MEFV gene (4). A high acute phase response with self-limiting inflammatory attacks of recurrent fever, peritonitis, pleuritic, and arthritis is typical for FMF (5). It is a clinically diagnosed disease supported by MEFV gene mutation analysis especially for atypical cases (6).
Patients carrying homozygous exon 10 mutations such as M694V and M680I are known to have a severe phenotype, but heterozygous mutations for V726A and E148Q are associated with a milder disease course. There are many studies on this subject, but the phenotype-genotype correlation is not yet clearly identified. For instance, controversy continues regarding the potential pathogenic role of the E148Q (7–9). Another challenge is that, although FMF is autosomal recessive, about a quarter of patients do not have a mutation in the second allele but have typical clinical findings (10, 11).
Colchicine is the gold standard of treatment for FMF, and in addition to suppressing inflammation, it also prevents formation of amyloidosis. However, 5–10% of FMF patients do not respond despite adequate doses of colchicine (12). Anti-IL-1 therapy, anakinra, canakinumab, and rilonacept emerged as an alternative treatment for colchicine-resistant FMF patients (13).
In this study, we aimed to determine clinical findings, phenotype-genotype correlation, and treatment outcomes in a large pediatric FMF cohort in the light of new information in the literature.
Medical files of 3,454 pediatric patients diagnosed with FMF according to the Turkish pediatric criteria (14) and followed up regularly in eight Pediatric Rheumatology Units were reviewed retrospectively. Demographic data, clinical features, and MEFV gene variant analysis were documented from medical charts. All patients in the cohort were analyzed for sequence variants in exon 2,3,5, and 10 of the MEFV gene. Patients without any mutations or with polymorphisms including R202Q were excluded from the cohort.
Alternative treatment options were also identified for resistant cases. Resistance to colchicine therapy was defined as ≥1 attack per month despite receiving the maximum tolerated dose for ≥6 months (13, 15). According to the recommendations (13), the colchicine dose was calculated from 1.2 mg/m2/day. The study has been approved by the institutional research ethics committee before was started and has been conducted by the principles outlined in the Helsinki Declaration. Written informed consents were taken from the legal guardians of the children.
Statistical analysis was performed by the SPSS software version 22 (SPSS Inc., Chicago, IL). The variables were investigated using visual (i.e., histograms and probability plots) and analytical methods (Kolmogorov-Smirnov/Shapiro-Wilk) to determine their distribution. Clinical and demographic characteristics were summarized by mean and standard deviation (SD) for continuous variables and count and percent for categorical variables. The categorical variables were compared with Chi-squared test. Bonferroni correction was used to adjust for multiple comparisons. Differences between independent samples were evaluated by the Student's T-test. A p < 0.05 was considered as statistically significant.
A total of 3,454 children (1,755 girls, 1,699 boys) were involved in the study. The mean ± SD of current age, age at symptom onset, and age at diagnosis were 12.1 ± 5.2, 5.1 ± 3.8, and 7.3 ± 4.0 years, respectively. The median (min-max) delay in diagnosis was 15 (0–230) months. Parental consanguinity was present in 30.5% of patients. One thousand nine hundred and eight patients (55.2%) had a family history of FMF. In addition, 8% of the patients had a family history of amyloidosis. It was determined that 97.3% of our patients met the Tel-Hashomer criteria (16) and 94.2% met the PRINTO/EuroFever 2019 criteria (17).
Of 3,454 patients, 88.2% had abdominal pain, 86.7% had fever, 27.7% had arthritis, 20.2% had chest pain, 23% had myalgia, 22.1% had exertional leg pain (ELP) and 13.1% had erysipelas-like erythema (ELE). The clinical findings of the patients are depicted in Table 1. When the patients were compared according to gender; arthritis and ELE were more frequent in females compared to males (n = 520 vs. n = 437, p = 0.01; n = 272 vs. n = 181, p < 0.001).
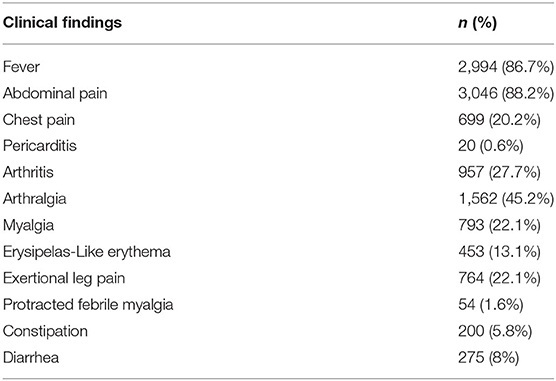
Table 1. The clinical findings of the familial Mediterranean fever patients (n = 3,454).
