
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 30 December 2021
Sec. Pediatric Immunology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.798959
 Aleksandra Szczawińska-Popłonyk1*
Aleksandra Szczawińska-Popłonyk1* Elzbieta Grześk2
Elzbieta Grześk2 Eyal Schwartzmann3Anna Materna-Kiryluk4Jadwiga Małdyk5
Eyal Schwartzmann3Anna Materna-Kiryluk4Jadwiga Małdyk5Autoimmune lymphoproliferative syndrome (ALPS) is a disorder characterized by a disruption of the lymphocyte apoptosis pathway, self-tolerance, and immune system homeostasis. Defects in genes within the first apoptosis signal (FAS)-mediated pathway cause an expansion of autoreactive double-negative T cells leading to non-malignant lymphoproliferation, autoimmune disorders, and an increased risk of lymphoma. The aim of the study was to show the diagnostic dilemmas and difficulties in the process of recognizing ALPS in the light of chronic active Epstein-Barr virus (CAEBV) infection. Clinical, immunological, flow cytometric, biomarkers, and molecular genetic approaches of a pediatric patient diagnosed with FAS-ALPS and CAEBV are presented. With the ever-expanding spectrum of molecular pathways associated with autoimmune lymphoproliferative disorders, multiple genetic defects of FAS-mediated apoptosis, primary immunodeficiencies with immune dysregulation, malignant and autoimmune disorders, and infections are included in the differential diagnosis. Further studies are needed to address the issue of the inflammatory and neoplastic role of CAEBV as a triggering and disease-modifying factor in ALPS.
Autoimmune lymphoproliferative syndrome (ALPS) also known as Canale-Smith syndrome is a disorder of the extrinsic first apoptosis signal (FAS)-mediated pathway of lymphocyte apoptosis characterized by marked genetic and clinical heterogeneity (1, 2). The hallmarks of the disease are childhood-onset chronic non-malignant, non-infectious lymphoproliferation manifesting as lymphadenopathy and/or splenomegaly, autoimmune cytopenia, and polyclonal IgG hypergammaglobulinemia. The defective lymphocyte apoptosis gives rise to non-malignant lymphoid hyperplasia with an expansion and an accumulation of autoreactive T cells which, in turn, triggers autoimmune phenomena. Clinical symptomatology in children most frequently includes multilineage cytopenia, but the involvement of solid organs such as hepatitis, uveitis, systemic lupus erythematosus, Guillain-Barre syndrome, and glomerulonephritis have also been noted. Affected individuals show a significantly increased risk for the development of malignancies, such as B-cell Hodgkin lymphoma, non-Hodgkin lymphoma, leukemia, or solid tumors of the breast, liver, and thyroid gland (3).
In ALPS, the immunopathology is associated with the FAS [tumor necrosis factor receptor superfamily 6 (TNFRSF6), CD95, APO1]-mediated process of activation-induced cell death (AICD). Upon T lymphocyte activation, an expression of Fas ligand (FASL) and its interaction with FAS receptors is triggered, leading subsequently to binding of FAS-associated protein with death domain (FADD). FADD then recruits pro-caspases 8 and 10 to engender the death-inducing signaling complex (DISC) and generates terminal caspase activation leading to cell death (1–3). This tightly regulated system of controlling lymphocyte stimulation plays a critical role in immune homeostasis thereby preventing an expansion of autoreactive T cells. Abnormal FAS-mediated activation-induced apoptosis pathway is caused by germline or somatic mutations in FAS which is the main genetic defect in the majority of ALPS patients (2, 4), FASLG, CASP8, and CASP10 genes, encoding crucial elements of AICD, Fas cell surface death receptor protein, FAS ligand protein, and caspases 8 and 10, respectively (5). While widely considered to be a primary immune deficiency presenting in early childhood, an ever-growing understanding of the immunogenetic background of ALPS has led to increasingly recognized adult-onset and ALPS-like disorders with mutations in NRAS and KRAS genes as well (5). The advances in ALPS delineation and definition have also been summarized by international investigators at the National Institutes of Health (NIH) in 2009, leading to the novel disease classification engaging genetic information and biomarkers (6). According to the revised nomenclature incorporating genetic variants, patients fulfilling the diagnostic criteria and having germline homozygous or heterozygous mutation in FAS, somatic mutations in FAS, germline mutations in FASLG, and germline mutations in CASP10 have been defined as ALPS-FAS, ALPS-sFAS, ALPS-FASLG, and ALPS-CASP10, respectively, while patients with the undetermined genetic defect have been classified as ALPS-U (6). Furthermore, a group of ALPS-related disorders has also been defined based on clinical symptomatology, diagnostic tests, and genetic variants in CASP8, NRAS, SH2D1A corresponding with CEDS (caspase 8 deficiency state), RALD (RAS-associated autoimmune leukoproliferative disease), XLP1 (X-linked lymphoproliferative syndrome), as well as DALD (Dianzani autoimmune lymphoproliferative disease) with unknown genetic background (6, 7). The proposed revised diagnostic criteria for ALPS have embraced extended clinical, immunological, histopathological features to maximize the accurateness of the diagnosis. The relative count of CD3+TCRαβ+CD4-CD8- double-negative (DN) T cells among total lymphocytes and CD3+ lymphocytes, defective lymphocytes apoptosis test, and elevated plasma sFASL levels have been underscored, and a combination of those two last-mentioned criteria proved to be the most important prognosticators for patients with FAS mutation (8). In 2019, ESID Working Definitions for Clinical Diagnosis of ALPS licensed a wider range of combinations of clinical and biomarker criteria with equal connotation and not including the ALPS genetic diagnosis (9). Nevertheless, sequential decisions on the multistep diagnostic process guided by NIH or ESID criteria leading to establishing a definitive ALPS diagnosis are still challenging and require a thorough differential diagnosis. Marked overlap of the clinical symptomatology with other disorders of immune dysregulation, namely primary immunodeficiencies, such as CVID (common variable immunodeficiency), RAG1 (recombination activation gene 1), IKBKG (inhibitor of nuclear factor kappa B (NF-κB) kinase regulatory subunit gamma) or NEMO (NF-κB essential modulator), STAT3 GOF (signal transducer and activator of transcription 3 gain-of-function), LRBA (Lipopolysaccharide (LPS) responsive beige-like anchor protein), CTLA-4 (Cytotoxic T lymphocyte antigen 4), MYO5B (myosin Vb) defects (3, 7, 10–14), primary or secondary HLH (hemophagocytic lymphohistiocytosis) as well as systemic lymphoproliferative disease in chronic active Epstein-Barr virus (EBV) infection (CAEBV) may confound the clinical diagnosis (15).
