 Edna N. Matjuda
Edna N. Matjuda Godwill Azeh Engwa
Godwill Azeh Engwa Constance R. Sewani-Rusike1
Constance R. Sewani-Rusike1 Benedicta N. Nkeh-Chungag
Benedicta N. Nkeh-Chungag- 1Department of Human Biology, Faculty of Health Sciences, Walter Sisulu University PBX1, Mthatha, South Africa
- 2Department of Biological and Environmental Sciences, Faculty of Natural Sciences, Walter Sisulu University PBX1, Mthatha, South Africa
The balance between dilatory and constrictive factors is important as it keeps blood vessels in a homeostatic state. However, altered physiological processes as a result of obesity, hypertension, oxidative stress, and other cardiovascular risk factors may lead to vascular damage, causing an imbalance of vasoactive factors. Over time, the sustained imbalance of these vasoactive factors may lead to vascular dysfunction, which can be assessed by non-invasive methods, such as flow-mediated dilation, pulse wave velocity, flow-mediated slowing, retinal vessel analysis, peripheral vascular reactivity, and carotid intima-media thickness assessment. Although there is increasing prevalence of cardiovascular risk factors (obesity and hypertension) in children in sub-Saharan Africa, little is known about how this may affect vascular function. This review focuses on vasoactive factors implicated in vascular (dys)function, highlighting the determinants and consequences of vascular dysfunction. It further describes the non-invasive methods used for vascular (dys)function assessments and, last, describes the impact of cardiovascular risk factors on vascular dysfunction in children of African ancestry.
Introduction
Cardiovascular diseases (CVDs) are a major cause of morbidity and mortality worldwide. In 2019, an estimated 17.9 million people died from CVDs, representing 32% of all global deaths (1). In sub-Saharan Africa (SSA), the disability-adjusted life years (DALYs) due to CVDs increased from 90.6 million in 1990 to 151.3 million in 2017 (2). CVDs in SSA are of major concern as they pose a challenge on an already strained health system (3). Although the prevalence of CVDs is higher in adults, the risk factors for CVDs, including obesity and hypertension, are increasing among children in SSA (4).
There is evidence that risk factors for CVDs, including obesity, hypertension, and hyperglycemia, begin early in life and may be associated with vascular dysfunction (5, 6). Also, it is reported that vascular dysfunction, an early initiator of CVD, begins in childhood and may lead to CVDs and associated complications in adulthood (7). Vascular dysfunction, which includes endothelial dysfunction, microvascular dysfunction, and stiffening of large arteries, results when the homeostatic function of relaxation and contraction of blood vessels is affected (8).
The endothelium is a major layer of blood vessels, and it is regulated by the release of potent vasodilators, such as nitric oxide (NO), prostaglandin I2, hydrogen sulfide, endothelium-derived hyperpolarizing factor as well as contracting factors, such as endothelin, prostacyclin, and thromboxane (9). A balance between vasodilatory and vasoconstrictive factors is important as it keeps blood vessels in a homeostatic state (10). Changes in the release of vasoactive factors, such as decreased bioavailability of NO, may lead to endothelial dysfunction. Endothelial dysfunction, along with other risk factors, such as aging, inflammation, obesity, increased salt intake, smoking, and alcohol consumption, could contribute to the development of arterial stiffness (8). Sustained arterial stiffening may predispose the intima layer of the affected blood vessels and may contribute to the development of atherosclerosis (11). Obesity is one of the major risk factors for the development of vascular dysfunction and CVDs (12). It increases the concentration of circulating free fatty acids and alters anti-inflammatory and pro-inflammatory cytokines that are released from visceral fat. These functional and structural changes affect the microvasculature, leading to vascular dysfunction and possibly CVDs (13). Also, oxidative stress is reported to affect vascular function as free radicals are shown to affect the availability of NO, leading to endothelial dysfunction (14). Free radicals can equally affect enzymes implicated in the regulation of the extracellular matrix of the blood vessel wall, leading to arterial stiffness (15).
It is reported that vascular dysfunction is central to the origin of CVDs (16). Moreover, there is increasing prevalence of cardiovascular risk factors, such as obesity and hypertension, in African children. A study conducted among adolescents in Fetakgomo Municipality, Limpopo Province of South Africa found that the prevalence of obesity was 35% (17). Another study carried out in the Eastern Cape Province of South Africa documented a 19.8% prevalence of obesity in children aged 6–9 years old (18). A recent meta-analysis study reports an increased prevalence of hypertension among African children aged 2–19 years (19). Although the prevalence of cardiovascular risk factors, such as obesity and hypertension, in children in SSA is on the rise, little is known about how these factors may affect vascular function. Hence, this review intends to give an overview of bioactive factors in the regulation of vascular function. It also discusses the causes of vascular dysfunction along with the methods used for assessment. It further highlights the key determinants of vascular dysfunction and the associated consequences and provides evidence of vascular dysfunction in children and adolescents of African ancestry.
Vascular Function
The vascular system is made up of blood vessels, such as arteries, veins, and capillaries (20). Blood vessels are organized in hierarchal levels with complex and different configurations designed to ensure efficient exchange of nutrients and waste in and between tissues throughout the body. Large arteries with diameters above 6 mm transport oxygenated blood from the heart to smaller arteries ranging between 1 and 6 mm in diameter, then to the arteriolar network with diameters of 100–1,000 μm, and last into capillary beds of 10–15 μm in diameter (21). The arterial wall is an organized structure composed of matrix proteins (collagen fibers oriented in various directions and elastic lamellae), vascular smooth muscle cells (VSMCs), and other matrix components, such as glycosaminoglycans and endothelial cells in the inner layer (22). The cross-sectional layers of the arterial wall are shown in Figure 1. The endothelium is a thin monolayer of simple squamous cells lining the inner surface of the whole cardiovascular system (23, 24). It was once thought to be just an inert layer wrapping all endovascular surfaces. However, over the last four decades, research on the endothelium has become enormous, and its results have led to an understanding of its complex functions (25). It forms a biocompatible barrier between the circulating blood and all the underlying tissues (26). The endothelium plays an essential role in vascular function through several mechanisms, including the synthesis and release of substances that act in an autocrine and/or paracrine form. It controls all cardiovascular activities by releasing several vasoactive agents (27). The endothelium-derived dilating and contracting factors are balanced under physiological conditions so that vascular homeostasis is maintained in favor of vasodilation. Dilatory factors include NO, hydrogen sulfide, prostacyclin (prostaglandin I2), and endothelium-derived hyperpolarizing factor, whereas contracting factors include endothelin, thromboxane, and asymmetric dimethyl arginine (ADMA) (27). Microcirculation is the terminal vascular network of the systemic circulation comprising microvessels with a diameter of <20 μm. These microvessels consist of arterioles, postcapillary venules, and capillaries. Microcirculation is regarded as the last destination of the cardiovascular system and is ultimately accountable for the transfer of oxygen from the red blood cells in the capillaries to the parenchymal cells where oxygen is delivered to fulfill the energy requirements of the tissue cells (28). The capillaries consist of a single layer of endothelial cells (29). The distensibility and elasticity of arteries keep a relatively fixed blood pressure regardless of the pulsating nature of blood flow by each heartbeat. Arteries expand as a result of receiving blood expelled from the heart during systolic contraction and eject it to the periphery during diastole to supply the peripheral circulation with a steady flow of blood during systole and diastole cycles (30). Some of the major vasoactive factors implicated in the vascular function of blood vessels are discussed below.
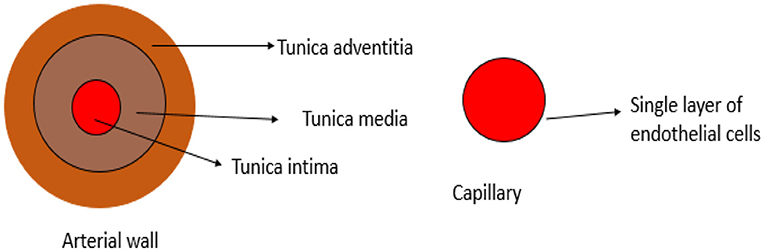
Figure 1. Cross-section of layers in the arterial wall.
Vasoactive Factors
Thromboxane and Prostacyclin
Prostacyclin and thromboxane are vasoactive factors implicated in the regulation of blood vessel relaxation and contraction. Although prostacyclin also known as prostaglandin I2 is a vasodilator, thromboxane is a vasoconstrictor. Prostacyclin and thromboxane are produced from the endothelium of blood vessels (31). Prostaglandin H2 is produced following the enzymatic degradation of phospholipid membrane in the endothelium by phospholipase enzyme to release arachidonic acid (AA) (32). AA is then metabolized by cyclooxygenase-1 (COX-1) or cyclooxygenase-2 (COX-2) to produce prostaglandin H2, which is a precursor for thromboxane synthase, prostaglandin synthase, and prostacyclin synthase. Under physiological conditions, COX-1 is expressed in most tissues, whereas COX-2 is expressed by inflammatory cells, such as macrophages, and it leads to the production of thromboxane, which plays a role in platelet aggregation, vasoconstriction, and proliferation (33, 34).
The platelets remain in their inactive state as they circulate through the blood vessels of the intact endothelium. This inactivated state is sustained by continuous secretion of prostacyclin as well as the absence of pro-inflammatory factors that can activate COX-2. Once there is a break in the endothelium, platelets become activated by thromboxane, which initiates the aggregation of platelets into a growing thrombus through the activation of G-protein. This activates phospholipase C to hydrolyze phosphatidylinositol phosphate to diacylglycerol and inositol triphosphate as well as increases calcium ion accumulation to directly heighten VSMC contraction (35, 36). Following the release of prostacyclin, it acts on VSMCs through prostacyclin receptors linked to the activation of membrane-bound adenylate cyclase, which converts adenosine triphosphate (ATP) to cyclic adenosine monophosphate (cAMP). Accumulation of cAMP as a result of prostacyclin leads to vasodilation and inhibition of platelets aggregation (37).
NO
The most important vasoactive factor is NO as it plays a crucial role in the vasculature stimulating VSMC relaxation and, thus, controlling vascular resistance and blood pressure. It also eliminates free radicals and prevents build-up of plaque (38). As blood flows through the vessels, endothelial cells detect shear stress exerted by the pressure of blood and respond by releasing acetylcholine to act on its endothelial receptor, which triggers excessive release of calcium ions from the endogenous storage sites (39). The released calcium ions attach to calmodulin protein in the cytoplasm of the cell to form a calcium-calmodulin complex, which activates the endothelial nitric oxide synthase (eNOS). The active form of this enzyme catalyzes the conversion of L-arginine and oxygen to citrulline and NO molecule. There are three isoforms of mammalian NOS, namely, neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS) of which the latter is the main source of NO in the endothelium (40). To apply its dilatory effects, NO diffuses to adjacent VSMCs, where it binds to the heme moiety of cytosolic guanylate cyclase (GC). This active enzyme, in turn, activates guanosine triphosphate to its active form, that is, cyclic guanosine monophosphate (cGMP) (41). It is the cGMP that facilitates the dephosphorylation of the myosin light chain, and this process induces the dissociation of myosin and actin filament resulting in VSMC relaxation (41).