The most common MEFV mutation patterns were homozygous (32.5%) and heterozygous (29.9%) exon 10 mutations. Homozygous M694V was present in 969 patients (28.1%) and allele frequencies of common mutations were M694V (n = 3,373, 55.3%), M680I (n = 782, 11.3%), V726A (n = 529, 7.6%), and E148Q (n = 503, 7.2%). The most common mutations of the cohort are summarized in Table 2. Genotype-phenotype analysis was performed by comparing the most common mutations with clinical findings. Accordingly, no relationship was found between different mutations and their association of fever and abdominal pain. However, chest pain was found to be common in patients with M694V/M680I (p < 0.001), M680I/M680I (p < 0.001), M694V/R761H (p < 0.001), and M680I/E148Q (p < 0.001) mutations. It was also shown that arthritis was more common in patients with M694V/M694V (p < 0.001) and R761H/- (p < 0.001) mutations. Mutations commonly seen with arthralgia, myalgia, and ELP, are shown in Table 3.

Table 2. The most common mutations in the familial Mediterranean fever cohort.
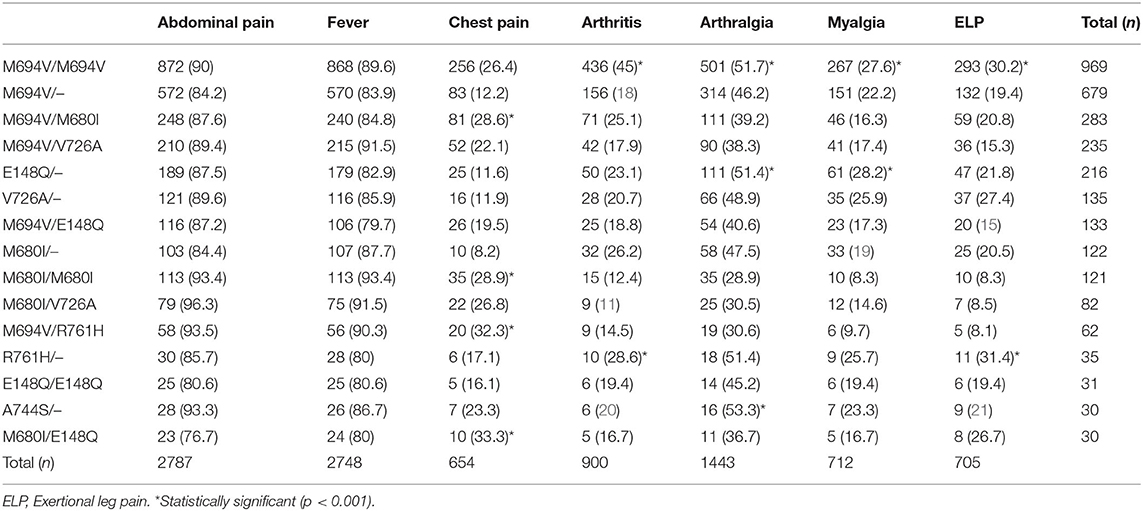
Table 3. Comparison of the most common mutations and clinical findings in familial Mediterranean fever.
When patients with and without homozygous or compound heterozygous mutations in exon 10 were compared, it was seen that children carrying homozygous or compound heterozygous mutations on exon 10 had an earlier age of disease onset (4.6 vs. 5.6 years, p < 0.001) and a higher number of attacks per year (11.1 vs. 9.6, p = 0.001). However, the delay in diagnosis was found to be longer in this group than in the other group (18 vs. 12 months, p < 0.001). In addition, it was found that fever (p < 0.001), abdominal pain (p < 0.001), chest pain (p < 0.001), arthritis (p < 0.001), and ELE (p < 0.001) were more common in children with homozygous or compound heterozygous mutations in exon 10. The comparison of patients carrying homozygous or compound heterozygous mutations in exon 10 and patients carrying other mutations are shown in Table 4. Although 8% of the patients had a family history of amyloidosis, amyloidosis was present in 0.3% (n = 11) of the cohort. All patients were diagnosed with kidney biopsy. M694V homozygosity was detected in nine patients who developed amyloidosis. The patients diagnosed with amyloidosis were treated with anti-IL-1 agents.
