
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 04 November 2021
Sec. Pediatric Surgery
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.766610
This article is part of the Research TopicInnovative Approaches in Pediatric Surgical OncologyView all 17 articles
 Gloria Pelizzo1,2*†
Gloria Pelizzo1,2*† Federica Riva3†
Federica Riva3† Stefania Croce4
Stefania Croce4 Maria Antonietta Avanzini4Gloria Acquafredda4
Maria Antonietta Avanzini4Gloria Acquafredda4 Annalisa de Silvestri5
Annalisa de Silvestri5 Emanuela Mazzon6Placido Bramanti6
Emanuela Mazzon6Placido Bramanti6 Gianvincenzo Zuccotti2,7Giuliano Mazzini8‡
Gianvincenzo Zuccotti2,7Giuliano Mazzini8‡ Valeria Calcaterra7,9‡
Valeria Calcaterra7,9‡The explanation for cancer recurrence still remains to be fully elucidated. Moreover, tumor dormancy, which is a process whereby cells enter reversible G0 cell cycle arrest, appears to be a critical step in this phenomenon. We evaluated the cell cycle proliferation pattern in pediatric tumor-derived mesenchymal stromal cells (MSCs), in order to provide a better understanding of the complex mechanisms underlying cancer dormancy. Specimens were obtained from 14 pediatric patients diagnosed with solid tumors and submitted to surgery. Morphology, phenotype, differentiation, immunological capacity, and proliferative growth of tumor MSCs were studied. Flow cytometric analysis was performed to evaluate the cell percentage of each cell cycle phase. Healthy donor bone marrow-derived mesenchymal stromal cells (BM-MSCs) were employed as controls. It was noted that the DNA profile of proliferating BM-MSC was different from that of tumor MSCs. All BM-MSCs expressed the typical DNA profile of proliferating cells, while in all tumor MSC samples, ≥70% of the cells were detected in the G0/G1 phase. In particular, seven tumor MSC samples displayed intermediate cell cycle behavior, and the other seven tumor MSC samples exhibited a slow cell cycle. The increased number of tumor MSCs in the G0–G1 phase compared with BM-MSCs supports a role for quiescent MSCs in tumor dormancy regulation. Understanding the mechanisms that promote dormant cell cycle arrest is essential in identifying predictive markers of recurrence and to promote a dedicated surgical planning.
The overall incidence rates of childhood cancer vary between 50 and 200 per million children across the world (1). A thorough study on this issue revealed an incidence rate of 138.5 children in every 1 million children worldwide (2). However, cancer is the third leading cause of death among children ages 1–4 and the second leading cause of death among children age 5–14 (3), representing about 8% of all pediatric deaths (4–6). Indeed, after surgical and medical treatment, recurrence, or cancer relapse after an initial diagnosis has been frequently recorded (4–6).
A proposed mechanism underlying the persistence of covert cancer cells during and after treatment is that some cancer stem cells enter a reversible quiescent or dormant state in which they are relatively resistant to radiation and chemotherapy. Conventional chemotherapy regimens include DNA-damaging agents and spindle poisons, and their effect is, therefore, dependent on the active cycling of tumoral cells through the S and M cell cycle phases, respectively. However, both cell intrinsic characteristics and extrinsic influences from surrounding normal cells determine tumor cell dormancy (7). Extrinsic factors include mesenchymal stromal cells (MSCs), endothelial cells (ECs), and immune cells that form the niche of the tumor. As described in in vitro leukemia models, blasts communicate closely with MSCs (8), and contact with MSCs has been demonstrated to provide key survival signals to leukemic blasts, rendering them resistant against the non-genotoxic components of leukemia treatment protocols (8, 9). Mesenchymal stromal cells play different roles in modulating tumor progression, growth, and metastasis. They are recruited to the tumor site in large numbers and subsequently have an important microenvironmental role in modulating tumor progression and drug sensitivity. However, the effects of the tumor microenvironment (TME) on MSCs remain poorly understood. It has been reported that a paracrine effect of cancer cells slows cycling and chemoresistance, through the secretion of soluble factors promoting a more stem-like state of MSCs (10). Additionally, the contact between cancer cells and MSCs in regulating cancer dormancy should not be excluded (11).