Endothelin
Endothelin is a vasoconstrictor that exists in three isoforms, namely, endothelin-1 (ET-1), endothelin-2 (ET-2), and endothelin-3 (ET-3). Three different genes encode endothelin, which gives rise to three different precursors of pre-pro-endothelin (42). Pre-pro-endothelin-1 is the first product encoded by the ET-1 gene (43). This precursor is transformed into pro-ET-1 by removal of a short sequence by a signal peptidase. The pro-ET-1 is then converted to big ET-1 through the activity of furin, a maturing enzyme. Mature ET-1 is obtained by proteolytic cleavage of big ET-1 by endothelin converting enzyme into a small active 21 residue ET-1 (44). Once ET-1 is formed and released from the endothelium, it acts through two types of receptors, namely, endothelin A (ETA) and endothelin B (ETB) receptors. Currently, ET-1 and ET-2 are known to have the strongest affinity for both receptors, whereas ET-3 binds only on ETB (42). ET-1 binds to these receptors on the VSMCs. ETA and ETB are coupled to G-protein to form inositol triphosphate (IP3). This IP3 accumulates in the sarcoplasmic reticulum, leading to the secretion of calcium ions, which, in turn, results in the contraction of VSMCs (45). It is documented that ET-1 is the most potent vasoconstrictor. Moreover, ET-1 is suggested to decrease endothelium-dependent vasodilation. This may be due to the combined effect of ET-1–induced vasoconstriction and, to a lesser extent, ET-1–mediated inhibition of NO production, which together affect the balance between dilatory and constrictive factors in favor of the latter (46). The normal vascular endothelium is considered as a gatekeeper of cardiovascular health, whereas harmful stimuli, such as oxidative stress and inflammation, alter the normal endothelium function, leading to the development of vascular dysfunction (45).
ADMA
Dimethyl arginines are formed during the methylation of L-arginine residues within specific proteins, a process that is catalyzed by arginine methyltransferase. ADMA is released following a cleavage of methylated proteins during physiological protein turnover (47). Under physiological conditions, ADMA is excreted in urine. However, under pathological conditions, its elimination may be blocked due to hypertension, hypercholesterolemia, diabetes mellitus, and chronic kidney failure (48). As such, there is increased ADMA concentrations in the circulation, which, in turn, competes with L-arginine for the NOS binding site, thereby inhibiting the production of NO (49). Furthermore, both ADMA and L-arginine are transported into the cell through a cationic amino acid transporter; therefore, they compete with each other at the transporter to enter the cell where they are being catalyzed by NOS. As such, the production of NO depends on the balance between L-arginine and ADMA because they both compete for NOS and cell transport (50).
Endothelium-Derived Hyperpolarizing Factor
Endothelial-derived hyperpolarizing factor (EDHF) plays an important role in controlling the vascular tone in the microvasculature (51). Whereas blood vessel relaxation is easily impaired as a result of decrease in NO, EDHF activity of relaxation is enhanced to preserve the homeostasis of blood vessels. This activity of EDHF induces the formation of a disulfide bond between two cysteine 42 residues of each of the adjacent chains in protein kinase G (PKG) (51). This leads to the opening of large Ca2+-dependent channels, resulting in hyperpolarizing and vasodilation (52). The vasoactive factors and their functions are summarized in Table 1.

Table 1. Vasoactive factors and their functions.
Vascular Dysfunction
Vascular dysfunction comprises dysfunction of the endothelium (endothelial dysfunction), microvascular dysfunction, and large artery dysfunction due to arterial stiffness (9). Endothelial dysfunction is characterized by an imbalance between constrictive factors and dilatory factors, increased concentration of reactive oxygen species (ROS), pro-inflammatory factors, and decreased NO bioavailability (41). The production of NO depends on its precursor, L-arginine, which is synthesized in healthy humans from l-citrulline by endogenous synthesis. This means that reduced levels of L-arginine and l-citrulline contribute to NO insufficiency. Also, free radicals, such as superoxide (O2·), may react with NO to form peroxynitrite (ONOO−) radicals, thereby reducing NO levels (40). A variety of ROS-producing systems, such as NADPH oxidase, xanthine oxidase, eNOS, and enzymes of the mitochondrial respiratory chain, are found within the vascular wall. Moderate levels of ROS have important signaling roles under physiological conditions. Excessive and persistent production of ROS, however, when exceeding the present antioxidant defense enzymes, leads to oxidative stress and decreased NO production (53). It is documented that NO production can also be decreased by ADMA, which competes with the substrate of eNOS, L-arginine, thus inhibiting NO production (54). Endothelial NO is one of the major dilatory factors, and its insufficiency contributes to elevated vascular constriction (55). A study documents that deterioration of NO results in increased levels of ET-1, which is a major vasoconstrictor, leading to a decrease in endothelial dilatory capacity (56). A study conducted in South Africa finds that ADMA is inversely correlated with carotid intima-media thickness (57). Another study documents that black men and women have higher central systolic blood pressure, higher plasma ADMA, and lower urinary nitrate than their white counterparts. This suggests potential increased chances for vascular damage and large arterial stiffness in people of African ancestry in the future as a result of endothelial dysfunction (58).
Microvascular dysfunction is a condition characterized by impaired endothelium-dependent dilation of isolated arterioles. It is documented that microvascular dysfunction precedes and predicts the development of conduit artery atherosclerosis and its determinants (59). Abnormal microvascular function may occur as a result of structural alterations in small arteries due to inward eutrophic remodeling without overall growth of the cell, leading to decreased vasodilator reserves and changes in distensibility of arterioles (60). A study reports that remodeling (damage) of the small artery plays a crucial role in the increase of vascular resistance. This damage in the small arteries, characterized by the thickening of the carotid intima, may be considered as the first manifestation of target organ damage before it occurs in the large arteries (61). More direct impairment of microvascular function occurs as a result of persistent ischemia, manifesting as reduced maximal flow on computerized tomography without the presence of conduit stenosis (59). Microvascular dysfunction is linked to several conditions, such as smoking, obesity, hypertension, and diabetes (62). As such, microcirculatory alteration noted in the renal and retinal systems are extensively studied to investigate the predictive role of glycemic variations early in diabetes (60).
The loss of arterial elasticity, also called arterial stiffness, describes the mechanical property of artery resistance to deformation (63). The stability, compliance, and resilience of the vascular wall are dependent on the activity of two major scaffolding proteins, namely, elastin and collagen (64). The content of these proteins is usually made stable by a dynamic but slow process of their synthesis and degradation. Dysregulation of this balance between their production and degradation commonly stimulated by inflammatory molecules leads to the overproduction of collagen at abnormal levels, which diminishes the normal elastin content. This affects the elasticity and resistance of the arteries, contributing to vascular stiffness (63). With every heartbeat, a pulse wave generated by the arteries travels through the vascular bed until it reaches peripheral resistance or any bifurcation point, producing a new reflected wave back to the heart (65, 66). The reflected wave velocity and the stage of the cardiac cycle in which it happens (during systole or diastole) depends on the peripheral vascular resistance, elasticity primarily of the large arteries, and central blood pressure (66). In healthy individuals, arteries are compliant, and therefore, the reflected wave is slow and returns to the heart during the diastole cycle. However, in individuals with arterial stiffness, the reflected wave reaches the heart early during systole cycle. As a result, this increases the systolic blood pressure with a subsequent increase in cardiac workload to overcome the augmented systolic blood pressure (30, 66).
Assessment of Vascular Function
Vascular function constitutes endothelial function and functioning of the microcirculation and macrocirculation. Endothelial function is mostly assessed by flow mediated dilation (FMD) techniques, which require occlusion. Retinal imaging is mostly used to assess the functioning microcirculation, and the macrocirculation function can be assessed by measuring the pulse wave velocity (PWV) as discussed below (67, 68).
FMD
Vascular function can be assessed by numerous methods, including invasive and non-invasive techniques (69). Among the non-invasive techniques, FMD is one of the validated methods for the assessment of vascular function. The method involves ultrasound imaging in stages, at baseline (before occlusion) and during reactive hyperemia (5 min after occlusion of the artery) (70). Endothelial cells lining the artery sense an increase in blood flow and react by generating NO, which causes the diameter of an artery to increase to accommodate the increased demand (71). Such a response is known as FMD. In this technique, a blood pressure cuff is inflated in the forearm to temporarily occlude the brachial artery for a few minutes. This is followed by deflation of the pressure cuff to restore blood flow to the forearm and using an ultrasound to measure the increased diameter of the brachial artery caused by the sudden increase in blood flow (69, 71).
Impaired FMD is linked with conditions predisposing CVDs and is known to be the earliest step in developing subclinical target organ damage (72). In addition, assessment of FMD can classify individuals at low, moderate, or high risk for future clinical events (69). FMD provides valuable prognostic data and is considered the gold standard for assessing endothelial dysfunction (72). However, it has a few limitations that are worth consideration. First, the absence of standardization and differences in placement or positioning of the cuff/probe makes comparison of results difficult. Results may be operator-dependent as the technique requires expertise in the placement of the probe on the arm to identify the pulsating artery. Moreover, changes in structure of the arteries and impaired dilation may be a limiting factor during an FMD test (69).
Flow-Mediated Slowing
Flow-mediated slowing (FMS) can be described as the minimum PWV during reactive hyperemia representing endothelial function (73). A vicorder device is used to perform this test, in which the participant is requested to rest in a supine position for at least 20 min before oscillometric cuffs are wrapped around the upper arm and wrist. FMS assessment commences with baseline measurement of PWV for 4 min followed by 5 min of blood pressure occlusion and finally, 4 min of a postocclusion in which the pressure cuff is released (74). At the end of the test, minimum PWV (m/s) during hyperemia is recorded. PWV is calculated by dividing the arterial length by transit time between the upper arm and wrist. Particularly, the length is measured directly using the device to bypass body contours between the two midpoints of the two cuffs (73). FMS is easier to perform than FMD and is less operator-dependent. As a result, some studies report that FMS seems to be a promising and feasible method for endothelial function assessments (75, 76).