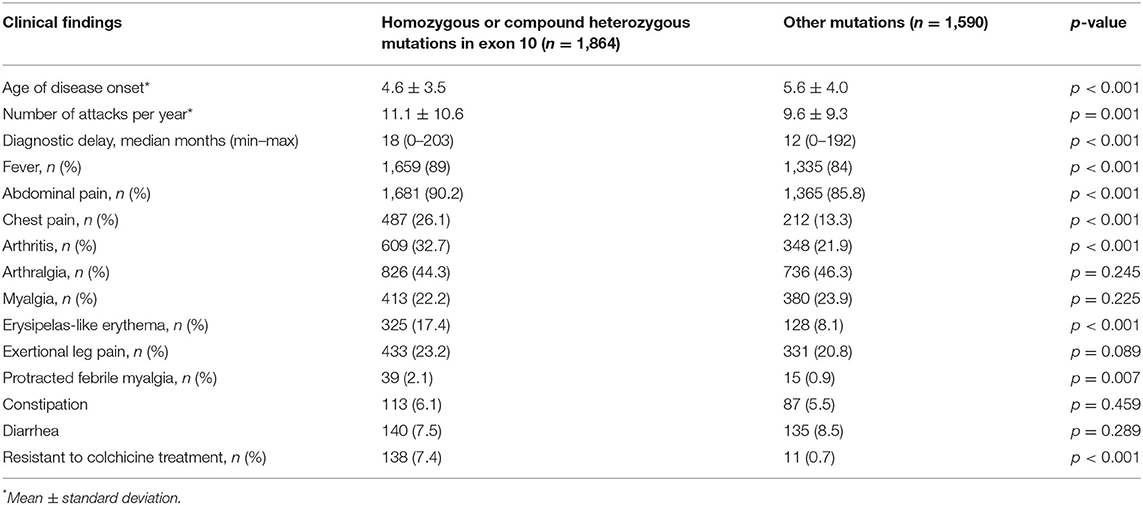
Table 4. The differences between Familial Mediterranean fever patients carrying homozygous or compound heterozygous mutations in exon 10 and patients carrying other mutations.
All patients in the cohort were on colchicine therapy, but 149 (4.3%) of them were colchicine-resistant. While exon 10 homozygous or compound heterozygous mutations were detected in 138 (91.6%) of these patients, the most common mutation was M694V homozygous (n = 121, 81.2%). Of the patients with colchicine resistance, 92 (61.7%) were girls and there was a significant difference between the two genders in terms of colchicine resistance (p = 0.006). Anti-IL 1 agents (Anakinra or Canakinumab) were added to the treatment of all these patients and remission was achieved.
This study investigated the relationship between demographics, clinical features, and genetic outcomes in a large cohort of FMF. In addition to the clinical findings, which are among the classification criteria, the relationship with the genetic results in other common findings was investigated. In addition, the most up-to-date information about the frequency of amyloidosis, which is the most important complication of the disease, is given.
It is well-known that exon 10 homozygous mutations cause more severe disease, and it has been shown that the symptoms of the disease appear earlier in patients with these mutations (8, 22). In our study, it was shown that disease symptoms started earlier in patients with exon 10 homozygous or combined heterozygous mutations.
Consistent with the literature in this study, the most common symptoms were abdominal pain (88.2%) and fever (86.7%). The incidence of fever in FMF has been reported as 82.9–93.1% (8, 20, 23, 24). Cases with afebrile FMF attacks have also been reported (18, 25). IL-1β is probably the main cause of fever, but attention should be paid to coexisting conditions such as microsomal prostaglandin E synthase-1 deficiency in afebrile FMF patients (26, 27). Therefore, additional research is needed to examine the mechanism in afebrile patients with FMF. The frequent occurrence of arthralgia, exertional leg pain, and myalgia, which are not among the classification criteria, suggests that these findings should also be paid attention to. Similar results have been found in the previous large series (8, 20, 28).