Therefore, the aim of this study was to characterize the proliferation pattern of the cell cycle in pediatric tumor-derived MSCs, in order to enhance our understanding of the complex mechanisms, implicated in the cancer dormancy process, that may influence therapeutic response.
Fourteen pediatric patients (eight females and six males; median age 5 years, range 9 months to 15 years), diagnosed with solid tumors (three neuroblastomas, three lymphomas, three nephroblastomas, and five others) and submitted to surgery were enrolled. Mesenchymal stromal cell isolation and expansion were performed starting from residual material for histological analysis. Samples were collected prior to chemotherapy. Stored bone marrow-derived mesenchymal stromal cells (BM-MSCs), obtained as previously described (12) from healthy donors (two females and two males; median age 5.5 years, range 4–7 years) enrolled for hematopoietic stem cell donation, were used as a control group.
The study was performed according to the Declaration of Helsinki and with the approval of the Institutional Review Board of the Children's Hospital “G. Di Cristina” (registry number 87 Civico 2017). Informed written consent was obtained from the parents and/or legal guardian after receiving information about the study.
Tumor tissue was mechanically dissociated and treated with collagenase type II as previously described (11). Tumor MSCs were expanded following the procedure normally used for BM-MSCs (12). Briefly, cells were plated in flasks or wells (Corning Costar, Corning, NY, USA) according to the cell number obtained, at a density of 160,000/cm2 in complete medium [D-MEM + GlutaMAX (Gibco), supplemented with 10% FBS (Euroclone), 50 mg/ml of gentamicin, and 1% penicillin (Sigma Aldrich)] and cultured at 37°C, 5% CO2.
Culture medium was changed twice a week until ≥80% confluence was reached; then tumor MSCs were trypsinized (Trypsin EDTA, Euroclone) and replated at a density of 4,000 cells/cm2 for expansion (12). Cells were propagated to reach senescence.
As defined by the Mesenchymal and Tissue Stem Cell Committee of the International Society for Cellular Therapy (ISCT), MSCs must be plastic adherent and exhibit a spindle-shape morphology in standard culture conditions. Proliferative capacity was evaluated as cumulative population doubling (cPD) resulting from the sum of PD at each passage calculated with the following formula PD = log10 (no. of harvested cells/no. of seeded cells)/log102.
Tumor MSCs were characterized by flow cytometry, using fluorescein isothiocyanate (FITC)- or phycoerythrin (PE)-labeled monoclonal antibodies specific for surface antigens: CD73, CD34, CD90, CD14, CD45, CD31, CD105, class I-HLA, and HLA-DR (Beckman Coulter, IL, Milan, Italy), as previously described (10). Analysis was performed by direct immunofluorescence with a FACS Navios flow cytometer (Beckman Coulter).
Tumor MSCs were cultured in osteogenic differentiation induction medium [αMEM, 10% FBS, 10−7 M dexamethasone, 50 mg/ml of L-ascorbic acid, and 5 mM β-glycerol phosphate (all from Sigma-Aldrich)], and in adipogenic differentiation medium [αMEM, 10% FBS, 10−7 M dexamethasone, 50 mg/ml of L-ascorbic acid, 5 mM β-glycerol phosphate, 100 mg/ml of insulin, 50 mM isobutyl methylxanthine (all from Sigma-Aldrich), and 0.5 mM indomethacin (MP Biomedica)]. The medium was replaced twice a week. After 21 days of culture, osteogenic differentiation was assessed by staining for alkaline phosphatase (AP) activity with Fast Blue and for calcium deposition, with Alizarin Red S stain (both from Sigma-Aldrich), while adipogenic differentiation was demonstrated by staining of fat droplets with Oil Red O (Bio Optica, Milan, Italy).
Tumor MSC senescence was defined by the β-galactosidase (SA-β-gal) staining Kit (Cell Signaling Technology, Danvers, MA, USA), according to the instructions of the manufacturer. The evaluation of senescence was performed by bright-field microscopy.