To expand the clinical and immunological phenotype of a pediatric patient showing a unique association of ALPS and CAEBV. We also aimed at showing the challenges of the clinical observations and difficulties in the diagnostic workup of ALPS with a concomitant EBV infection.
A 5-year-old boy was referred to our pulmonology, allergy, and clinical immunology unit of the pediatric university hospital with a suspicion of primary immunodeficiency due to pancytopenia, splenomegaly, and lymphadenopathy.
He was born to a young, healthy, non-consanguineous couple, at 39 weeks gestational age (WGA), by cesarean section, in a good general condition, with an Apgar score of 10 and birth weight of 4310 g. During the first year of life, his psychomotor development was normal, he did not suffer from infections. He received all live attenuated (BCG after birth and measles/mumps/rubella in the 13th month of life) and inactivated vaccines (against poliomyelitis, pertussis, tetanus, 10-valent polysaccharide conjugated pneumococcal vaccine), without adverse effects following immunization (AEFI). At the age of 2 years, he was hospitalized in a regional pediatric hospital due to pneumonia with thrombocytopenia 100 × 10/mcL, splenomegaly (length 14.4 cm), and positive EBV anti-VCA IgM. After 3 months he presented hepatosplenomegaly and petechiae accompanied by pancytopenia: the platelet count decreased to 70 × 10/mcL, anemia, leukopenia with neutropenia and eosinophilia were also observed. At that time, the diagnostic workup included: infections (serological tests against CMV, HCV, Toxocara canis, Toxoplasma gondii, Ascaris lumbricoides), autoimmune disorders (antiglobulin Coombs tests, ANA, ANCA autoantibodies), and metabolic diseases (Niemann-Pick A/B, Gaucher disease), all negative. Due to the suspicion of a malignant process, the boy was referred to the hematology and oncology unit of our university hospital, where a bone marrow aspiration was performed and features of malignant lymphoproliferation were not detected. Further monitoring of the patient's hematological parameters by a specialist in pediatric hematology/oncology for the next almost 3 years in ambulatory care revealed persistent splenomegaly and lymphadenopathy with pancytopenia (Supplementary Figure 1).
Clinical evaluation and a multidirectional diagnostic workup were performed in the patient aged 5 years old, including:
1. In-depth pediatric history concerning the onset of lymphadenopathy and the number of groups of enlarged lymph nodes as well as the onset of splenomegaly together with the physical examination to enable assessment of these crucial clinical data according to the NIH and ESID diagnostic criteria for ALPS
2. Thoracic X-ray and abdominal ultrasound examination to assess mediastinal and abdominal lymphadenopathy along with splenomegaly
3. Analysis of basic hematology and biochemistry, with the complete blood count (CBC), differential white blood count (WBC), ferritin, lactic dehydrogenase (LDH) activity, fasting triglycerides (TG), cholesterol, and fibrinogen
4. Advanced immunology including serum immunoglobulin G, M, and A levels, flow cytometric analysis of B and T cell subsets with the particular emphasis on evaluation of the relative and absolute counts of CD3+CD4-CD8- double-negative (DN) T cells, which constitute an important NIH and ESID diagnostic criterion for ALPS
5. Screening for markers of autoimmunity: indirect and direct Coombs test, antinuclear (ANA) and antineutrophil cytoplasmic (ANCA) antibodies, anti-thyroid antibodies
6. Serological tests and RT-PCR identification of viral and bacterial etiology of symptoms as an element of the differential diagnosis
7. Evaluation of biomarkers recommended both by NIH and ESID as diagnostic predictors for ALPS: cobalamin (vitamin B12), interleukin (IL)-10
8. Lymph node biopsy for immunohistological findings of ALPS
9. The molecular genetic testing of 11 genes contained in Invitae ALPS Panel
The patient presented clinical symptoms of chronic, more than 3-year-lasting lymphoproliferation, with splenomegaly and cervical, submandibular, retroauricular, supra- and subclavicular, axillar, and inguinal lymphadenopathy, accompanied by enlarged abdominal lymph nodes forming a conglomerative mass assessed in the ultrasound examination. The spleen reached maximal diameters 17.2 × 6.6 cm (length × width), corresponding with the 3rd degree of splenomegaly according to Hackett's WHO grading system for palpable examination. In peripheral blood, a variable decrease of all cell lineages, with hemoglobin ranging from 7.1 to 9.9 g/dL, RBC from 2.75 to 3.89 × 10/mcL, WBC from 1.51 to 3.02 × 10/mcL, and PLT count from 71 to 86 × 10/mcL and neutropenia from 0.1 to 0.48 × 10/mc were noted. Both serum screening tests useful in diagnosing malignancy, LDH activity and ferritin level were not increased, as well as fasting TG, cholesterol, and plasma fibrinogen levels as screening biochemical tests toward hemophagocytic lymphohistiocytosis (HLH) were normal.