More than 300 MEFV mutations have been identified to date, and the pathogenic mutations are mostly located in exon 10 such as M694V, M680I, V726A, and M694I. Among these mutations, M694V is the most common with a frequency of 20–65% (3). In this study, the allele frequency was calculated, and the most common mutations were found to be M694V, M680I, V726A, and E148Q, respectively. In line with the literature, the M694V homozygous mutation (28.1%) was identified as the most common variant in our study. To make a genotype-phenotype correlation, patients were divided into those carrying and not carrying homozygous or compound heterozygous exon 10 mutations, which are known to be pathogenic and accepted as confirmatory mutations according to the new classification criteria (17). In this study, it was shown that abdominal pain, fever, chest pain, and arthritis were more common in patients with homozygous or compound heterozygous exon 10 mutations compared to the other group. However, ELE, which is not among the diagnostic criteria, was also found more frequently in patients with homozygous or combined heterozygous exon 10 mutations. It has been shown that this symptom, which is less common than other findings, should be paid attention to and questioned.
Whether the E148Q variant is a disease-causing mutation, or a polymorphism is still a matter of debate. It has been reported that the E148Q variant has unknown pathogenic significance and carrying this variant alone cannot support the diagnosis of FMF (9, 10). E148Q was found to be the fourth most common mutation in our cohort. In a recent study by Tirosh et al. (29), it was reported that the presence of the E148Q variant with the M694V variant was not worsening the clinical phenotype. However, M694V/E148Q mutation was found in three of the patients who did not respond to colchicine in our study. Of course, the pathogenic role of M694V cannot be denied here, but it is clear that the discussions about E148Q are not over yet. Therefore, it should be noted that the diagnosis of FMF is still based on clinical evaluation and genetic analysis can be supportive.
Amyloidosis is still the most serious complication of FMF and only 11 of our patients had amyloidosis. While M694V homozygous mutation was found in nine of the patients who developed amyloidosis, M694V heterozygous mutation was found in one and M694V/M680I mutations were detected in the other patient. This rate (0.3%) was lower than the previous reports regarding amyloidosis (19, 21, 23, 30–32). Akse-Onal et al. (33) evaluated the distribution of amyloidosis frequency by years and they demonstrated a significant decline in the frequency of secondary amyloidosis from 12.1% (1978–1990) to 2% (after 2000; p < 0.001). This may be a result of increased awareness of the disease, not delaying the diagnosis, and presence of new treatment options in cases resistant to colchicine. Proven anti-IL-1 treatments were used in these patients and clinical response was obtained in accordance with the literature (34, 35).
Although this study is limited in its retrospective design, to our knowledge it is the largest FMF series of children evaluating the phenotype-genotype correlation as a real-life data. In this study, it has been demonstrated that exon 10 mutations, particularly the M694V homozygous mutation, are important in disease severity and outcome. Although E148Q is considered as a polymorphism in some populations, it was identified as a disease-causing mutation in our cohort. Secondary amyloidosis still occurs in adults, but is extremely rare among children, probably due to increased awareness, tight control, and availability of anti-IL1 agents in colchicine-resistant cases.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by TC Saglik Bakanligi Istanbul Saglik Bilimleri Üniversitesi Ümraniye Egitim ve Araştirma Hastanesi Klinik Araştirmalar Etik Kurulu. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
KÖ, NA, and BS designed the study. KÖ, TC, EB, HS, GY, FÇ, FGD, AT, SK, SÖ, FD, and MÇ collected and analyzed data. KÖ, HS, NA, and BS wrote the manuscript. All authors have read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer EB declared a past co-authorship with several of the authors KÖ, EB, HS, GY, FÇ, SK, SO, FD, MÇ, NA, and BS and reviewer ES declared a past collaboration with several of the authors HS, SK, SO, FD, NA, and BS to the handling editor at the time of the review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med. (1967) 43:227–53. doi: 10.1016/0002-9343(67)90167-2
2. French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. (1997) 17:25–31. doi: 10.1038/ng0997-25
3. The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. (1997) 90:797–807. doi: 10.1016/S0092-8674(00)80539-5
4. Touitou, I,. The Registry of Hereditary Auto- Inflammatory Disorders Mutations. Available online at: https://infevers.umai-montpellier.fr/web/search.php?n=1 (accessed November 29, 2020).