Tumor MSCs at P3–P4, were collected after trypsinization. Cell suspensions were centrifuged at 1,200 rpm for 5 min, and then pellets were rinsed twice with phosphate-buffered solution (PBS). After the last centrifugation, 1 × 106 cells were resuspended in 2 ml of DNA staining solution (50 μg/ml of propidium iodide in PBS, 0.1% Igepal, 100 U/ml of RNase type 1A; all reagents from Sigma-Aldrich) and left for 2 h at room temperature before measurement. For the four MSC samples at P3–P4, from healthy hematopoietic stem cell donor (HD), bone marrow was used as a control group.
The cell percentage at each cell cycle phase was evaluated by flow cytometry, as previously described (13). Monoparametric conventional analysis was performed with a Partec PAS II flow cytometer (Sysmex, Milan) using a blue laser and with data recorded on a dedicated computer integrated in the system.
To ensure the best instrumental analytical performance, preliminary alignment, and control were always set up using standard calibration fluorescence beads (Sysmex Ref-4018 KW 160317). The best histogram resolution was achieved measuring a minimum of 50,000 cells. All measurements were performed blindly.
To excite and intercalate propidium iodide into the double-stranded nuclear DNA, the laser line was set at 488 nm, while a 610-nm-long pass filter permitted the selection and measurement of the red fluorescence emission. Data analyses were displayed as frequency histograms of red fluorescence intensities (equivalent to DNA content). Cell cycle analysis and estimation of the three G0/G1, S, and G2/M phases were analyzed with FlowMax software. Cell number in all phases was expressed as cell percentage frequency.
Qualitative variables were described as count and percentage. Comparison between BM-MSCs and tumor MSCs was performed by Fisher exact test. Values of p < 0.05 were considered statistically significant. Analyses were performed using the SPSS statistical package (SPSS, Chicago, IL, USA) and Stata 8.0.
Residual material from solid pediatric tumor biopsies taken for histological analysis was used as the starting material. In each case, MSC expansion was possible, and all MSC cultures met the minimal criteria defined by the ISCT (14).
As preliminarily reported for NB-derived MSCs (NB) (11), tumor MSCs exhibit spindle-shape morphology and are plastic adherent (Figure 1A). Our tumor MSCs expressed the following surface antigens: CD73, CD90, CD105, and HLA-class I ≥95% and CD34, CD14, CD45, CD31, and HLA-DR ≤ 5% (Figure 1B). They presented the capacity to differentiate into osteoblast and adipocytes (Figure 1C). Senescence was reached at a median passage of P11 (range P6–P23) (Figure 1D).
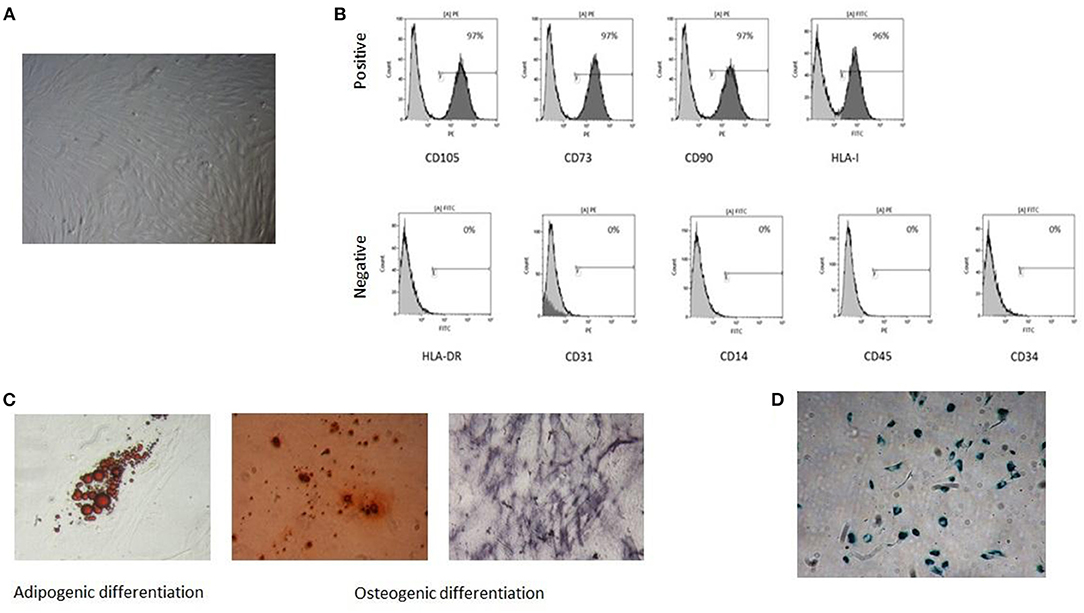
Figure 1. Characterization of tumor mesenchymal stromal cells (MSCs). A representative sample is reported in the figure: (A) spindle-shape morphology, (B) immunophenotype with positive and negative surface antigens, (C) in vitro adipogenic and osteogenic differentiation capacity, (D) senescent tumor MSC at P13.