Peripheral Vascular Reactivity Assessment
Endothelial dysfunction can also be measured non-invasively by using a quantitative magnetic resonance imaging (MRI) technique that measures the peripheral vascular reactivity in the superficial femoral artery and vein (77). In this method, participants are required to lie in a supine position on the imager table whereby an eight-channel extremity transmitter–receiver coil is used for assessment. Following 2 min of a baseline period, a sphygmomanometer cuff is applied to the upper right thigh proximal to the targeted vessels, and then it is quickly inflated with a pneumatic pump for a 5-min occlusion period to the target pressure of 220 mmHg. This is followed by a post-occlusion period of 5 min (78). Vessel-wall imaging is done at baseline, occlusion, and post-occlusion to quantify superficial femoral artery luminal flow-mediated dilation, venous oxygen saturation, and arterial blood flow velocity (78). A study reports that methods of quantitative MRI can detect endothelial dysfunction in the presence of overt cardiovascular disease. However, so far, the use of this instrument is limited to research to identify biomarkers for disease progression (77).
Retinal Microvasculature Assessment
The retina is rich with blood vessels and, thus, shares similar anatomical features and physiological properties with blood vessels in the body. As such, visualization of the retinal vasculature allows direct non-invasive assessment of the microvasculature in relation to health and diseases of the vascular system (79). Retinal microvascular changes, such as arteriolar narrowing, arteriovenous nicking, focal arteriolar narrowing, and changes in static retinal vascular caliber, are reported to be early signs of hypertensive retinopathy and atherosclerosis (80). Analysis of the retinal image is of importance as it assists in early diagnosis of diabetic and hypertensive retinopathy and CVDs (80). A portable and easily movable fundus camera is a tool used to assess changes in the retina, retinal vasculature, and macula of the eye using a low-power intricate microscope in a cost-efficient manner (80, 81). Furthermore, dynamic measurements, such as maximal retina vessel dilation, can also be used to further assess retinal microcirculation (77). The digital interior imaging of the eye through a fundus camera has sensors that convert a light signal into an electric signal, and the result is stored in the form of a pixel (80). Static digital photographs of the retina are taken from both eyes, and computer-based software is used to measure the diameter of arterioles and venules (79). The diameter of the central retinal artery (CRAE) and central renal vein equivalent (CRVE) are calculated. Also, other structural changes, including arteriovenous nicking (AVN) and focal arteriolar narrowing (FAN), are assessed (79). To perform this test, the patient is required to sit in front of the camera with the patient's forehead against the bar. The trainer focuses and aligns the fundus camera on the pupil, and the shutter button is released, thus, firing a flash that forms a photograph of the interior surface of the eye (82). A fundus camera can assist health workers to control vascular diseases affecting both the central and peripheral retina, and it can help patients understand the extent of their cardiovascular health condition (82). An observational study among 40- to 60-year-old adults in the United Kingdom shows that retinal fundus imaging alone may predict multiple cardiovascular risk factors, such as age, gender, and systolic blood pressure (83).
Pulse Wave Velocity
At the end of the ventricular ejection phase, a pressure wave generated from the heart propagates along the arterial tree (69). PWV is defined as a measure of the speed of the arterial pressure wave traveling from the heart along the aorta to the large arteries. It is calculated as the distance of the pressure wave between the arteries/transit time. PWV is the most widely used measure for arterial stiffness (84). There are different types of PWV measurements with carotid-femoral PWV (cfPWV) and brachial-ankle (baPWV) being the most commonly used methods in clinical settings and research (84). PWV can be assessed non-invasively using a vicorder device, and it is referred to as the “gold standard” measurement for arterial stiffness because it is a reliable, inexpensive, and simple non-invasive tool to identify or detect CVD risk in its earliest stages (84). A study finds that the 10th, 50th, and 90th percentiles of cfPWV assessed using a vicorder were, respectively, 4.8, 5.57, and 6.6 m/s as reference values for adolescents aged 18 years old (85).
Apart from the vicorder, the sphygmocor cardiovascular management suite (CvMS) has been used in the field as a non-invasive method for PWV and aortic pressure waveform assessment. This device depends on applanation tonometry to detect radial, carotid, and femoral blood pressure waveforms (86). Studies utilize this device to measure PWV (87, 88). A study in South Africa has equally utilized this device to assess PWV in pre-eclamptic women (89). Although this device is reported to be effective in assessing PWV, its major disadvantage is difficulty in obtaining the peripheral waveform. Also, the device is technically difficult to use, and it is operator-dependent in identifying the peripheral signal (86, 90).
Recently, a new device called the Sphygmocor XCEL, which makes use of the volumetric displacement (cuff-based) technique to obtain pulse information, was developed (86). It is used to measure arterial stiffness and wave reflection strength (91). A study in South Africa reports that further studies are required to investigate the accuracy of PWV measurements by Sphygmocor XCEL (89). This device is preferable over the Sphygmocor CvMS because it is not operator-dependent (92). Furthermore, there is no need for an electrocardiogram to be aligned sequentially to acquire signals when assessing cfPWV using Sphygmor XCEL. However, Sphygmocor CvMS is more suitable in research than Sphygmocor XCEL in measuring high-frequency components of the waveform (86).
Another device for the measurement of PWV and central systolic blood pressure is the Complior. This device measures the PWV between the carotid and radial arteries using piezoelectric clips (sensors) placed around the neck and the wrist (93). This device is suggested to be accurate and reliable in the non-invasive assessment of PWV and is utilized in studies in South Africa to measure PWV (94–97). However, one of the limitations of this device is that it is operator-dependent in accurately positioning the sensors in the various arteries to measure the waveform. This may lead to discrepancies between the distance measured between the sensors and the actual path length traveled by the pulse wave. Furthermore, the sensors are highly sensitive to motion and may be affected by the positioning of the arteries (94, 98).
Carotid Intima-Media Thickness Assessment
Carotid intima-media thickness (cIMT) is the thickness of the intimal and medial layers of the carotid arterial wall, and it can be measured non-invasively using a scanner imaging device (99). The test is performed using a sonography with a high frequency of 7.5 MHz linear array transducer. The patient is required to lie in a supine position, and the common carotid artery is visualized at 1 cm proximal to its bifurcation (100). The cIMT is described as the length between the leading edge of the luminal echo to the leading edge of the adventitia of the media (101). It is documented that cIMT >0.9 mm is denoted as a marker of asymptomatic organ damage. Moreover, intima media thickness (IMT) is accepted as an earliest marker of atherosclerotic vascular disease, and screening of IMT can help physicians to classify patients with cardiovascular risk into lower or higher risk categories (102). A study conducted in South Africa reveals that cIMT is elevated in females with HIV aged 35–45 years old in Elandsdoorn, Limpopo (103). A study among a group of individuals from Johannesburg and Limpopo, South Africa, finds that increased cIMT is associated with cholesterol (104). In the North West Province of South Africa, lower cIMT was associated with physical activity among female teachers (105).
Determinants of Endothelial Dysfunction
It is known that risk factors for CVDs begin early in life (5, 6). A study finds that carotid bifurcation regions depicted widespread intimal lipid accumulation among newborn cadavers (106). Moreover, bifurcation anatomy affects blood flow, which causes endothelial injury (106). This indicates that endothelial dysfunction begins early in life. A study confirms that offspring have a distinct endothelial regulatory micro RNA profile at birth, which is associated with altered endothelial cell behavior during the first 3 months of life (107). It is documented that maternal total cholesterol (TC) concentrations increase in human pregnancy to meet the demands of the growing fetus (108). In some pregnancies, however, TC increases excessively mainly due to low-density lipoprotein cholesterol levels, a condition called maternal supraphysiological hypercholesterolemia in pregnancy, which is associated with endothelial dysfunction of the umbilical vein and early development of atherosclerosis in the fetal aorta (109). Furthermore, endothelial dysfunction is associated with various obstetrical syndromes, such as fetal growth restriction (FGR) (110). Evidence shows that FGR fetuses alter their cardiovascular function in utero to adjust to persisting suboptimal conditions, mainly chronic hypoxia (111). Changes in cardiovascular function secondary to utero-placental deficiency may result in permanent alterations in vascular structure (112). Fetal growth restriction leads to low birth weight. Children born with low birth weight experience catch up growth during their first years of life, thus, accumulating greater visceral adiposity, exposing them to an adverse metabolic outcome (110). All these findings suggest that maternal cardiovascular risk factors may affect the vascular function of the fetus and neonates.
Obesity, a multifactorial condition characterized by excess adipose tissue is a major determinant of vascular dysfunction and constitutes a serious worldwide health problem (113). The adipose tissue, where fat is stored in the body, is a type of connective tissue comprising lipid-filled cells (adipocytes) surrounded by a matrix of collagen fibers, blood vessels, immune cells, and fibroblasts. It consists of several cells with adipocytes being the most abundant. Other cells include stromal vascular fraction (SVF), endothelial cells, macrophages, stem cells, fibroblasts, and lymphocytes (114). Persistent accumulation of fat in the adipose tissue leads to adipocyte hypertrophy and hyperplasia (113). Adipose tissue hypertrophy (adipocyte cell size increases) and hyperplasia (increase in adipocyte number) occurs in childhood (115). The expansion of adipocytes leads to an increased release of free fatty acids and necrotic cell death due to hypoxia and inflammation (116). During physiological conditions, inflammation is regarded as a protective mechanism. However, obesity is accompanied by some degree of inflammation called low-grade inflammation (117) whereby the adipose tissue secretes high levels of pro-inflammatory adipocytokines, including tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), resistin, and leptin, due to cell death by necrosis following hypoxia (113). This causes an infiltration of neutrophils, eosinophils, monocytes, and lymphocytes to clean up the dead cells (117). The resident macrophages in the adipose tissue release chemo-attractants for macrophages, which results in the persistent nature of chronic inflammation. This, in turn, promotes the inhibition of the production of adiponectin, an anti-inflammatory adipokine (117). Adiponectin is regarded as a beneficial adipokine in relation to metabolism with plasma concentration indirectly associated with trunk obesity, type 2 diabetes risk, and insulin resistance, whereas leptin positively correlates with waist circumference and is associated with the onset of insulin resistance (95, 118). TNF-α is known to trigger insulin resistance in obese individuals. IL-6 is known to be implicated in the pathways of insulin sensitivity, lipoprotein lipase downregulation and triglyceride synthesis (119). Persistent release of these pro-inflammatory markers, such as TNF-α and IL-6 results in decreased production of adiponectin (120). Decreased plasma levels of adiponectin promote the synthesis of arginase, a metalloprotease that catalyzes the conversion of L-arginine to L-orthinine and urea. The increased concentrations of arginase compete with eNOS for the substrate L-arginine. Increased arginase activity uncouples eNOS for the synthesis of NO, thereby leading to reduced production of NO (121). A decreased bioavailability of NO leads to endothelial dysfunction. Defect in the synthesis of NO can also be caused by high concentrations of ADMA in the plasma (122). ADMA is an endogenous competitive inhibitor of L-arginine for all three isoforms of NOS. High levels of ADMA block the synthesis of NO and limit the cellular uptake of L-arginine, thereby further disrupting the production of NO. In this manner, ADMA further affects the endothelial function (123).