Concerning for ALPS, immunological tests were performed according to the suggested NIH algorithm, and showed polyclonal IgG hypergammaglobulinemia, elevated relative and absolute TCR alpha/beta CD3+CD4-CD8- DN T cells among total and CD3+ lymphocytes along with markedly elevated serum levels of the most discriminatory biomarkers, cobalamin and IL-10. A summary of the clinical evaluation and laboratory workup of the patient, according to the NIH and ESID diagnostic criteria are displayed in Table 1. The recommended FAS-mediated apoptosis assay and soluble FASL blood level were not examined due to the lack of availability of these tests. The flow cytometric analysis of the B and T cell peripheral blood compartments is displayed in Table 2. Because of the high index of suspicion, molecular genetic testing of 11 genes included in Invitae ALPS Panel was performed and showed a germline heterozygous pathogenic mutation in FAS, consistent with the diagnosis of ALPS-FAS. The panel of genes analyzed and a description of the variant found and its pathogenicity are shown in Table 3. Of note, neither the presumed autoimmune etiology of anemia or neutropenia was documented nor ANA, ANCA, anti-tissue transglutaminase (anti-tTG), or anti-thyroid peroxidase (anti-TPO), anti-thyroglobulin (anti-TG), and anti-thyrotrophin (TSH) (anti-TR) antibodies were detected. The results of the differential diagnostics are displayed in Table 4. In search of results supporting the ALPS diagnosis, bone marrow aspirate microscopic and flow cytometric evaluation were done, which, besides the increased number of CD3+HLA-DR+ activated T cells, did not show impaired development of lymphocytic, myeloid, and erythroid lines or DN TCR alpha/beta T cells. While recommended by NIH, histopathological examination of the lymph node was performed, not indicating for ALPS or HLH, but of CAEBV NK-cell type. In the biopsied lymph node, regressive lumps showing proliferation of atypical CD3+ CD57+ CD5+ NK cells in the deep cortex and numerous reactive T helper CD4+ lymphocytes were found. The expression of the nuclear Ki67 antigen, a marker of proliferation, was present in 70–80% of cells with the Epstein Barr virus-encoded small RNAs (EBERs) seen in sparse cells (Figure 1).
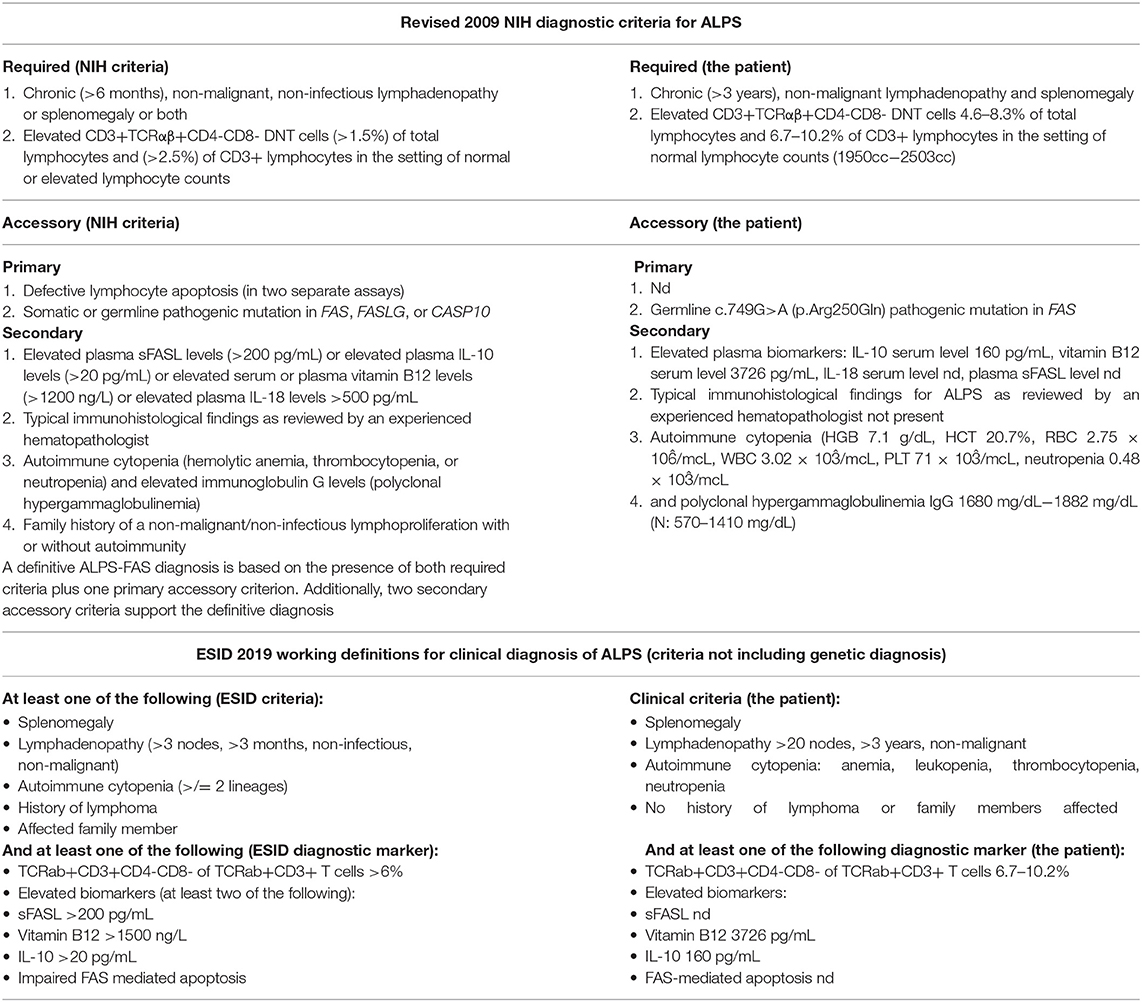
Table 1. Clinical and laboratory workup according to the revised 2009 NIH and 2019 ESID diagnostic criteria for ALPS.