Tumor MSCs did not exhibit any differences in phenotypical or functional characteristics among the different kinds of tumors.
Blindly evaluated data related to MSC DNA content allowed us to define three different classes of cell cycle behavior. Proliferating samples were defined as cells at the following concentrations: 55–65, 20–25, and 20%, respectively, in G0/G1, S, G2/M. Samples were considered to be in an intermediate cell cycle condition when 70–75% of cells were observed in G1, 10–15% in S, and 10–15% in G2/M. A slow cell cycle had a high cell number in the G1 phase (80–90%) and lower cell percentage in S (5–10%) and G2/M (10–15%) phases, respectively (Figure 2; Table 1).
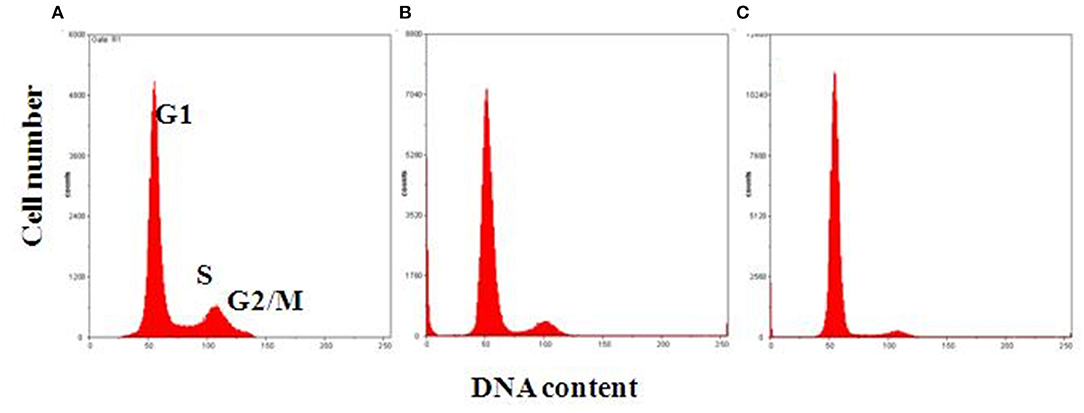
Figure 2. Representative flow cytometric analysis of MSC DNA profiles. Histograms show cell cycle progression in G1, S, and G2/M phases for each representative condition. (A) Proliferating cells with 60% of cells in G1, 20% in S, and 20% in G2/M phase. (B) Intermediate condition of proliferation, with 70% of cells in G1, 15% in S, and 15% in G2/M phase. (C) Slow cell proliferation, with 85% of cells in G1, 5% in S, and 10% in G2/M phase on the x-, y-axes, respectively: DNA content vs. cell number.

Table 1. Definition of the three classes of cell cycle behavior.
Different proliferating cell DNA profiles were noted when comparing BM-MSCs with tumor MSCs. All of the BM-MSCs had a typical DNA profile of proliferating cells, while all tumor-MSC samples had ≥70% of cells detected in the G0/G1 phase. In particular, seven tumor MSC samples (one neuroblastoma, two lymphomas, and four others) displayed intermediate behavior, and the other seven tumor MSC samples (two neuroblastomas, two nephroblastomas, one lymphoma, and one other) were in a slow cell cycle. The distribution of proliferating BM-MSCs and tumor MSCs resulted significantly different between the two groups (p < 0.001) (Figure 3).