Secreted inflammatory molecules, including pro-inflammatory cytokines, contribute to the generation of ROS (124). Since adipose tissue are known to secrete pro-inflammatory cytokines, they may promote the generation of ROS. As such, adipose tissue is regarded as an independent factor for the development of oxidative stress (125). ROS are highly reactive radicals derived from molecular oxygen, such as O2−, hydrogen peroxide (H2O2), hydroxyl radical (OH·), and ONOO−, that impair structural conformation of protein, DNA, and RNA in the cell, resulting in cellular dysfunction and cell death (126). Under physiological conditions, ROS contribute to cellular growth regulation, differentiation, and apoptosis (114). Furthermore, they are produced from endothelial cells by several enzymes, including NADPH oxidases, xanthine oxidoreductase (XOR), and mitochondrial enzymes, among many other sources (127). It is known that H2O2 has vasodilatory effects, whereas O2− is a vasoconstrictor and leads to endothelial dysfunction (128). High levels of O2− may react with NO to form an unstable free radical called ONOO− (129). Furthermore, ROS can be produced from the uncoupling of eNOS (129). eNOS uncoupling may occur due to limited availability of the substrate L-arginine (128). As a result, eNOS may produce O2− instead of NO, leading to more defect in the synthesis of NO and, hence, endothelial dysfunction (129). Also, small, dense, low-density lipoprotein (LDL) in the lumen is deposited into the subendothelial space where it becomes oxidized by ROS to become ox-LDL, which activates endothelial cells, causing expressed receptors for white blood cells on the surface (130). It is reported that ox-LDL induces the expression of ICAM-1 and VCAM-1, increasing the adhesive properties of the endothelium. The production of NO by endothelial cells is inhibited by ox-LDL. It is documented that ox-LDL leads to oxidative stress, producing high amounts of O2−, which inactivates NO to form ONOO− (131). The decrease in NO as a result of ox-LDL leads to endothelial dysfunction.
Although hypertension is generally known be a consequence of endothelial dysfunction (132, 133), recent data suggest that hypertension may be a cause of endothelial dysfunction. There are reports that hypertension-induced endothelial dysfunction may be a result of hypertension-induced oxidative and inflammation (134). Hypertension-associated oxidative stress regulated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and mitochondria show reductions in endothelium-dependent vasodilation to acetylcholine in carotid arteries of mice exposed to increasing intraluminal pressure as a result of increase in NADPH oxidase activity and vascular O2− production (135). Also, obese hypertensive rats with perivascular inflammation show impaired endothelial function (136). Further, the activation of the innate immunity complement pathway, which regulates inflammation, is negatively associated with vascular endothelial function in hypertensives (137). All these studies support the notion that hypertension may be the cause of endothelial dysfunction.
Consequences of Vascular Dysfunction
Endothelial dysfunction is a crucial risk factor for the development of high blood pressure as it not only impairs the control of the vascular tonus, but also alters structural function, such as the tunica intima of blood vessels (138). LDL as a result of hyperlipidemia, which is associated with obesity, may be deposited into the intima of blood vessels where they may be oxidized by ROS. This oxidized LDL (ox-LDL) activates the endothelial cells to induce monocyte recruitment into the endothelial wall (139). The recruited monocytes differentiate into macrophages that take up the ox-LDL via scavenger factors, resulting in intracellular lipid accumulation and subsequently the formation of foam cells (139, 140). Foam cells produce growth factors that cause the synthesis of collagen and VSMC to migrate into the intima, which begins to proliferate and secrete extracellular matrix, resulting in thickening of the arterial intima. Thickening of the intima can lead to severe CVDs, such as stroke, ischemic disease, and congestive heart failure later in life (139, 141).
It is known that early endothelial dysfunction decreases vascular relaxation and causes the infiltration of inflammatory cells, leading to mild inflammation in blood vessels (142). eNOS is formed in high concentrations in endothelial cells, specifically in the renal medulla, where it maintains medullary blood flow in response to renal vasoconstrictors, such as angiotensin II. Impaired activity of eNOS may be due to endothelial damage or extrinsic free radical activity altering NO activity (143). ROS may influence the effects of dilatory and constrictive factors, thus leading to elevated vascular resistance and acute kidney injury (144).
Sustained damage by hyperglycemia or other factors, such as hypertension in the microvessels of the retina results in diabetic retinopathy (145). Diabetic retinopathy is the main cause of blindness in high- and middle-income countries (109). Hyperglycemia increases hypoxia induced factor 1 (HIF-1) and insulin-like growth factor-1 (IGF-1). The overexpression of HIF-1 and IGF-1 and other factors activate Müller cells to transform into chronic inflammatory cells. Moreover, this induces overexpression and buildup of vascular endothelial growth factor (VEGF) causing fibroblast growth, thereby initiating fibrosis (146). VEGF is documented to stimulate angiogenesis and neovascularization, which are involved in the pathogenesis of proliferative retinopathy (145). Microvascular dysfunction can also result from arterial stiffness (147). Arterial stiffness is associated with normal and accelerated aging (147). The consequence of arterial stiffness includes augmented systolic blood pressure, which is characterized by pulse pressure (30, 148). Greater pulsatile pressure increases the pulsatile flow to penetrate deeper into the periphery and damage the microvasculature specifically in the brain and kidney (30).
Vascular Dysfunction in Children and Adolescents of African Ancestry
The increasing prevalence of cardiovascular risk factors, such as hypertension, in SSA children has implications on their vascular health (4). However, very few studies assess the vascular function of children of African ancestry. A study in Kwa-Zulu Natal Province of South Africa shows that age and resting heart rate were positively associated with arterial stiffness among children aged 10–13 years old (149). Age could play an important role when assessing arterial stiffness (150). However, for a deeper understanding, it should be examined in conjunction with growth and maturation, given that body height at the transition from childhood to adolescence is documented to affect arterial stiffness. An association between resting heart rate and arterial stiffness in children is still lacking in the literature (149). A study conducted in the Eastern Cape Province, South Africa, among 6- to 9-year-old children finds that blood pressure parameters, such as mean arterial and diastolic blood pressure, increased with increasing PWV (151). This suggests that hypertension may result in vascular impairment in children. Another study conducted in Potchefstroom, North West Province of South Africa, in 6- to 8-year-old boys shows that oxidative stress is positively associated with cfPWV and carotid dorsalis pedis PWV in boys exposed to maternal cardiovascular risk compared with the non-maternal risk group (152). This suggests that oxidative stress may be an early mediator of vascular changes in children exposed to maternal cardiovascular risk. PWV significantly correlates with ADMA and systolic blood pressure (SBP) in a study conducted among 13- to 16-year-old children in the Eastern Cape Province of South Africa, suggesting that ADMA might be considered as a major risk factor of vascular dysfunction in adolescents (153). The PWV increased with cumulative time on ART in children living with HIV among primary school children in Cape Town, Western Cape Province of South Africa (154). In Mozambique, a study conducted among children with perinality-acquired HIV finds that PWV is higher in participants with increased visceral fat, elevated lipids, and insulin resistance (155). A study carried out in Egypt among 74 obese children aged 6–18 years finds a significant positive correlation between cIMT and BMI. cIMT equally shows a significant positive correlation with triglycerides and TC (156). Another study conducted in Egypt among 5- to 14-year-old children finds that cIMT is higher in obese children as compared with non-obese children. Further, obese children with elevated LDL and TC show increased risk for endothelial dysfunction and early signs of atherosclerosis (157). Thus, higher cIMT in obese children denotes increased risk for early vascular dysfunction. Exposure to risk factors of CVDs, such as hypertension and hyperlipidemia in obese children may induce alterations in the arteries, thus contributing to impaired endothelial function (156, 157). Higher PWV (carotid-radial, carotid-femoral, and carotid-dorsalis), diastolic blood pressure, and cIMT are reported in black boys than in white boys aged 6–8 years old in Potchefstroom, North West Province of South Africa. Moreover, black boys had increased levels of pentosidine, which is a biomarker for microvascular complications. However, arterial stiffness was not associated with pentosidine in both groups of boys, suggesting that vascular aging begins early in black population (158). Risk factors associated with vascular dysfunction in African children are summarized in Table 2.
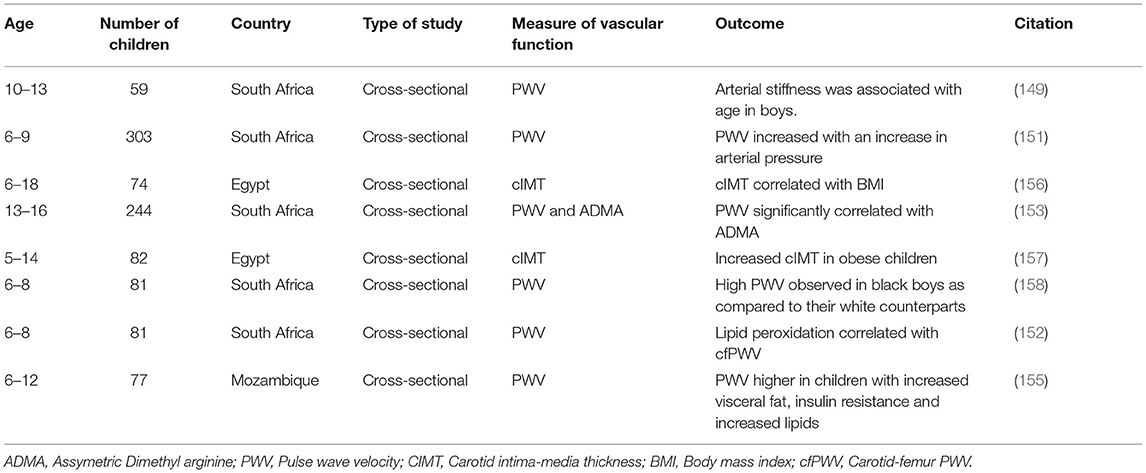
Table 2. Vascular dysfunction and their associated risk factors in African children.