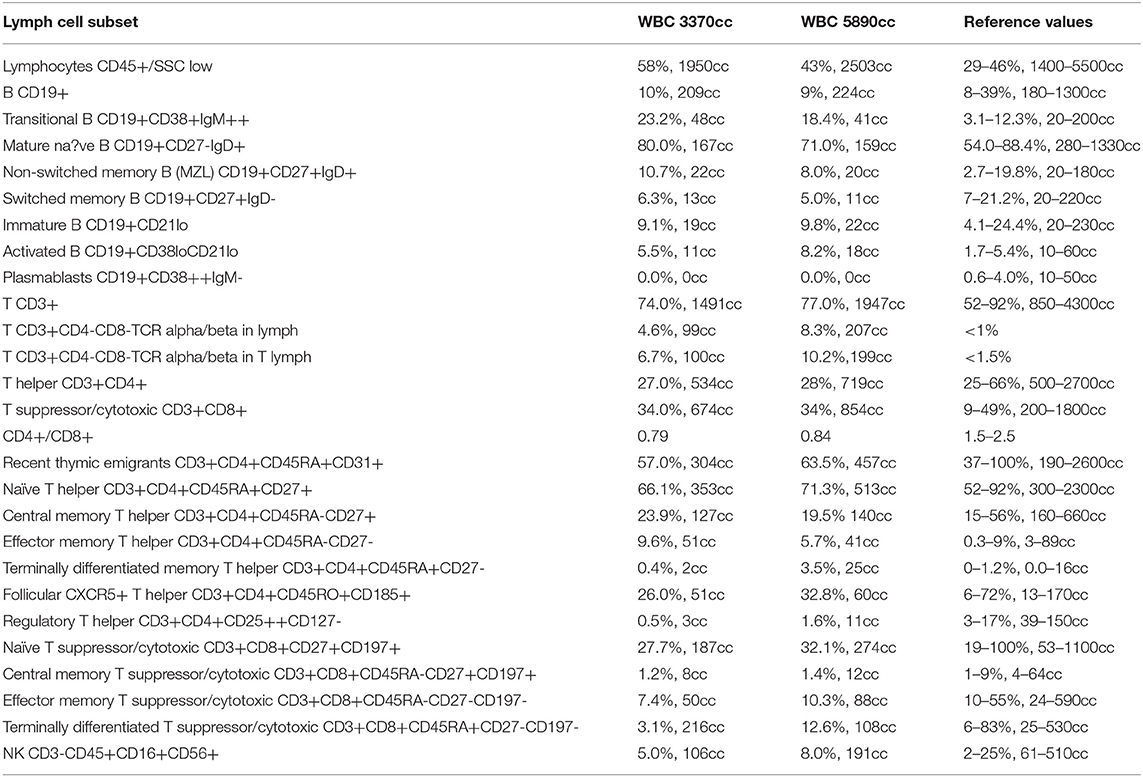
Table 2. Peripheral blood flow cytometric immunophenotyping.

Table 3. Genetic testing comprising 11 genes involved in immune dysregulation disorders.

Table 4. Differential diagnosis of ALPS and overlapping lymphoproliferative conditions.
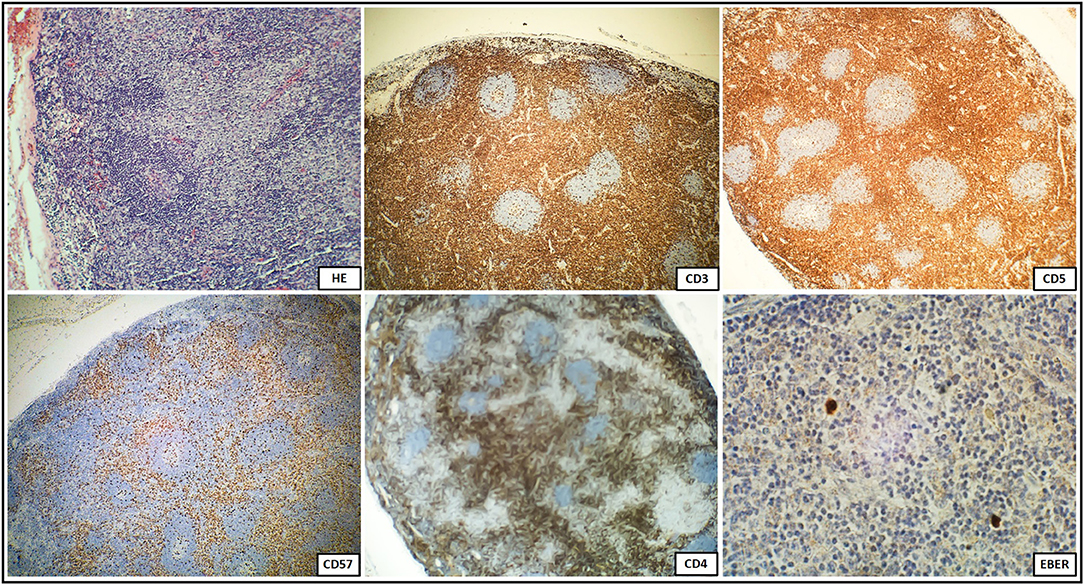
Figure 1. Immunohistochemical staining of a biopsied lymph node with regressive lumps showing diffusely positive CD3+CD57+CD5+NK cells and numerous reactive CD4 positive T cells demonstrating a high proliferation index, and sparse EBER positive cells.