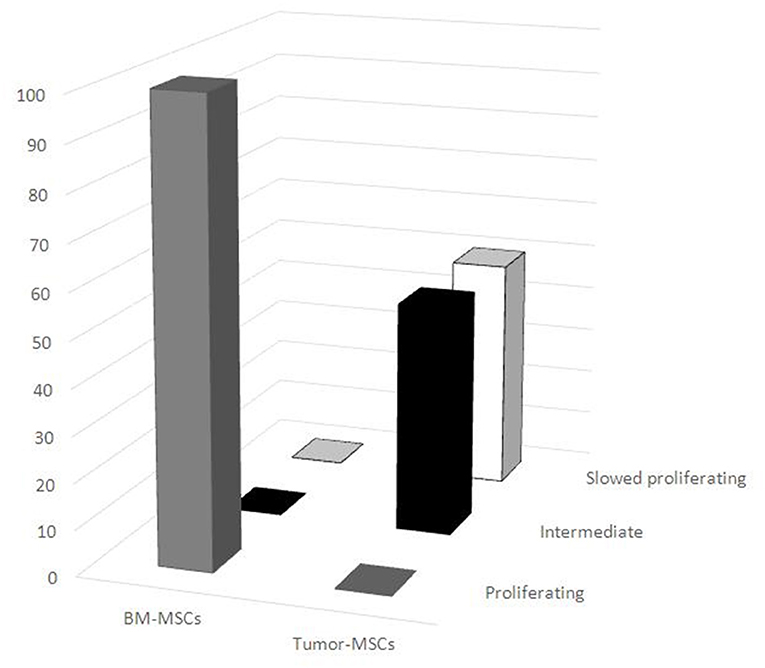
Figure 3. Distribution of proliferating mesenchymal stromal cells (MSCs). Different DNA proliferating cell profiles were noted between BM-MSCs and tumor MSCs. All BM-MSCs expressed the typical DNA profile of proliferating cells, while all tumor MSC samples had a higher percentage of cells detected in the G0/G1 phase. BM-MSC, bone marrow-mesenchymal stromal cells; tumor MSCs, tumor-derived mesenchymal stromal cells.
Tumor relapse and metastasis in some cancers can arise years or decades after initial surgical and medical treatment and are responsible for the majority of cancer-related deaths (15). The identification of predictive markers for recurrence should be crucial to identify a correct surgical strategy. Cancer recurrence has not been fully elucidated. Moreover, tumor dormancy seems to be a critical condition in this phenomenon.
Cancer cell dormancy is defined as a process in which cells enter reversible G0 cell cycle arrest (16), called quiescence. Quiescent cells may acquire additional mutations, survive in a new environment and initiate metastasis, become resistant to chemotherapeutic drugs, and evade immune destruction, thereby influencing cancer progression (16). Different factors have been suggested as contributors to cell dormancy, including complex interactions between metastatic cells and the microenvironment (17–19).
The TME includes endothelial cells, fibroblasts, MSCs, and various immune cells, which are together with cytokines and growth factors embedded in the tumor stroma endowed with specific physical and biomechanical cues (20). It is widely accepted that MSCs participate in each step of tumor development, including relapse and metastasis, due to tumor-homing ability, dynamic phenotype, and immunoregulatory activity (21). It has been reported that tumor MSCs may be driven by the tumor secretome determining their molecular and functional behavior. In vitro tumor MSC angiogenic capacity and tumor growth have been described to be supported by melanoma cells. On the contrary, glioblastoma cells reduced protumorigenic effect (22). Moreover, medullary thyroid carcinoma, human breast carcinoma, and glioblastoma cells determined changes in MSCs, stimulating their inhibitory effect on cell proliferation and tumor growth (23–25).
Several cells, including MSCs, are recruited to the stroma of tumors where they acquire important microenvironmental roles in modulating tumor progression and drug sensitivity (26–28). Even though clear evidence has not been reported, it is known that the reaction of the host to tumors includes the release of proinflammatory cytokines and chemokines, which modulate the TME, support tumor growth, invasion, and metastasis (29–33). However, the crosstalk between tumor and tumor MSCs is sustained by a more complex pattern determined also by the tumor nature (34). It has been shown that tumor cells secreted several factors exerting different effects on the MSC activity and molecular changes bringing alterations in capacity to stimulate tumor growth (22). El-Badawy et al. (10) have recently reported that co-culture of tumor cell lines with BM-MSCs resulted in their phenotypic and functional alteration.