Conclusion
Cardiovascular risk factors, such as obesity and hypertension, are known to be major contributors to the development of vascular dysfunction in children of African ancestry. Parameters of vascular function, such as PWV, cIMT, and ADMA, are used to assess cardiovascular risk in children of African ancestry. The presence of vascular dysfunction triggered by obesity, hypertension, oxidative stress, and inflammation in these children suggest a future risk of CVDs, such as stroke and heart attack in adulthood. However, only a few studies assess vascular changes in children of African ancestry, and such assessments are mostly limited to arterial stiffness and cIMT, as non-invasive methods along with a few vasoactive factors. Moreover, limited or no studies utilize FMD, FMS, retinal vascular assessments, and other recent PWV techniques to assess vascular function. These findings are, therefore, not sufficient to clearly describe the state of vascular dysfunction in children of African ancestry, and thus, additional studies with more robust methods for the assessment of vascular function, such as FMD and retinal microvasculature measurements are needed to provide sufficient information on vascular function in children of African ancestry and its implication.
Author Contributions
GE and BN-C were involved in the development and conceptualization of this review. EM developed the literature with the assistance from GE, BN-C, and CS-R. All authors mentioned contributed to the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. World Health Organisation. Cardiovascular diseases (CVDs). (2021). Available obline at: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed August 05, 2021).
2. Roth GA, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. (2018) 10:1736–88. doi: 10.1016/S0140-6736(18)32203-7
3. Gouda HN, Charlson F, Sorsdahl K, Ahmadzada S, Ferrari AJ, Erskine H, et al. Burden of non-communicable diseases in sub-Saharan Africa, 1990–2017: results from the Global Burden of Disease Study 2017. Lancet Glob Health. (2019) 7:e1375–87. doi: 10.1016/S2214-109X(19)30374-2
4. Chikafu H, Chimbari MJ. Cardiovascular disease healthcare utilization in sub-Saharan Africa: a scoping review. Int J Environ Res Public Health. (2019) 16:419. doi: 10.3390/ijerph16030419
5. Tam WH, Ma RC, Ozaki R, Li AM, Chan MH, Yuen LY, et al. In utero exposure to maternal hyperglycemia increases childhood cardiometabolic risk in offspring. Diabetes Care. (2017) 40:679–86. doi: 10.2337/dc16-2397
6. Yang IV, Zhang W, Davidson EJ, Fingerlin TE, Kechris K, Dabelea D. Epigenetic marks of in utero exposure to gestational diabetes and childhood adiposity outcomes: the EPOCH study. Diabetic Med. (2018) 35:612–20. doi: 10.1111/dme.13604
7. Peairs AD, Shah AS, Summer S, Hess M, Couch SC. Effects of the dietary approaches to stop hypertension (DASH) diet on glucose variability in youth with Type 1 diabetes. Diabetes Manag Lond. (2017) 7:383–91.
8. Van Sloten TT. Vascular dysfunction: at the heart of cardiovascular disease, cognitive impairment and depressive symptoms. Artery Res. (2017) 19:18–23. doi: 10.1016/j.artres.2017.05.002
9. Peairs AD, Shah AS, Summer S, Hess M, Couch SC. Effects of the dietary approaches to stop hypertension (DASH) diet on glucose variability in youth with Type 1 diabetes. Diabetes Manag Lond. (2017) 7:383–91.
10. Imig JD. Eicosanoid blood vessel regulation in physiological and pathological states. Clin Sci. (2020) 134:2707–27. doi: 10.1042/CS20191209
11. Rafieian-Kopaei M, Setorki M, Doudi M, Baradaran A, Nasri H. Atherosclerosis: process, indicators, risk factors and new hopes. Int J Prev Med. (2014) 5:927.
12. Chester AH, Yacoub MH, Moncada S. Nitric oxide and pulmonary arterial hypertension. Glob Cardiol Sci Pract. (2017) 2017:14. doi: 10.21542/gcsp.2017.14
13. Lyle AN, Raaz U. Killing me unsoftly: causes and mechanisms of arterial stiffness. Arterioscler Thromb Vasc Biol. (2017) 37:e1–1. doi: 10.1161/ATVBAHA.116.308563
14. Higashi Y, Maruhashi T, Noma K, Kihara Y. Oxidative stress and endothelial dysfunction: clinical evidence and therapeutic implications. Trends Cardiovasc Med. (2014) 24:165–9. doi: 10.1016/j.tcm.2013.12.001
15. Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. (2005) 25:932–43. doi: 10.1161/01.ATV.0000160548.78317.29
16. Cheung YF. Arterial stiffness in the young: assessment, determinants, and implications. Korean Circ J. (2010) 40:153–62. doi: 10.4070/kcj.2010.40.4.153
17. Debeila S, Modjadji P, Madiba S. High prevalence of overall overweight/obesity and abdominal obesity amongst adolescents: an emerging nutritional problem in rural high schools in Limpopo Province, South Africa. Afr J Prim Health Care Fam Med. (2021) 13:e1–e9. doi: 10.4102/phcfm.v13i1.2596
18. Matjuda EN, Engwa GA, Letswalo PB, Mungamba MM, Sewani-Rusike CR, Nkeh-Chungag BN. Association of hypertension and obesity with risk factors of cardiovascular diseases in children aged 6–9 years old in the eastern Cape Province of South Africa. Children. (2020) 7:25. doi: 10.3390/children7040025
19. Noubiap JJ, Essouma M, Bigna JJ, Jingi AM, Aminde LN, Nansseu JR. Prevalence of elevated blood pressure in children and adolescents in Africa: a systematic review and meta-analysis. Lancet Public Health. (2017) 2:e375–86. doi: 10.1016/S2468-2667(17)30123-8
20. Gnidovec T, Žemlja M, Dolenec A, Torkar G. Using augmented reality and the structure– behavior–function model to teach lower secondary school students about the human circulatory system. J Sci Educ Technol. (2020) 29:774–84. doi: 10.1007/s10956-020-09850-8
21. Fleischer S, Tavakol DN, Vunjak-Novakovic G. From arteries to capillaries: approaches to engineering human vasculature. Adv Funct Mater. (2020) 30:1910811. doi: 10.1002/adfm.201910811
22. Segers P, Rietzschel ER, Chirinos JA. How to measure arterial stiffness in humans. Arterioscler Thromb Vasc Biol. (2020) 40:1034–43. doi: 10.1161/ATVBAHA.119.313132
23. Jamwal S, Sharma S. Vascular endothelium dysfunction: a conservative target in metabolic disorders. Inflamm Res. (2018) 67:391–405. doi: 10.1007/s00011-018-1129-8
24. McCarron JG, Wilson C, Heathcote HR, Zhang X, Buckley C, Lee MD. Heterogeneity and emergent behaviour in the vascular endothelium. Curr Opin Pharmacol. (2019) 45:23–32. doi: 10.1016/j.coph.2019.03.008
25. Moncada S. The vascular endothelium. Endothelium and Cardiovascular Diseases. Academic Press. (2018). p. 5–10.
26. Pulous FE, Petrich BG. Integrin-dependent regulation of the endothelial barrier. Tissue Barriers. (2019) 7:1685844. doi: 10.1080/21688370.2019.1685844
27. Jourde-Chiche N, Fakhouri F, Dou L, Bellien J, Burtey S., Frimat M, et al. Endothelium structure and function in kidney health and disease. Nat Rev Nephrol. (2019) 15:87–108. doi: 10.1038/s41581-018-0098-z
28. Guven G, Hilty MP, Ince C. Microcirculation: physiology, pathophysiology, and clinical application. Blood Purif. (2020) 49:143–50. doi: 10.1159/000503775
29. Pi X, Xie L, Patterson C. Emerging roles of vascular endothelium in metabolic homeostasis. Circ Res. (2018) 123:477–94. doi: 10.1161/CIRCRESAHA.118.313237
30. Mozos I, Malainer C, Horbańczuk J, Gug C, Stoian D., Luca CT, et al. Inflammatory markers for arterial stiffness in cardiovascular diseases. Front Immunol. (2017) 8:1058. doi: 10.3389/fimmu.2017.01058
31. Li Z, Zhang Y, Liu B, Luo W, Li H, Zhou Y. Role of E-type prostaglandin receptor EP3 in the vasoconstrictor activity evoked by prostacyclin in thromboxane-prostanoid receptor deficient mice. Sci Rep. (2017) 7:1–1. doi: 10.1038/srep42167
32. Chen H. Role of thromboxane A2 signaling in endothelium-dependent contractions of arteries. Prostaglandins Other Lipid Mediat. (2018) 134:32–7. doi: 10.1016/j.prostaglandins.2017.11.004
33. Chen P, Gao H, Lu Y, Nie H, Liu Z, Zhao Y, et al. Altered expression of eNOS, prostacyclin synthase, prostaglandin G/H synthase, and thromboxane synthase in porcine aortic endothelial cells after exposure to human serum—relevance to xenotransplantation. Cell Biol Int. (2017) 41:798–808. doi: 10.1002/cbin.10782
34. da Costa TJ, Echem C, Colli LG, Akamine EH, Dantas AP, de Carvalho MH. Characteristics of the endothelium in both sexes. Endothelium and Cardiovascular Diseases. Academic Press (2018). p. 63–81.