5. Ben-Chetrit E, Levy M. Familial Mediterranean fever. Lancet. (1998) 351:659–66 doi: 10.1016/S0140-6736(97)09408-7
6. Ozen S, Bilginer Y. A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next-of-kin. Nat Rev Rheumatol. (2014) 10:135–47. doi: 10.1038/nrrheum.2013.174
7. Gangemi S, Manti S, Procopio V, Casciaro M, Di Salvo E, Cutrupi M, et al. Lack of clear and univocal genotype-phenotype correlation in familial Mediterranean fever patients: a systematic review. Clin Genet. (2018) 94:81–94. doi: 10.1111/cge.13223
8. Ayaz NA, Tanatar A, Karadag SG, Çakan M, Keskindemirci G, Sönmez HE. Comorbidities and phenotype-genotype correlation in children with familial Mediterranean fever. Rheumatol Int. (2021) 41:113–20. doi: 10.1007/s00296-020-04592-7
9. Park YH, Remmers EF, Lee W, Ombrello AK, Chung LK, Shilei Z et al. Ancient familial Mediterranean fever mutations in human pyrin and resistance to Yersinia pestis. Nat Immunol. (2020) 21:857–67. doi: 10.1038/s41590-020-0705-6
10. Shinar Y, Obici L, Aksentijevich I. Guidelines for the genetic diagnosis of hereditary recurrent fevers. Ann Rheum Dis. (2012) 71:1599–605. doi: 10.1136/annrheumdis-2011-201271
11. Procopio V, Manti S, Bianco G, Conti G, Romeo A, Maimone F, et al. Genotype-phenotype correlation in FMF patients: a “non classic” recessive autosomal or “atypical” dominant autosomal inheritance? Gene. (2018) 641:279–86. doi: 10.1016/j.gene.2017.10.068
12. Ben-Chetrit E, Ozdogan H. Non-response to colchicine in FMF definition, causes and suggested solutions. Clin Exp Rheumatol. (2008) 26:49–51
13. Ozen S, Demirkaya E, Erer B, Livneh A, Ben-Chetrit E, Giancane G, et al. EULAR recommendations for the management of familial Mediterranean fever. Ann Rheum Dis. (2016) 75:644–51. doi: 10.1136/annrheumdis-2015-208690
14. Yalçinkaya F, Ozen S, Ozçakar ZB, Aktay N, Cakar N, Düzova A, et al. A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology (Oxford). (2009) 4:395–8. doi: 10.1093/rheumatology/ken509
15. Özen S, Sag E, Ben-Chetrit E, Gattorno M, Gül A, Hashkes PJ, et al. Defining colchicine resistance/intolerance in patients with familial Mediterranean fever: a modified-Delphi consensus approach. Rheumatology. (2021) 60:3799–808. doi: 10.1093/rheumatology/keaa863
16. Pras M. Familial Mediterranean fever: from the clinical syndrome to the cloning of the pyrin gene. Scand J Rheumatol. (1998) 27:92–7. doi: 10.1080/030097498440949
17. Gattorno M, Hofer M, Federici S, Vanoni F, Bovis F, Aksentijevich I, et al. Classification criteria for autoinfammatory recurrent fevers. Ann Rheum Dis. (2019) 78:1025–32. doi: 10.1136/annrheumdis-2019-215048
18. Öztürk K, Çakan M. Protracted febrile myalgia syndrome as the first manifestation of familial Mediterranean fever in children: case-based review. Rheumatol Int. (2021) 41:213–8. doi: 10.1007/s00296-020-04696-0
19. Ozturk C, Halicioglu O, Coker I, Gulez N, Sutcuoglu S, Karaca N, et al. Association of clinical and genetical features in FMF with focus on MEFV strip assay sensitivity in 452 children from western Anatolia, Turkey. Clin Rheumatol. (2012) 31:493–501 doi: 10.1007/s10067-011-1876-1
20. Barut K, Sahin S, Adrovic A, Sinoplu AB, Yucel G, Pamuk G, et al. Familial Mediterranean fever in childhood: a single-center experience. Rheumatol Int. (2018) 38:67–74. doi: 10.1007/s00296-017-3796-0
21. Moradian MM, Sarkisian T, Ajrapetyan H, Avanesian N. Genotype-phenotype studies in a large cohort of armenian patients with familial Mediterranean fever suggest clinical disease with heterozygous MEFV mutations. J Hum Genet. (2010) 55:389–93. doi: 10.1038/jhg.2010.52
22. Özdel S, Özçakar ZB, Kunt SS, Elhan AH, Yalçinkaya F. Late-onset disease is associated with a mild phenotype in children with familial Mediterranean fever. Clin Rheumatol. (2016) 35:1837–40. doi: 10.1007/s10067-016-3196-y
23. Tunca M, Akar S, Onen F, Turkish FMF Study Group. Familial Mediterranean fever (FMF) in Turkey: results of a nationwide multicenter study. Medicine. (2005) 84:1–11. doi: 10.1097/01.md.0000152370.84628.0c
24. Öztürk K, Çakan M. The analysis of genotype-phenotype correlation in familial Mediterranean fever. Pediatr Int. (2021). doi: 10.1111/ped.15017. [Epub ahead of print].