The histopathological features corresponded with anti-VCA (viral capsid antigen) and anti-EBNA (Epstein-Barr virus nuclear antigen) IgG antibodies and EBV-DNA expression in peripheral blood as shown in Table 4. Considering the expanding peripheral blood EBV-DNA expression, the therapy with anti-CD20 was instituted, resulting in complete waning of EBV-DNA copies in peripheral blood. The patient is being regularly monitored by specialists in pediatric immunology and hematology/oncology considering the potential administration of an mTOR (mammalian target of rapamycin) inhibitor, rapamycin. The results of DN T cells and EBV-DNA expression monitoring over time are displayed in Table 5.
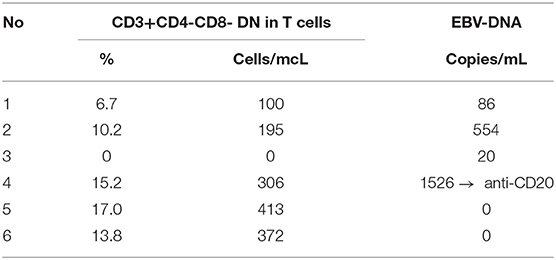
Table 5. DN T-cells and EBV-DNA monitoring at 2-month intervals.
Chronic lymphadenopathy and splenomegaly reflecting lymphoproliferation along with cytopenia in children pose a clinical and diagnostic challenge and exemplifies the need for a thorough differential diagnosis, including ALPS and clinically overlapping immunodeficiencies (16–20), malignancies (20, 21), infections (15, 20), autoimmune (22), and metabolic disorders (23, 24). Furthermore, the recommended diagnostic algorithm of ALPS developed by NIH experts (6) introduces required and accessory clinical and laboratory immunological as well as genetic criteria, engaging primarily the relative count of TCR alpha/beta CD3+CD4-CD8- T cells, levels of IL-10, IL-18, vitamin B12 as biomarkers, and examination of FAS-mediated apoptosis and soluble FASL level. The ESID diagnostic criteria for ALPS are guided by the discriminatory laboratory markers but their construction does not include genetic testing and leaves more space for the individual disease phenotype (9).
However, the analyses of the predictive value of biomarkers and double-negative T cells in patients with lymphoproliferation and autoimmune cytopenia proved their limited efficiency as correlates with germline FAS mutations and the role of lymphocyte apoptosis assays as a key diagnostic marker for ALPS (8) and sFASL assessment as the initial diagnostic step were underscored (25). A combination of autoimmune cytopenias and hypergammaglobulinemia was also shown to have a predictive value of ALPS in patients with lymphoproliferation and elevated double-negative T cells (26). In this setting, in vitro examination of Fas-mediated lymphocyte apoptosis requires a multistep, labor-intensive assay with mitogen activation, IL-2 stimulated culture, and anti-Fas IgM monoclonal antibody exposure, which is a fine, but expensive test with limited availability and unsuitability for routine clinical use (26, 27). Instead, despite the highly variable expressivity of ALPS mutations, the increasing number of achievements in immunogenetics allow for an accurate molecular genetic diagnosis in more than 70% of affected patients and thereby enable establishing a definitive clinical diagnosis of ALPS (26). Given these challenges to be able to accurately diagnose ALPS, we sought to determine, which key predictive markers are found in our patient studied. Chronic non-malignant lymphoproliferation, cytopenia, polyclonal hypergammaglobulinemia, elevated TCR alpha/beta CD3+CD4-CD8- T cells, biomarker Il-10 and cobalamin levels, though their variable sensitivity and specificity as sole or combined indicators (8, 25, 27, 28), were observable thereby fulfilling ESID diagnostic criteria (9). Confirming germline FAS pathogenic variant in our patient, the revised NIH criteria for the definitive diagnosis were also accomplished (6).