35. Erdman MK, Leary MC. Antiplatelet Agents: Mechanisms and Their role in Stroke Prevention. In: Primer on Cerebrovascular Diseases. Academic Press (2017). p. 874–81. doi: 10.1016/B978-0-12-803058-5.00166-1
36. Mitchell JA, Kirkby NS. Eicosanoids, prostacyclin and cyclooxygenase in the cardiovascular system. Br J Pharmacol. (2019) 176:1038–50. doi: 10.1111/bph.14167
37. Smyth EM. Thromboxane and the thromboxane receptor in cardiovascular disease. Clin Lipidol. (2010) 5:209–19. doi: 10.2217/clp.10.11
38. Sriram K, Laughlin JG, Rangamani P, Tartakovsky DM. Shear-induced nitric oxide production by endothelial cells. Biophys J. (2016) 111:208–21. doi: 10.1016/j.bpj.2016.05.034
39. Zhao Y, Vanhoutte PM, Leung SW. Vascular nitric oxide: Beyond eNOS. J Pharmacol Sci. (2015) 129:83–94. doi: 10.1016/j.jphs.2015.09.002
40. Ghimire K, Zaric J, Alday-Parejo B, Seebach J, Bousquenaud M, Stalin J, et al. MAGI1 mediates eNOS activation and NO production in endothelial cells in response to fluid shear stress. Cells. (2019) 8:388. doi: 10.3390/cells8050388
41. Klinger JR, Kadowitz PJ. The nitric oxide pathway in pulmonary vascular disease. Am J Cardiol. (2017) 120:S71–9. doi: 10.1016/j.amjcard.2017.06.012
42. Genovesi S, Giussani M, Orlando A, Lieti G, Viazzi F, Parati G. Relationship between endothelin and nitric oxide pathways in the onset and maintenance of hypertension in children and adolescents. Pediatr Nephrol. (2021) 3:1–9. doi: 10.1007/s00467-021-05144-2
43. Nepal G, Ojha R, Dulal HP, Yadav BK. Association between Lys198Asn polymorphism of endothelin-1 gene and ischemic stroke: a meta-analysis. Brain Behav. (2019) 9:e01424. doi: 10.1002/brb3.1424
44. Eroglu E, Kocyigit I, Lindholm B. The endothelin system as target for therapeutic interventions in cardiovascular and renal disease. Clin Chim Acta. (2020) 506:92–106. doi: 10.1016/j.cca.2020.03.008
45. Sun J, Qiao YN, Tao T, Zhao W, Wei LS, Li YQ, et al. Distinct roles of smooth muscle and non-muscle myosin light chain-mediated smooth muscle contraction. Front Physiol. (2020) 11:1610. doi: 10.3389/fphys.2020.593966
46. Nishiyama SK, Zhao J, Wray DW, Richardson RS. Vascular function and endothelin-1: tipping the balance between vasodilation and vasoconstriction. J Appl Physiol. (2017) 122:354–60. doi: 10.1152/japplphysiol.00772.2016
47. Hannemann J, Zummack J, Hillig J, Böger R. Metabolism of asymmetric dimethylarginine in hypoxia: from bench to bedside. Pulm Circ. (2020) 10:31–41. doi: 10.1177/2045894020918846
48. Sibal L, Agarwal SC, Home PD, Boger RH. The role of asymmetric dimethylarginine (ADMA) in endothelial dysfunction and cardiovascular disease. Curr Cardiol Rev. (2010) 6:82–90. doi: 10.2174/157340310791162659
49. Reyhani A, Celik Y, Karadag H, Gunduz O, Asil T, Sut N. High asymmetric dimethylarginine, symmetric dimethylarginine and L-arginine levels in migraine patients. Neurol Sci. (2017) 38:1287–91. doi: 10.1007/s10072-017-2970-1
50. Gambardella J, Khondkar W, Morelli MB, Wang X, Santulli G, Trimarco V. Arginine and endothelial function. Biomedicines. (2020) 8:277. doi: 10.3390/biomedicines8080277
51. Matsumoto T, Takayanagi K, Kojima M, Katome T, Taguchi K, Kobayashi T. Direct impairment of the endothelial function by acute indoxyl sulfate through declined nitric oxide and not endothelium-derived hyperpolarizing factor or vasodilator prostaglandins in the rat superior mesenteric artery. Biol Pharm Bull. (2019) 42:1236–42. doi: 10.1248/bpb.b19-00177
52. Bautista-Niño PK, van der Stel M, Batenburg WW, de Vries R, Roks AJ, Danser AJ. Endothelium-derived hyperpolarizing factor and protein kinase G Iα activation: H2O2 versus S-nitrosothiols. Eur J of Pharm. (2018) 827:112–6. 43. doi: 10.1016/j.ejphar.2018.03.019
53. Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. (2017) 120:713–35. doi: 10.1161/CIRCRESAHA.116.309326
54. Tsikas D, Bollenbach A, Hanff E, Kayacelebi AA. Asymmetric dimethylarginine (ADMA), symmetric dimethylarginine (SDMA) and homoarginine (hArg): the ADMA, SDMA and hArg paradoxes. Cardiovasc Diabetol. (2018) 17:1–4. doi: 10.1186/s12933-017-0656-x
55. Yuyun MF, Ng LL, Ng GA. Endothelial dysfunction, endothelial nitric oxide bioavailability, tetrahydrobiopterin, and 5-methyltetrahydrofolate in cardiovascular disease. Where are we with therapy? Microvasc Res. (2018) 119:7–12. doi: 10.1016/j.mvr.2018.03.012
56. Sepulveda C, Palomo I, Fuentes E. Mechanisms of endothelial dysfunction during aging: predisposition to thrombosis. Mech Ageing Dev. (2017) 164:91–9. doi: 10.1016/j.mad.2017.04.011
57. Mokhaneli MC, Fourie CM, Botha-Le Roux S, Böger RH, Schwedhelm E, Mels CM. Asymmetric dimethylarginine and L-homoarginine prospectively relate to carotid wall thickness in a South African cohort. Amino Acids. (2020) 52:965–73. doi: 10.1007/s00726-020-02866-9
58. Craig A, Mels CM, Tsikas D, Boeger RH, Schwedhelm E, Schutte AE, et al. Central systolic blood pressure relates inversely to nitric oxide synthesis in young black adults: the African-PREDICT study. J Hum Hypertens. (2020) 35:985–93. doi: 10.1097/01.hjh.0000744688.00397.48
59. Gutterman DD, Chabowski DS, Kadlec AO, Durand MJ, Freed JK, Ait-Aissa K, et al. The human microcirculation: regulation of flow and beyond. Circ Res. (2016) 118:157–72. doi: 10.1161/CIRCRESAHA.115.305364
60. Strain WD, Paldánius PM. Diabetes, cardiovascular disease and the microcirculation. Cardiovasc Diabetol. (2018) 17:1–0. doi: 10.1186/s12933-018-0703-2
61. Rizzoni D, Agabiti Rosei C, De Ciuceis C, Semeraro F, Rizzoni M, Docchio F. New methods to study the microcirculation. Am J Hypertens. (2018) 31:265–73. doi: 10.1093/ajh/hpx211
62. Chen C, Wei J, AlBadri A, Zarrini P, Bairey Merz CN. Coronary microvascular dysfunction- epidemiology, pathogenesis, prognosis, diagnosis, risk factors and therapy. Circ J. (2016) 81:3–11. doi: 10.1253/circj.CJ-16-1002
63. Chen Y, Shen F, Liu J, Yang GY. Arterial stiffness and stroke: de-stiffening strategy, a therapeutic target for stroke. Stroke Vasc Neurol. (2017) 2:65–72. doi: 10.1136/svn-2016-000045
64. Zarkovic K, Larroque-Cardoso P, Pucelle M, Salvayre R, Waeg G, Nègre-Salvayre A, et al. Elastin aging and lipid oxidation products in human aorta. Redox Biol. (2015) 4:109–17. doi: 10.1016/j.redox.2014.12.008
65. DuPont JJ, Kenney RM, Patel AR, Jaffe IZ. Sex differences in mechanisms of arterial stiffness. Br J Pharmacol. (2019) 176:4208–25. doi: 10.1111/bph.14624
66. Mikael LD, Paiva AM, Gomes MM, Sousa AL, Jardim PC, Vitorino PV, et al. Vascular aging and arterial stiffness. Arq Bras Cardiol. (2017) 109:253–8. doi: 10.5935/abc.20170091
67. Tomiyama H, Yamashina A. Non-invasive vascular function tests: their pathophysiological background and clinical application. Circ J. (2010):0911120515. doi: 10.1253/circj.CJ-09-0534
68. Paterson EN, Cardwell C, MacGillivray TJ, Trucco E, Doney AS, Foster P, et al. Investigation of associations between retinal microvascular parameters and albuminuria in UK Biobank: a cross-sectional case-control study. BMC Nephrol. (2021) 22:1. doi: 10.1186/s12882-021-02273-6
69. Storch AS, Mattos JD, Alves R, Galdino ID, Rocha HN. Methods of endothelial function assessment: description and applications. Int J Cardiovasc Sci. (2017) 30:262–73. doi: 10.5935/2359-4802.20170034
70. Harris RA, Nishiyama SK, Wray DW, Richardson RS. Ultrasound assessment of flow-mediated dilation. Hypertens. (2010) 55:1075–85. doi: 10.1161/HYPERTENSIONAHA.110.150821
71. Nabavizadeh P, Liu J, Havel CM, Ibrahim S, Derakhshandeh R, Jacob Iii P, et al. Vascular endothelial function is impaired by aerosol from a single IQOS HeatStick to the same extent as by cigarette smoke. Tob Control. (2018) 27:s13–9. doi: 10.1136/tobaccocontrol-2018-054325
72. Thijssen DH, Bruno RM, van Mil AC, Holder SM, Faita F, Greyling A, et al. Expert consensus and evidence-based recommendations for the assessment of flow-mediated dilation in humans. Eur Heart J. (2019) 40:2534–47. doi: 10.1093/eurheartj/ehz350
73. Stoner L, Stone K, Zieff G, Blackwell J, Diana J, Credeur DP, et al. Endothelium function dependence of acute changes in pulse wave velocity and flow-mediated slowing. Vasc Med. (2020) 25:419–26. doi: 10.1177/1358863X20926588
74. Cauwenberghs N, Heyrman Y, Thijs L, Yang WY, Wei FF, Zhang ZY, et al. Flowmediated slowing of brachial-radial pulse wave velocity: Methodological aspects and clinical determinants. Artery Res. (2018) 21:29–37. doi: 10.1016/j.artres.2017.12.001
75. Basgaran A, Maki-Petaja K, Wilkinson I, McEniery C. Flow-mediated slowing as a novel method for the non-invasive assessment of endothelial function. Artery Res. (2016) 16:70. doi: 10.1016/j.artres.2016.10.076
76. Pereira T, Almeida A, Conde J. Flow-mediated slowing as a methodological alternative to the conventional echo-tracking flow-mediated dilation technique for the evaluation of endothelial function: a preliminary report. Mayo Clin Proc Innov Qual Outcomes. (2018) 2:199–203. doi: 10.1016/j.mayocpiqo.2018.02.002
77. Englund EK, Langham MC. Quantitative and dynamic MRI measures of peripheral vascular function. Front Physiol. (2020) 11:120. doi: 10.3389/fphys.2020.00120
78. Caporale A, Langham MC, Guo W, Johncola A, Chatterjee S, Wehrli FW. Acute effects of electronic cigarette aerosol inhalation on vascular function detected at quantitative MRI. Radiology. (2019) 293:97–106. doi: 10.1148/radiol.2019190562
79. Cheung CY, Ikram MK, Sabanayagam C, Wong TY. Retinal microvasculature as a model to study the manifestations of hypertension. Hypertens. (2012) 60:1094–103. doi: 10.1161/HYPERTENSIONAHA.111.189142
80. Al-Fiadh AH, Farouque O, Kawasaki R, Nguyen TT, Uddin N, Freeman M, et al. Retinal microvascular structure and function in patients with risk factors of atherosclerosis and coronary artery disease. Atherosclerosis. (2014) 233:478–84. doi: 10.1016/j.atherosclerosis.2013.12.044
81. Badar M, Haris M, Fatima A. Application of deep learning for retinal image analysis: a review. Comp Sci Rev. (2020) 35:100203. doi: 10.1016/j.cosrev.2019.100203
82. Kubin AM, Wirkkala J, Keskitalo A, Ohtonen P, Hautala N. Handheld fundus camera performance, image quality and outcomes of diabetic retinopathy grading in a pilot screening study. Acta Ophthalmol. (2021) 99:e1415–20. doi: 10.1111/aos.14850
83. Poplin R, Varadarajan AV, Blumer K, Liu Y, McConnell MV, Corrado GS, et al. Prediction of cardiovascular risk factors from retinal fundus photographs via deep learning. Nat Biomed Eng. (2018) 2:158–64. doi: 10.1038/s41551-018-0195-0
84. Kim HL, Kim SH. Pulse wave velocity in atherosclerosis. Front Cardiovasc Med. (2019) 6:41. doi: 10.3389/fcvm.2019.00041
85. Thurn D, Doyon A, Sözeri B, Bayazit AK, Canpolat N, Duzova A, et al. Aortic pulse wave velocity in healthy children and adolescents: reference values for the Vicorder device and modifying factors. Am J Hypertens. (2015) 28:1480–8. doi: 10.1093/ajh/hpv048
86. Butlin M, Qasem A. Large artery stiffness assessment using SphygmoCor technology. Pulse. (2016) 4:180–92. doi: 10.1159/000452448
87. Ehrenthal DB, Goldstein ND, Wu P, Rogers S, Townsend RR, Edwards DG. Arterial stiffness and wave reflection 1 year after a pregnancy complicated by hypertension. J Clin Hypertens. (2014) 16:695–9. doi: 10.1111/jch.12398
88. Lopez-Sublet M, Girerd N, Bozec E, Machu JL, Ferreira JP, Zannad F, et al. Nondipping pattern and cardiovascular and renal damage in a population-based study (The STANISLAS Cohort Study). Am J Hypertens. (2019) 32:620–8. doi: 10.1093/ajh/hpz020
89. Kolkenbeck-Ruh A, Soepnel LM, Kim AW, Naidoo S, Smith W, Davies J, et al. Pulse wave velocity in South African women and children: comparison between the Mobil-O-Graph and SphygmoCor XCEL devices. J Hypertens. (2021) 39:1–11. doi: 10.1097/HJH.0000000000002976
90. Barroso WK, Gonçalves CF, Berigó JA, Melo MA, Arantes AC, Lelis ED, et al. Tonometric and oscillometric methods for measurement of central blood pressure parameters: a comparison in patients with borderline hypertension or stage 1 hypertension. Int J Cardiovas Sci. (2020) 33:145–50.