Additionally, regulation and modulation of the cytokine repertoire produced by various tissue-derived MSCs may affect the cancer cell cycle. Fathi et al. (35), reported that cell cycle progression of K562 co-cultured with BM-MSCs resulted in an accumulation of cells in the G0/G1 phase, with slowed entry into the S phase. Fonseka et al. (36) demonstrated that the arrest of K562 growth in the G0/G1 phase was due to the anti-proliferative effect of human umbilical cord blood-derived MSCs.
The role of cancer cells in modulating the cell cycle of MSCs derived from the TME has not been previously considered. A better knowledge of the mechanisms that promote dormant cell cycle arrest could allow to identify new perspectives of study in pediatric surgery. A dedicated surgical strategy for the prevention of cancer recurrence could be defined. In the present study, we noted that tumor MSCs represent a population with phenotypical and functional characteristics of BM-MSCs, with a slow or intermediate cell cycle.
Considering the hypothesis that cytokines and growth factors might be highly involved in the anti-tumor effect mediated by MSCs, a high content of MSCs blocked in the G1 phase, as observed in our study, supports a scenario of “selective” cytokine secretion able to regulate tumor cell arrest in the G0/G1 phase, thereby inducing cancer dormancy (37). As documented by Li et al. (38), senescent MSCs may alter the tissue microenvironment and affect nearby malignant cells via cytokine secretion. In the same way, quiescent MSCs could have tumor-regulating effects.
Additionally, as reported by El-Badawy et al. (10), similar to cancer-induced stem cells, tumor-derived MSCs are slow cycling upon exposure to cancer cell-secreted factors. However, further studies are mandatory to support these hypotheses.
We recognize that this study has some limitations. The sample size was small, but the results were consistent with those of other studies investigating this specific population. The study was limited to characterizing pediatric tumor-derived MSCs, without investigation of the regulation mechanisms involved in cancer cellular processes. Additionally, our cases were not age-matched with controls; as reported (39), the same and identical results were found irrespective of whether matching or not matching was applied. Thus, it is possible that age and underlying condition may affect the outcomes studied. Finally, the follow-up of patients was not adequate to provide any correlation between the tumor MSC effect and tumor prognosis.
Despite these limitations, our data support a role for tumor MSCs in the cross talk between cancer cells and their microenvironment and promotion of cancer dormancy.
In conclusion, we characterized the proliferation pattern of pediatric tumor-derived MSCs. The increased number of tumor MSCs in the G0–G1 phase compared with BM-MSCs supports the role of quiescent MSCs in tumor dormancy regulation. Further studies focusing on the mechanisms, which enable a dormant cell cycle, are needed.
New predictive markers of risk recurrence should be evaluated as an innovative perspective of surgical strategy in children.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Istitutional Review Board of the Children's Hospital G. Di Cristina (registry number 87 Civico 2017). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
GP, FR, MA, GM, and VC conceptualized the study. GP, FR, SC, MA, GA, EM, AS, PB, GZ, GM, and VC developed the methodology. AS performed the formal analysis. SC, MA, and GA were in charge of the investigation. GP, FR, MA, SC, GA, AS, EM, PB, GM, and VC prepared and wrote the original draft. GP, FR, MA, EM, PB, GZ, GM, and VC wrote, reviewed, and edited the manuscript. GP, FR, MA, EM, PB, GZ, GM, and VC supervised the study. All authors have read and agreed to the published version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank Dr. E. Lenta for technical assistance, Dr. L. Kelly for English revision, and the Fondazione Sicilia, Palermo, Italy, for supporting research in Pediatric Surgery.