According to the NIH criteria, the differential diagnosis in pediatric patients with lymphoproliferative disorders presenting clinical phenotypes overlapping with ALPS also requires tissue biopsy and distinctive histopathology to exclude several distinguishable autoimmune, malignant, and infectious disorders. This spectrum encompasses Castleman disease which shares a common mTOR activation pathway with ALPS (29–31), Kikuchi-Fujimoto disease (32), Rosai-Dorfman disease (33), RALD (7), X-linked lymphoproliferative disease (XLP) (34), or caspase eight deficiency syndrome (CEDS) (35). In both ALPS and CAEBV, the histological features of the biopsied lymph node may be similar. The most prominent finding in an EBV infection is the marked paracortical expansion of small lymphocytes, immunoblasts, and plasma cells. Many of the paracortical lymphocytes express markers of associated with cytotoxicity, such as perforin, TIA1 (cytotoxic granule-associated RNA binding protein, associated with apoptosis), CD57, and demonstrate a high proliferative index. In an EBV infection, the presence of an EBER-positive lymphocyte subset and reactive T cells expressing the gamma/delta TCR are common histological features. In our patient, the histopathology of the biopsied lymph node was not confirmatory for ALPS, but instead, it showed features of CAEBV with EBER-positive cells. This indicates potential diagnostic pitfalls associated with an EBV infection in face of ALPS favoring hematology, flow cytometric, biomarkers, and genetic approach.
As an impaired antiviral signaling and a defective controlling of an EBV infection may play an adjunctive role in the immunopathogenesis of these syndromes, yet the link between the EBV infection and the development of lymphoproliferative disease due to malfunctioning apoptosis pathway in ALPS requires further studies (15).
CAEBV is a life-threatening EBV-associated T or NK cell lymphoproliferative disease, characterized by an increased EBV viral load in peripheral blood and expansion of EBV-infected T and/or NK cells (36–39). The clinical course of CAEBV is heterogeneous, ranging from an inactive course and stable patient condition and occasionally a self-limiting disease to an aggressive course and with the fatal outcome due to hemophagocytic lymphohistiocytosis, multiple organ failure, or progression to leukemia or lymphoma (39–41). The immunopathogenesis of CAEBV is complex showing features of both malignancy and immunodeficiency (36). The systemic form of CAEBV is clinically characterized by lymphadenopathy, splenomegaly, hepatomegaly, and fevers. Additional CAEBV complications may include pulmonary, neurological, digestive, cardiovascular, dermal, and ocular disorders (39–41), which may also constitute overlapping symptomatology of ALPS (26) making the clinical presentation of lymphoproliferation misleading. Finally, cytopenia, hypergammaglobulinemia, autoimmunity, an elevated IL-10 production as a result of Th2-skewed response essential for ALPS pathogenesis, with an increased risk of malignant lymphoma are further common features attributable to both ALPS and CAEBV. In children, T-cell CAEBV has been more often reported than in adults, with a less aggressive clinical course. It requires a differential diagnosis with either NK-cell or B-cell CAEBV as well as other EBV-related lymphoproliferative diseases to establish an appropriate therapeutic approach. Careful monitoring and observation are required for early recognition of HLH or lymphoma in affected patients (40–45).
Several inborn errors of immunity are particularly burdened with a high risk of EBV-induced immune dysregulation and malignant lymphoproliferation. Mutations in genes encoding costimulatory molecules, involved in T-cell mediated immunity, such as members of the tumor necrosis factor (TNF) receptor superfamily (TNFRSF), CD137, CD27, and its ligand CD70, as well as coronin 1A (CORO1A), cytidine triphosphate synthase 1 (CTPS1), IL2-inducible T-cell kinase (ITK), magnesium transporter 1 (MAGT1), Ras guanyl releasing protein 1 (RASGRP1), serine/threonine kinase 4 (STK4), capping protein Arp 2/3 and myosin-I linker 2 (CARMIL2), SH2 domain-containing protein 1A (SH2D1A), X-linked inhibitor of apoptosis (XIAP) are associated with extreme susceptibility to the EBV-induced tumorigenesis (38).
In conclusion, symptomatology of ALPS may parallel CAEBV thereby posing the risk of misdiagnosis and incorrect therapy. In light of this diagnostic challenge to differentiate between these mutually mimicking disorders, the role of the precise analysis of NIH and ESID criteria, including biomarker approach and genetic molecular investigations as the basis of the definitive diagnosis, needs to be highlighted. Further studies are required to address the issue of the assumptive role of an EBV infection as a triggering immunopathogenetic factor for ALPS.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
AS-P was responsible for the conception and design of the study, acquisition, interpretation of data, and drafted the manuscript. EG contributed to the design of the study, acquisition, and interpretation of data. ES helped in the acquisition and analysis of data and drafting the manuscript. JM and AM-K contributed to the diagnosing the patient and interpretation of data. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.798959/full#supplementary-material
Supplementary Figure 1. Timeline showing the clinical course of the disease and results of immunodiagnostic investigations.