91. Hwang MH, Yoo JK, Kim HK, Hwang CL, Mackay K, Hemstreet O, et al. Validity and reliability of aortic pulse wave velocity and augmentation index determined by the new cuff-based SphygmoCor Xcel. J Hum Hypertens. (2014) 28:475–81. doi: 10.1038/jhh.2013.144
92. Savant JD, Furth SL, Meyers KE. Arterial stiffness in children: pediatric measurement and considerations. Pulse. (2014) 2:69–80. doi: 10.1159/000374095
93. Benas D, Kornelakis M, Triantafyllidi H, Kostelli G, Pavlidis G, Varoudi M, et al. Pulse wave analysis using the Mobil-O-Graph, Arteriograph and Complior device: a comparative study. Blood Press. (2019) 28:107–13. doi: 10.1080/08037051.2018.1564236
94. Fiori G, Fuiano F, Scorza A, Conforto S, Sciuto SA. Non-invasive methods for PWV measurement in blood vessel stiffness assessment. IEEE Rev Biomed Eng. (2021). doi: 10.1109/RBME.2021.3092208. [Epub ahead of print].
95. Dieden A, Malan L, Mels CM, Lammertyn L, Wentzel A, Nilsson PM, et al. Exploring biomarkers associated with deteriorating vascular health using a targeted proteomics chip: The SABPA study. Medicine. (2021) 100:e25936. doi: 10.1097/MD.0000000000025936
96. Kruger R, Schutte R, Huisman HW, Hindersson P, Olsen MH, Eugen-Olsen J, et al. NT-proBNP, C-reactive protein and soluble uPAR in a bi-ethnic male population: the SAfrEIC study. PLoS ONE. (2013) 8:e58506. doi: 10.1371/journal.pone.0058506
97. van Rooyen JM, Schutte AE, Huisman HW, Schutte R, Fourie CM, Malan NT, et al. End-organ damage in urbanized Africans with low plasma renin levels: the SABPA study. Clin Exp Hypertens. (2014) 36:70–5. doi: 10.3109/10641963.2013.789044
98. van Velzen MH, Stolker RJ, Loeve AJ, Niehof SP, Mik EG. Comparison between pulse wave velocities measured using complior and measured using biopac. J Clin Monit Comput. (2019) 33:241–7. doi: 10.1007/s10877-018-0165-9
99. Willeit P, Tschiderer L, Allara E, Reuber K, Seekircher L, Gao LU, et al. Carotid intimamedia thickness progression as surrogate marker for cardiovascular risk: meta-analysis of 119 clinical trials involving 100 667 patients. Circulation. (2020) 142:621–42. doi: 10.1161/CIRCULATIONAHA.120.046361
100. Khutan H, Aggarwal S, Kajal KS, Garg R, Kaur R, Kaur A. Study of carotid intimal medial thickness in essential hypertension with or without left ventricular hypertrophy. Ann Afr Med. (2017) 16:192–5. doi: 10.4103/aam.aam_9_17
101. Okeahialam BN, Alonge BA, Pam SD, Puepet FH. Carotid intima media thickness as a measure of cardiovascular disease burden in Nigerian Africans with hypertension and diabetes mellitus. Int J Vasc Med. (2011) 2011:327171. doi: 10.1155/2011/327171
102. Simova I. Intima-media thickness: appropriate evaluation and proper measurement. J Cardiol Pract. (2015) 13:1–4.
103. Schoffelen AF, De Groot E, Tempelman HA, Visseren FL, Hoepelman AI, Barth RE. Carotid intima media thickness in mainly female HIV-infected subjects in rural South Africa: association with cardiovascular but not HIV-related factors. Clin Infect Dis. (2015) 61:1606–14. doi: 10.1093/cid/civ586
104. Roozen GV, Vos AG, Tempelman HA, Venter WD, Grobbee DE, Scheuermaier K, et al. Cardiovascular disease risk and its determinants in people living with HIV across different settings in South Africa. HIV Med. (2020) 21:386–96. doi: 10.1111/hiv.12831
105. Veldsman T, Swanepoel M, Monyeki MA, Brits JS, Malan L. Relationship between physical activity and carotid intima–media thickness among teachers in South Africa: the SABPA study. Cardiovas J Afr. (2020) 2020:31. doi: 10.5830/CVJA-2020-024
106. Uslu B, Cakmak YO, Sehirli Ü, Keskinoz EN, Cosgun E, Arbak S, et al. Early onset of atherosclerosis of the carotid bifurcation in newborn cadavers. J Clin Diagn Res. (2016) 10:AC01–5. doi: 10.7860/JCDR/2016/19827.7706
107. Yu GZ, Reilly S, Lewandowski AJ, Aye CY, Simpson LJ, Newton LD, et al. Neonatal microRNA profile determines endothelial function in offspring of hypertensive pregnancies. Hypertens. (2018) 72:937–45. doi: 10.1161/HYPERTENSIONAHA.118.11343
108. Okala SG, Sise EA, Sosseh F, Prentice AM, Woollett LA, Moore SE. Maternal plasma lipid levels across pregnancy and the risks of small-for-gestational age and low birth weight: a cohort study from rural Gambia. BMC Pregnancy Childbirth. (2020) 20:1–6. doi: 10.1186/s12884-020-2834-1
109. Fuenzalida B, Sobrevia B, Cantin C, Carvajal L, Salsoso R, Gutiérrez J, et al. Maternal supraphysiological hypercholesterolemia associates with endothelial dysfunction of the placental microvasculature. Sci Rep. (2018) 8:1–0. doi: 10.1038/s41598-018-25985-6
110. Tomimatsu T, Mimura K, Matsuzaki S, Endo M, Kumasawa K, Kimura T. Preeclampsia: maternal systemic vascular disorder caused by generalized endothelial dysfunction due to placental antiangiogenic factors. Int J Mol Sci. (2019) 20:4246. doi: 10.3390/ijms20174246
111. Sehgal A, Allison BJ, Gwini SM, Menahem S, Miller SL, Polglase GR. Vascular aging and cardiac maladaptation in growth-restricted preterm infants. J Perinatol. (2018) 38:92–7. doi: 10.1038/jp.2017.135
112. Pisaneschi S, Boldrini A, Genazzani AR, Coceani F, Simoncini T. Feto-placental vascular dysfunction as a prenatal determinant of adult cardiovascular disease. Intern Emerg Med. (2013) 8:41–5. doi: 10.1007/s11739-013-0925-y
113. Castro-Barquero S, Lamuela-Raventós RM, Doménech M, Estruch R. Relationship between Mediterranean dietary polyphenol intake and obesity. Nutrients. (2018) 10:1523. doi: 10.3390/nu10101523
114. Giabicani E, Pham A, Brioude F, Mitanchez D, Netchine I. Diagnosis and management of postnatal fetal growth restriction. Best Pract Res Clin Endocrinol Metab. (2018) 32:52334. doi: 10.1016/j.beem.2018.03.013
115. Freemark M. Determinants of risk for childhood obesity. N Engl J Med. (2018) 379:1371–2. doi: 10.1056/NEJMe1811305
116. Longo M, Zatterale F, Naderi J, Parrillo L, Formisano P, Raciti GA, et al. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int J Mol Sci. (2019) 20:2358. doi: 10.3390/ijms20092358
117. Castro AM. Macedo-De la Concha LE, Pantoja-Meléndez CA. Low-grade inflammation and its relation to obesity and chronic degenerative diseases. Rev Med Hosp Gen Mex. (2017) 80:101–5. doi: 10.1016/j.hgmx.2016.06.011
118. Nicholson T, Church C, Baker DJ, Jones SW. The role of adipokines in skeletal muscle inflammation and insulin sensitivity. J Inflamm. (2018) 15:1. doi: 10.1186/s12950-018-0185-8
119. Pîrsean C, Negu? C. Stefan-van Staden RI, Dinu-Pirvu CE, Armean P, Udeanu DI. The salivary levels of leptin and interleukin-6 as potential inflammatory markers in children obesity. PLoS ONE. (2019) 14:e0210288. doi: 10.1371/journal.pone.0210288
120. Kwaifa IK, Bahari H, Yong YK, Noor SM. Endothelial dysfunction in obesity-induced inflammation: molecular mechanisms and clinical implications. Biomolecules. (2020) 10:291. doi: 10.3390/biom10020291
121. Virdis A, Masi S, Colucci R, Chiriacò M, Uliana M, Puxeddu I, et al. Microvascular endothelial dysfunction in patients with obesity. Curr Hypertens Rep. (2019) 21:1–7. doi: 10.1007/s11906-019-0930-2
122. Triches CB, Mayer S, Quinto BM, Batista MC, Zanella MT. Association of endothelial dysfunction with cardiovascular risk factors and new-onset diabetes mellitus in patients with hypertension. J Clin Hypertens. (2018) 20:935–41. doi: 10.1111/jch.13269
123. Dymara-Konopka W, Laskowska M. The role of nitric oxide, ADMA, and homocysteine in the etiopathogenesis of preeclampsia. Int J Mol Sci. (2019) 20:2757. doi: 10.3390/ijms20112757
124. Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-κB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol. (2007) 292:L567–76. doi: 10.1152/ajplung.00308.2006