1. Gatta G, Capocaccia R, Coleman MP, Ries LA, Berrino F. Childhood cancer survival in Europe and the United States. Cancer. (2002) 95:1767–72. doi: 10.1002/cncr.10833
2. Kaatsch P. Epidemiology of childhood cancer. Cancer Treat Rev. (2010) 36:277–85. doi: 10.1016/j.ctrv.2010.02.003
4. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics 2010. CA Cancer J Clin. (2011) 61:133–4. doi: 10.3322/caac.20106
5. Edwards BK, Howe HL, Ries LA, Thun MJ, Rosenberg HM, Yancik R, et al. Annual report to the nation on the status of cancer, 1973-1999, featuring implications of age and aging on U.S. Cancer burden. Cancer. (2002) 94:2766–92. doi: 10.1002/cncr.10593
6. Bhakta N, Liu Q, Ness KK, Baassiri M, Eissa H, Yeo F, et al. The cumulative burden of surviving childhood cancer: an initial report from the St Jude Lifetime Cohort Study (SJLIFE). Lancet. (2017) 390:2569–82. doi: 10.1016/S0140-6736(17)31610-0
7. Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. (2010) 9:665–74. doi: 10.1038/nrc2714
8. Polak R, de Rooij B, Pieters R, den Boer ML. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood. (2015) 126:2404–14. doi: 10.1182/blood-2015-03-634238
9. Iwamoto S, Mihara K, Downing JR, Pui CH, Campana D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest. (2007) 117:1049–57. doi: 10.1172/JCI30235
10. El-Badawy A, Ghoneim MA, Gabr MM, Salah RA, Mohamed IK, Amer M, et al. Cancer cell-soluble factors reprogram mesenchymal stromal cells to slow cycling, chemoresistant cells with a more stem-like state. Stem Cell Res Ther. (2017) 8:254. doi: 10.1186/s13287-017-0709-9
11. Pelizzo G, Veschi V, Mantelli M, Croce S, Di Benedetto V, D'Angelo P, et al. Microenvironment in neuroblastoma: isolation and characterization of tumor-derived mesenchymal stromal cells. BMC Cancer. (2018) 18:1176. doi: 10.1186/s12885-018-5082-2
12. Bernardo ME, Avanzini MA, Perotti C, Cometa AM, Moretta A, Lenta E, et al. Optimization of in vitro expansion of human multipotent mesenchymal stromal cells for cell-therapy approaches: further insights in the search for a fetal calf serum substitute. J Cell Physiol. (2007) 211:121–30. doi: 10.1002/jcp.20911
13. Danova M, Riccardi A, Ucci G, Luoni R, Giordano M, Mazzini G. Ras Oncogene expression and DNA content in plasma cell dyscrasias. A flow cytometric study. Br J Cancer. (1990) 62:781–5. doi: 10.1038/bjc.1990.379
14. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. (2006) 8:315–7. doi: 10.1080/14653240600855905
15. Gao XL, Zhang M, Tang YL, Liang XH. Cancer cell dormancy: mechanisms and implications of cancer recurrence and metastasis. Onco Targets Ther. (2017) 10:5219–28. doi: 10.2147/OTT.S140854
16. Recasens A, Munoz L. Targeting cancer cell dormancy. Trends Pharmacol Sci. (2019) 40:128–41. doi: 10.1016/j.tips.2018.12.004
17. Bahmad HF, Chamaa F, Assi S, Chalhoub RM, Abou-Antoun T, Abou-Kheir W. Cancer stem cells in neuroblastoma: expanding the therapeutic frontier. Front Mol Neurosci. (2019) 12:131. doi: 10.3389/fnmol.2019.00131
18. Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer. (2018) 18:669–80. doi: 10.1038/s41568-018-0056-x
19. Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci. (2018) 25:20. doi: 10.1186/s12929-018-0426-4
20. Ye J, Wu D, Wu P, Chen Z. Huang J. The cancer stem cell niche: cross talk between cancer stem cells and their microenvironment. Tumor Biol. (2014) 35:3945–51. doi: 10.1007/s13277-013-1561-x
21. Han Z, Jing Y, Zhang S, Liu Y, Sh Y, Wei L. The role of immunosuppression of mesenchymal stem cells in tissue repair and tumor growth. Cell Biosci. (2012) 2:8. doi: 10.1186/2045-3701-2-8
22. Kucerova L, Zmajkovic J, Toro L, Skolekova S, Demkova L, Matuskova M. Tumor-driven molecular changes in human mesenchymal stromal cells. Cancer Microenviron. (2015) 8:1–14. doi: 10.1007/s12307-014-0151-9