1. Bride K, Teachey D. Autoimmune lymphoproliferative syndrome: more than a FAScinating disease. F1000Res. (2017) 6:1928. doi: 10.12688/f1000research.11545.1
2. Magerus A, Bercher-Brayer C, Rieux-Laucat F. The genetic landscape of the FAS pathway deficiencies. Biomed J. (2021) 44:388–99. doi: 10.1016/j.bj.2021.06.005
3. Matson DR, Yang DT. Autoimmune lymphoproliferative syndrome. Arch Pathol Lab Med. (2020) 144:245–51. doi: 10.5858/arpa.2018-0190-RS
4. Hafezi N, Zaki-Dizaji M, Nirouei M, Asadi G, Sharifinejad N, Jamee M, et al. Clinical, immunological, and genetic features in 780 patients with autoimmune lymphoproliferative syndrome (ALPS) and ALPS-like diseases-a systematic review. Pediatr Allergy Immunol. (2021) 32:1519–32. doi: 10.1111/pai.13535
5. Li P, Huang P, Yang Y, Hao M, Peng H, Li F. Updated understanding of autoimmune lymphoproliferative syndrome (ALPS). Clin Rev Allergy Immunol. (2016) 50:55–63. doi: 10.1007/s12016-015-8466-y
6. Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. (2010) 116:35–40. doi: 10.1182/blood-2010-04-280347
7. Blanchard-Rohner G, Ragotte RJ, Junker AK, Sharma M, Del Bel KL, Lu HY, et al. Idiopathic splenomegaly in childhood and the spectrum of RAS-associated lymphoproliferative disease: a case report. BMC Pediatr. (2021) 21:45. doi: 10.1186/s12887-021-02508-3
8. Molnar E, Radwan N, Kovacs G, Androkovics H, Zarafov A, Hayman A, et al. Key diagnostic markers for autoimmune lymphoproliferative syndrome with molecular genetic diagnosis. Blood. (2020) 136:1933–45. doi: 10.1182/blood.2020005486
9. Abinun MA, Beaussant Cohen S, Buckland M, Bustamante J, Cant A, Casanova JL, et al. ESID Registry—Working Definitions for Clinical Diagnosis of PID. ESID. (2019). Available online at: https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria (accessed December 15, 2021)
10. Cabral-Marques O, Schimke LF, de Oliveira EB, El Khavanky N, Ramos RN, Al-Ramadi BK, et al. Flow cytometry contributions for the diagnosis and immunopathological characterization of primary immunodeficiency diseases with immune dysregulation. Front Immunol. (2019) 10:2742. doi: 10.3389/fimmu.2019.02742
11. Gamez-Diaz L, Grimbacher B. Immune checkpoint deficiencies and autoimmune lymphoproliferative syndromes. Biomed J. (2021) 44:400–11. doi: 10.1016/j.bj.2021.04.005
12. Lopez-Nevado M, Gonzalez-Granado LI, Ruiz-Garcia R, Pieguezuelo D, Cabrera-Marantre O, Salmon N, et al. Primary immune regulatory disorders with an autoimmune lymphoproliferative syndrome-like phenotype: immunologic evaluation, early diagnosis and management. Front immunol. (2021) 12:671755. doi: 10.3389/fimmu.2021.671755
13. Rieux-Laucat F, Kanellopoulos JM, Ojcius DM. Scaling the tips of the ALPS. Biomed J. (2021) 44:383–7. doi: 10.1016/j.bj.2021.08.002
14. Casamayor-Pollo L, Lopez-Nevado L, Paz-Artal E, Anel A, Rieux-Laucat F, Allende LM. Immunologic evaluation and genetic defects of apoptosis in patients with autoimmune lymphoproliferative syndrome (ALPS). Crit Rev Clin Lab Sci. (2020) 58:253–74. doi: 10.1080/10408363.2020.1855623
15. Nomura K, Kanegane H, Otsubo K, Wakiguchi H, Noda Y, Kasahara Y, et al. Autoimmune lymphoproliferative syndrome mimicking chronic active Epstein-Barr virus infection. Int J Hematol. (2011) 93:760–4. doi: 10.1007/s12185-011-0877-9
16. Cagdas D, Halacli SO, Tan C, Lo B, Cetinkaya PG, Esenboga S, et al. A spectrum of clinical findings from ALPS to CVID: several novel LRBA defects. J Clin Immunol. (2019) 39:726–38. doi: 10.1007/s10875-019-00677-6
17. Fernandez KS, Antony R, Kumar A. Patients with ALPS-like phenotype diagnosed with immune dysregulation due to LRBA deficiency. Pediatr Blood Cancer. (2019) 66:e27558. doi: 10.1002/pbc.27558
18. Barzaghi F, Minniti F, Mauro M, De Bortoli M, Balter R, Bonetti E, et al. ALPS-like phenotype caused by ADA2 deficiency rescued by allogeneic hematopoietic stem cell transplantation. Front Immunol. (2019) 9:2767. doi: 10.3389/fimmu.2018.02767
19. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. (2015) 125:591–9. doi: 10.1182/blood-2014-09-602763
20. Natkunam J, Gratzinger D, Chadburn A, Goodland JR, Chan JKC, et al. Immunodeficiency-associated lymphoproliferative disorders: time for reappraisal? Blood. (2018) 132:1871–8. doi: 10.1182/blood-2018-04-842559
21. Price S, Shaw PA, Seitz A, Joshi G, Davis J, Niemela JE, et al. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood. (2014) 123:1989–99. doi: 10.1182/blood-2013-10-535393
22. Li Y, Li G, Liu H, Shi Y, Guan W, Zhang T, et al. Genetic heterogeneity of pediatric systemic lupus erythematosus with lymphoproliferation. Medicine (Baltimore). (2020) 99:e20232. doi: 10.1097/MD.0000000000020232
23. Jeyaratnam J, Frenkel J. Management of mevalonate kinase deficiency: a pediatric perspective. Front Immunol. (2020) 11:1150. doi: 10.3389/fimmu.2020.01150