125. Fernández-Sánchez A, Madrigal-Santillán E, Bautista M, Esquivel-Soto J, Morales-González Á, Esquivel-Chirino C, et al. Inflammation, oxidative stress, and obesity. Int J Mol Sci. (2011) 12:3117–32. doi: 10.3390/ijms12053117
126. Vaka VR, McMaster KM, Cunningham MW Jr, Ibrahim T, Hazlewood R, Usry N, et al. Role of mitochondrial dysfunction and reactive oxygen species in mediating hypertension in the reduced uterine perfusion pressure rat model of preeclampsia. Hypertens. (2018) 72:703–11. doi: 10.1161/HYPERTENSIONAHA.118.11290
127. Oliveira-Paula GH, Pinheiro LC, Tanus-Santos JE. Mechanisms impairing blood pressure responses to nitrite and nitrate. Nitric Oxide. (2019) 85:35–43. doi: 10.1016/j.niox.2019.01.015
128. Togliatto G, Lombardo G, Brizzi MF. The future challenge of reactive oxygen species (ROS) in hypertension: from bench to bed side. Int J Mol Sci. (2017) 18:1988. doi: 10.3390/ijms18091988
129. Aldosari S, Awad M, Harrington EO, Sellke FW, Abid MR. Subcellular reactive oxygen species (ROS) in cardiovascular pathophysiology. Antioxidants. (2018) 7:14. doi: 10.3390/antiox7010014
130. Boren J, Chapman MJ, Krauss RM, Packard CJ, Bentzon JF, Binder CJ, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. (2020) 41:2313–30. doi: 10.1093/eurheartj/ehz962
131. Leiva E, Wehinger S, Guzmán L, Orrego R. Role of oxidized LDL in atherosclerosis. Hypercholesterolemia. (2015) 17:55–78. doi: 10.5772/59375
132. Brandes RP. Endothelial dysfunction and hypertension. Hypertens. (2014) 64:924–8. doi: 10.1161/HYPERTENSIONAHA.114.03575
133. Virdis A, Bacca A, Colucci R, Duranti E, Fornai M, Materazzi G, et al. Endothelial dysfunction in small arteries of essential hypertensive patients: role of cyclooxygenase-2 in oxidative stress generation. Hypertens. (2013) 62:337–44. doi: 10.1161/HYPERTENSIONAHA.111.00995
134. Dharmashankar K, Widlansky ME. Vascular endothelial function and hypertension: insights and directions. Curr Hypertens Rep. (2010) 12:448–55. doi: 10.1007/s11906-010-0150-2
135. Marchesi C, Ebrahimian T, Angulo O, Paradis P, Schiffrin EL. Endothelial nitric oxide synthase uncoupling and perivascular adipose oxidative stress and inflammation contribute to vascular dysfunction in a rodent model of metabolic syndrome. Hypertens. (2009) 54:1384–92. doi: 10.1161/HYPERTENSIONAHA.109.138305
136. Vecchione C, Carnevale D, Di Pardo A, Gentile MT, Damato A, Cocozza G, et al. Pressure-induced vascular oxidative stress is mediated through activation of integrin-linked kinase 1/betaPIX/Rac-1 pathway. Hypertens. (2009) 54:1028–34. doi: 10.1161/HYPERTENSIONAHA.109.136572
137. Magen E, Feldman A, Cohen Z, Alon DB, Linov L, Mishal J, et al. Potential link between C3a, C3b and endothelial progenitor cells in resistant hypertension. Am J Med Sci. (2010) 339:415–9. doi: 10.1097/MAJ.0b013e3181d7d496
138. Lobato NS, Filgueira FP, Akamine EH, Tostes RC, Carvalho MH, Fortes ZB. Mechanisms of endothelial dysfunction in obesity-associated hypertension. Braz J Med Biol Res. (2012) 45:392–400. doi: 10.1590/S0100-879X2012007500058
139. Engin A. Endothelial dysfunction in obesity. Obes Lipotoxicity. (2017) 2017:345–79. doi: 10.1007/978-3-319-48382-5_15
140. Yan P, Xia C, Duan C, Li S, Mei Z. Biological characteristics of foam cell formation in smooth muscle cells derived from bone marrow stem cells. Int J Biol Sci. (2011) 7:937. doi: 10.7150/ijbs.7.937
141. Xie N, Chen M, Dai R, Zhang Y, Zhao H, Song Z. SRSF1 promotes vascular smooth muscle cell proliferation through a Δ133p53/EGR1/KLF5 pathway. Nat Commun. (2017) 8:1–9. doi: 10.1038/ncomms16016
142. Satoh M. Endothelial dysfunction as an underlying pathophysiological condition of chronic kidney disease. Clin Exp Nephrol. (2012) 16:518–21. doi: 10.1007/s10157-012-0646-y
143. Basile DC, Yoder M. Renal endothelial dysfunction in acute kidney ischemia reperfusion injury. Cardiovasc Haematol Disord Drug Targets. (2014) 14:3–14. doi: 10.2174/1871529X1401140724093505
144. Xu N, Jiang S, Persson PB, Persson EA, Lai EY, Patzak A. Reactive oxygen species in renal vascular function. Acta Physiol. (2020) 229:e13477. doi: 10.1111/apha.13477
145. Gui F, You Z, Fu S, Wu H, Zhang Y. Endothelial dysfunction in diabetic retinopathy. Front Endocrinol. (2020) 11:591. doi: 10.3389/fendo.2020.00591
146. Mrugacz M, Bryl A, Zorena K. Retinal vascular endothelial cell dysfunction and neuroretinal degeneration in diabetic patients. J Clin Med. (2021) 10:458. doi: 10.3390/jcm10030458
147. Miyoshi T, Ito H. Arterial stiffness in health and disease: the role of cardio–ankle vascular index. J Cardiol. (2021) 78:493–501. doi: 10.1016/j.jjcc.2021.07.011
148. Lee JG, Joo SJ. Arterial stiffness and cardiovascular risk. Korean J Internal Med. (2019) 34:504. doi: 10.3904/kjim.2019.110
149. van Biljon A, Erasmus H, Mathunjwa ML. Associations between arterial stiffness and cardiovascular disease risk factors among black South African children. S Afr J Child Health. (2021) 15:14–7. doi: 10.7196/SAJCH.2021.v15i1.01743
150. Kohn JC, Lampi MC, Reinhart-King CA. Age-related vascular stiffening: causes and consequences. Front Genet. (2015) 6:112. doi: 10.3389/fgene.2015.00112
151. Matjuda EN, Engwa GA, Anye SN, Nkeh-Chungag BN, Goswami N. Cardiovascular risk factors and their relationship with vascular dysfunction in South African Children of African Ancestry. J Clin Med. (2021) 10:354. doi: 10.3390/jcm10020354
152. Bollenbach A, Schutte AE, Kruger R, Tsikas D. An ethnic comparison of arginine dimethylation and cardiometabolic factors in healthy black and white youth: the ASOS and African-PREDICT studies. J Clin Med. (2020) 9:844. doi: 10.3390/jcm9030844
153. Letswalo BP, Schmid-Zalaudek K, Brix B, Matjuda EN, Klosz F, Obernhumer N, et al. Cardiometabolic risk factors and early indicators of vascular dysfunction: a cross- sectional cohort study in South African adolescents. BMJ Open. (2021) 11:e042955. doi: 10.1136/bmjopen-2020-042955
154. Innes S, Cotton MF, Otwombe K, Laughton B, Herbst PG, Magogotya Z, et al. (2015). Does pulse-wave velocity normalize with increasing time on ART in children? Evidence from the CHER Trial Cohort. Abstract retrieved from conference on retroviruses and opportunistic infections. (Abstract No. 658).
155. Dobe IS, Mocumbi AO, Majid N, Ayele B, Browne SH, Innes S. Earlier antiretroviral initiation is independently associated with better arterial stiffness in children living with perinatally acquired HIV with sustained viral suppression in Mozambique. South Afr J HIV Med. (2021) 22:6. doi: 10.4102/sajhivmed.v22i1.1282
156. Al-Drawny Z, Saleh SH, El-Sammak AA, Attia HM. Carotid Intima Media Thickness in Obese Egyptian Children and Adolescent. Egypt J Hosp Med. (2020) 80:672–7. doi: 10.21608/ejhm.2020.95264
157. Kandil ME, Anwar GM, Fatouh A, Salama N, Ahmed A, Elabd E, et al. Relation between serum homocysteine and carotid intima-media thickness in obese Egyptian children. J Clin Basic Cardiol. (2011) 13:8–11.
Keywords: vascular dysfunction, obesity, cardiovascular risk factors, African children, vascular function
Citation: Matjuda EN, Engwa GA, Sewani-Rusike CR and Nkeh-Chungag BN (2021) An Overview of Vascular Dysfunction and Determinants: The Case of Children of African Ancestry. Front. Pediatr. 9:769589. doi: 10.3389/fped.2021.769589
Received: 02 September 2021; Accepted: 08 November 2021;
Published: 10 December 2021.
Edited by:
Ruan Kruger, North-West University, South AfricaReviewed by:
Gontse Gratitude Mokwatsi, North-West University, South AfricaLebo F. Gafane-Matemane, North-West University, South Africa
Copyright © 2021 Matjuda, Engwa, Sewani-Rusike and Nkeh-Chungag. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benedicta N. Nkeh-Chungag, Ym5rZWhjaHVuZ2FnQHdzdS5hYy56YQ==