23. Kucerova L, Matuskova M, Hlubinova K, Altanerova V, Altaner C. Tumor cell behaviour modulation by mesenchymal stromal cells. Mol Cancer. (2010) 9:129. doi: 10.1186/1476-4598-9-129
24. Kucerova L, Matuskova M, Hlubinova K, Bohovic R, Feketeova L, Janega P, et al. Bystander cytotoxicity in human medullary thyroid carcinoma cells mediated by fusion yeast cytosine deaminase and 5-fluorocytosine. Cancer Lett. (2011) 311:101–12. doi: 10.1016/j.canlet.2011.07.014
25. Kucerova L, Skolekova S, Matuskova M, Bohac M, Kozovska Z. Altered features and increased chemosensitivity of human breast cancer cells mediated by adipose tissue-derived mesenchymal stromal cells. BMC Cancer. (2013) 13:535. doi: 10.1186/1471-2407-13-535
26. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
27. Pisco AO, Huang S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: ‘what does not kill me strengthens me’. Brit J Cancer. (2015) 112:1725–32. doi: 10.1038/bjc.2015.146
28. Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. (2013) 32:303–15. doi: 10.1007/s10555-012-9415-3
29. Shalapour S, Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest. (2015) 125:3347–55. doi: 10.1172/JCI80007
30. Hanahan D. Weinberg RA. The hallmarks of cancer. Cell. (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
31. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
32. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. (2009) 30:1073–81. doi: 10.1093/carcin/bgp127
33. Brown J M., Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev. Cancer. (2004) 4:437–47. doi: 10.1038/nrc1367
34. Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE. (2009) 4:e4992. doi: 10.1371/journal.pone.0004992
35. Fathi E, Farahzadi R, Valipour B, Sanaat Z. Cytokines secreted from bone marrow derived mesenchymal stem cells promote apoptosis and change cell cycle distribution of K562 cell line as clinical agent in cell transplantation. PLoS ONE. (2019) 14:e0215678. doi: 10.1371/journal.pone.0215678
36. Fonseka M, Ramasamy R, Tan BC, Seow HF. Human umbilical cord blood-derived mesenchymal stem cells (hUCB-MSC) inhibit the proliferation of K562 (human erythromyeloblastoid leukaemic cell line). Cell Biol Int. (2012) 36:793–801. doi: 10.1042/CBI20110595
37. Ingangi V, Minopoli M, Ragone C, Motti ML, Carriero MV. Role of microenvironment on the fate of disseminating cancer stem cells. Front Oncol. (2019) 9:82. doi: 10.3389/fonc.2019.00082
38. Li G, Zhang R, Zhang X, Shao S, Hu F, Feng Y. Human colorectal cancer derived-MSCs promote tumor cells escape from senescence via P53/P21 pathway. Clin Transl Oncol. (2019) 22:503–11. doi: 10.1007/s12094-019-02152-5
Keywords: mesenchymal stromal cells, proliferation pattern, cell cycle, tumor, children
Citation: Pelizzo G, Riva F, Croce S, Avanzini MA, Acquafredda G, de Silvestri A, Mazzon E, Bramanti P, Zuccotti G, Mazzini G and Calcaterra V (2021) Proliferation Pattern of Pediatric Tumor-Derived Mesenchymal Stromal Cells and Role in Cancer Dormancy: A Perspective of Study for Surgical Strategy. Front. Pediatr. 9:766610. doi: 10.3389/fped.2021.766610
Received: 29 August 2021; Accepted: 01 October 2021;
Published: 04 November 2021.
Edited by:
Luca Pio, Giannina Gaslini Institute (IRCCS), ItalyReviewed by:
Francesco Molinaro, University of Siena, ItalyCopyright © 2021 Pelizzo, Riva, Croce, Avanzini, Acquafredda, de Silvestri, Mazzon, Bramanti, Zuccotti, Mazzini and Calcaterra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gloria Pelizzo, Z2xvcmlhcGVsaXp6b0BnbWFpbC5jb20=
†These authors have contributed equally to this work
‡These authors have jointly supervised this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.