25. Hotta Y, Kawasaki T, Kotani T, Okada H, Ikeda K, Yamane S, et al. Familial Mediterranean fever without fever. Intern Med. (2020) 59:1267–70. doi: 10.2169/internalmedicine.3175-19
26. Blomqvist A, Engblom D. Neural mechanisms of inflammation-induced fever. Neuroscientist. (2018) 24:381–99. doi: 10.1177/1073858418760481
27. Engström L, Ruud J, Eskilsson A, Larsson A, Mackerlova L, Kugelberg U, et al. Lipopolysaccharide-induced fever depends on prostaglandin E2 production specifically in brain endothelial cells. Endocrinology. (2012) 153:4849–61. doi: 10.1210/en.2012-1375
28. Demir F, Bolac GL, Merter T, Canbek S, Dogan OA, Demirkol YK, et al. The musculoskeletal system manifestations in children with familial Mediterranean fever. North Clin Istanb. (2020) 7:438–42. doi: 10.14744/nci.2020.96636
29. Tirosh I, Yacobi Y, Vivante A, Barel O, Ben-Moshe Y, Erez Granat O, et al. Clinical significance of E148Q heterozygous variant in paediatric familial Mediterranean fever. Rheumatology. (2021) 9:keab128. doi: 10.1093/rheumatology/keab128
30. Yalcinkaya F, Cakar N, Misirlioglu M, Tumer N, Akar N, Tekin M, et al. Genotype-phenotype correlation in a large group of Turkish patients with familial mediterranean fever: evidence for mutation-independent amyloidosis. Rheumatology. (2000) 39:67–72. doi: 10.1093/rheumatology/39.1.67
31. Dusunsel R, Dursun I, Gunduz Z, Poyrazoglu MH, Gurgoze MK, Dundar M. Genotype-phenotype correlation in children with familial Mediterranean fever in a Turkish population. Pediatr Int. (2008) 50:208–12. doi: 10.1111/j.1442-200X.2008.02554.x
32. Ben-Zvi I, Herskovizh C, Kukuy O, Kassel Y, Grossman C, Livneh A. Familial Mediterranean fever without MEFVmutations: a case-control study. Orphanet J Rare Dis. (2015) 10:34. doi: 10.1186/s13023-015-0252-7
33. Akse-Onal V, Sag E, Ozen S, Bakkaloglu A, Cakar N, Besbas N, et al. Decrease in the rate of secondary amyloidosis in Turkish children with FMF: are we doing better? Eur J Pediatr. (2010) 69:971–4. doi: 10.1007/s00431-010-1158-y
34. De Benedetti F, Gattorno M, Anton J, Ben-Chetrit E, Frenkel J, Hoffman HM, et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med. (2018) 378:1908–19. doi: 10.1056/NEJMoa1706314
Keywords: familial Mediterranean fever, phenotype, genotype-phenotype correlation, pediatric, amyloidosis
Citation: Öztürk K, Coşkuner T, Baglan E, Sönmez HE, Yener GO, Çakmak F, Demirkan FG, Tanatar A, Karadag SG, Ozdel S, Demir F, Çakan M, Aktay Ayaz N and Sözeri B (2022) Real-Life Data From the Largest Pediatric Familial Mediterranean Fever Cohort. Front. Pediatr. 9:805919. doi: 10.3389/fped.2021.805919
Received: 31 October 2021; Accepted: 27 December 2021;
Published: 20 January 2022.
Edited by:
Teresa Giani, University of Florence, ItalyReviewed by:
Ezgi Deniz Batu, Hacettepe University, TurkeyCopyright © 2022 Öztürk, Coşkuner, Baglan, Sönmez, Yener, Çakmak, Demirkan, Tanatar, Karadag, Ozdel, Demir, Çakan, Aktay Ayaz and Sözeri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kübra Öztürk, b3p0dXJrMTIwOUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.