24. Miano M, Madeo A, Cappelli E, Lanza F, Lanza T, Stroppiano M, et al. Defective FAS-mediated apoptosis iand immune dysregulation in Gaucher disease. J Allergy Clin Immunol Pract. (2020) 8:3535–42. doi: 10.1016/j.jaip.2020.06.065
25. Rensing-Ehl A, Janda A, Lorenz MR, Gladstone BP, Fuchs I, Abinun M, et al. Sequential decisions on FAS sequencing guided by biomarkers in patients with lymphoproliferation and autoimmune cytopenia. Haematologica. (2013) 98:1948–55. doi: 10.3324/haematol.2012.081901
26. Teachey DT. New advances in the diagnosis and treatment of autoimmune lymphoproliferative syndrome (ALPS). Curr Opin Pediatr. (2012) 24:1–8. doi: 10.1097/MOP.0b013e32834ea739
28. Liapis K, Tsagarakis NJ, Panitsas F, Taparkou A, Liapis I, Roubakis C, et al. Causes of double-negative T-cell lymphocytosis in children and adults. J Clin Pathol. (2019) 73:431–8. doi: 10.1136/jclinpath-2019-206255
29. Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. (2017) 129:1646–57. doi: 10.1182/blood-2016-10-746933
30. Arenas DJ, Floess K, Kobrin D, Pai RL, Srkalovic MB, Tamakloe MA, et al. Increased mTOR activation in idiopathic multicentric Castleman disease. Blood. (2020) 135:1673–84. doi: 10.1182/blood.2019002792
31. Volkl S, Rensing-Ehl A, Allgauer A, Schreiner E, Lorenz MR, Rohr J, et al. Hyperactive mTOR pathway promotes lymphoproliferation and abnormal differentiation in autoimmune lymphoproliferative syndrome. Blood. (2016) 128:227–38. doi: 10.1182/blood-2015-11-685024
32. Kim L, Tatarina-Numlan O, Yin YD, John M, Sundaram R. A case of Kikuchi-Fujimoto disease in a 7-year-old African American patient: a case report and review of the literature. Am J Case Rep. (2020) 21:e922784. doi: 10.12659/AJCR.922784
33. Allen CE, Kelly KM, Bollard CM. Pediatric lymphomas and histiocytic disorders of childhood. Pediatr Clin North Am. (2015) 62:139–65. doi: 10.1016/j.pcl.2014.09.010
34. Morra M, Howie D, Grande MS, Sayos J, Wang NY, Wu C, et al. X-linked lymphoproliferative disease: a progressive immunodeficiency. Annu Rev Immunol. (2001) 19:657–82. doi: 10.1146/annurev.immunol.19.1.657
35. Oliveira JB. The expanding spectrum of the autoimmune lymphoproliferative syndromes. Curr Opin Pediatr. (2013) 25:722–9. doi: 10.1097/MOP.0000000000000032
36. Fujiwara S, Nakamura H. Chronic active Epstein-Barr virus infection: is it immunodeficiency, malignancy, or both? Cancers. (2020) 12:3202. doi: 10.3390/cancers12113202
37. Kimura H, Cohen JI. Chronic active Epstein-Barr virus disease. Front Immunol. (2017) 8:1867. doi: 10.3389/fimmu.2017.01867
38. Lino CNR, Ghosh S. Epstein-Barr virus in inborn immunodeficiency—more than infection. Cancers. (2021) 13:4752. doi: 10.3390/cancers13194752
39. Montes-Mojarro IA, Kim WY, Fend F, Quintanilla-Martinez L. Epstein-Barr virus positive T and NK-cell lymphoproliferations: morphological features and differential diagnosis. Semin Diagn Pathol. (2020) 37:32–46. doi: 10.1053/j.semdp.2019.12.004
40. Sawada A, Inoue M, Kawa K. How we treat chronic active Epstein-Barr virus infection. Int J Hematol. (2017) 105:406–18. doi: 10.1007/s12185-017-2192-6
41. Okano M. Recent concise viewpoints of chronic active Epstein-Barr virus infection. Curr Pediatr Rev. (2015) 11:5–9. doi: 10.2174/1573396311666150501002809
42. Kim WY, Montes-Mojarro, Fend F, Quintanilla-Martinez L. Epstein-Barr virus-associated T and NK cell lymphoproliferative diseases. Front Pediatr. (2019) 15:71. doi: 10.3389/fped.2019.00071
43. Arai A. Chronic active Epstein-Barr virus infection: a bi-faceted disease with inflammatory and neoplastic elements. Immunol Med. (2018) 41:162–9. doi: 10.1080/25785826.2018.1556030
44. Arai A. Advances in the study of chronic active Epstein-Barr virus infection: clinical features under the 2016 WHO classification and mechanisms of development. Front Pediatr. (2019) 7:14. doi: 10.3389/fped.2019.00014
Keywords: autoimmune lymphoproliferative syndrome, chronic active Epstein-Barr virus infection, lymphoproliferation, children, immunodeficiency–primary
Citation: Szczawińska-Popłonyk A, Grześk E, Schwartzmann E, Materna-Kiryluk A and Małdyk J (2021) Case Report: Autoimmune Lymphoproliferative Syndrome vs. Chronic Active Epstein-Barr Virus Infection in Children: A Diagnostic Challenge. Front. Pediatr. 9:798959. doi: 10.3389/fped.2021.798959
Received: 20 October 2021; Accepted: 07 December 2021;
Published: 30 December 2021.
Edited by:
Elizabeth Secord, Wayne State University, United StatesReviewed by:
Eric McGrath, Wayne State University, United StatesCopyright © 2021 Szczawińska-Popłonyk, Grześk, Schwartzmann, Materna-Kiryluk and Małdyk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aleksandra Szczawińska-Popłonyk, YXN6Y3phd2luc2thQHVtcC5lZHUucGw=; a2xpbmlrYXBhZEBza3AudW1wLmVkdS5wbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.