
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 05 January 2022
Sec. Pediatric Neurology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.755344
 Juan Xiong1,2
Juan Xiong1,2 Zhonghua Liu3
Zhonghua Liu3 Shimeng Chen1,2
Shimeng Chen1,2 Miriam Kessi1,2
Miriam Kessi1,2 Baiyu Chen1,2
Baiyu Chen1,2 Haolin Duan1,2
Haolin Duan1,2 Xiaolu Deng1,2Lifen Yang1,2
Xiaolu Deng1,2Lifen Yang1,2 Jing Peng1,2
Jing Peng1,2 Fei Yin1,2*
Fei Yin1,2*Objective: Vitro functional analyses of KCNB1 variants have been done to disclose possible pathogenic mechanisms in KCNB1-related neurodevelopmental disorder. “Complete or partial loss of function (LoF),” “dominant-negative (DN) effect” are applied to describe KCNB1 variant's molecular phenotypes. The study here aimed to investigate clinical presentations and variant effects associations in the disorder.
Methods: We reported 10 Chinese pediatric patients with KCNB1-related neurodevelopmental disorder here. Functional experiments on newly reported variants, including electrophysiology and protein expression, were performed in vitro. Phenotypic, functional, and genetic data in the cohort and published literature were collected. According to their variants' molecular phenotypes, patients were grouped into complete or partial LoF, and DN effect or non-dominant-negative (non-DN) effect to compare their clinical features.
Results: Nine causative KCNB1 variants in 10 patients were identified in the cohort, including eight novel and one reported. Epilepsy (9/10), global developmental delay (10/10), and behavior issues (7/10) were common clinical features in our patients. Functional analyses of 8 novel variants indicated three partial and five complete LoF variants, five DN and three non-DN effect variants. Patient 1 in our series with truncated variants, whose functional results supported haploinsufficiency, had the best prognosis. Cases in complete LoF group had earlier seizure onset age (64.3 vs. 16.7%, p = 0.01) and worse seizure outcomes (18.8 vs. 66.7%, p = 0.03), and patients in DN effect subgroup had multiple seizure types compared to those in non-DN effect subgroup (65.5 vs. 30.8%, p = 0.039).
Conclusion: Patients with KCNB1 variants in the Asian cohort have similar clinical manifestations to those of other races. Truncated KCNB1 variants exhibiting with haploinsufficiency molecular phenotype are linked to milder phenotypes. Individuals with complete LoF and DN effect KCNB1 variants have more severe seizure attacks than the other two subgroups.
KCNB1 encodes α-subunit of the Kv2.1 channel, which consists of six transmembrane segments (S1–S6), with a voltage-sensing domain (VSD, S1–S4) and ion-conducting pore formed by domain S5 and S6 (1). In vivo, homotetrameric Kv2.1 channels (assembled only by α-subunits) contribute a large portion of delayed rectifier potassium currents in various neuron subtypes, including pyramidal neurons of the hippocampus and cortex (2, 3), while heterotetrameric Kv2.1 channels (co-assembled with α and β-subunits) are also involved in physiological processes such as regulating excitability in mouse dorsal root ganglia neurons (4–6). Since Torkamani et al. (7) firstly correlated epileptic encephalopathy with KCNB1 variants in 2014, about 80 patients with KCNB1-related neurodevelopmental disorders have been reported due to extensive clinical genetic testing (8, 9). Now, the gene is one of the most mutated potassium genes in children with neurodevelopmental disorders. Most patients with KCNB1-related neurodevelopmental disorders have variable epilepsy, intellectual disabilities, psychiatric and behavior problems (9, 10).
Most functional studies in vitro focused on the potassium conductance properties of KCNB1 protein. Researchers have successfully applied high-through automated electrophysiology to analyze the pathogenicity of KCNB1 variants (11). Nevertheless, the differences between the automated and manual methods in the channel have never been studied. Unlike in vivo, assembling Kv2.1 channels with solely KCNB1 wild-type or variant subunits is considered homomeric configuration, while co-assembling with wild-type and variant is considered heteromeric configuration in vitro (11, 12). Now, it has been proven that causative KCNB1 variants result in loss of Kv2.1 channel function, which is classified into “partial loss of function (LoF)” and “complete LoF” according to the amplitude of Kv2.1 currents in homotetrameric expression models (7, 11–13). Like other genetic variants of ionic channels (14), the dominant-negative (DN) phenomenon that variants could interfere with the co-expressed wild type, has also been confirmed as the pathogenesis of the disorder (15). Conversely, those variants that do not have sufficient evidence to support the DN phenomenon could be subgrouped as the non-dominant-negative (non-DN) effect. However, what genetic mechanisms underlying them are unclear. Notwithstanding, whether patients could benefit from the above functional classification remains unknown.
Here, we described detailed clinical and genetic information of 10 patients with KCNB1-related neurodevelopmental disorders from the Chinese population [1 had been described briefly before (16)]. Functional experiments, including manual electrophysiology and western blotting of eight novel KCNB1 variants, were done in vitro to disclose their functional alterations. Combined with previously published phenotypic, functional, and genetic data of the disorder, we linked variant effects to clinical presentations, which was innovative attempts to study the disease.
Children with KCNB1-related neurodevelopmental disorders were enrolled in the study at Xiangya Hospital, Central South University from May 2013 to January 2021. Detailed clinical data of all patients were evaluated by two experienced pediatric neurological doctors, including perinatal history, family history, developmental history, seizure types, neuropsychiatric symptoms, genetic results, video electroencephalography (EEG), neuroimaging, developmental scales, and therapeutic schedules. Follow-up work was undertaken by outpatient or phone call.
Next-generation sequencing of epilepsy panel (17) for 2 cases and whole-exome sequencing for other 8 patients was adopted in the cohort (Table 1). Candidate variants were validated by Sanger sequencing. The data were analyzed as described in previous studies (17, 18). The pathogenicity of these variants was interpreted according to the American College of Medical Genetics (ACMG) guidelines (19).
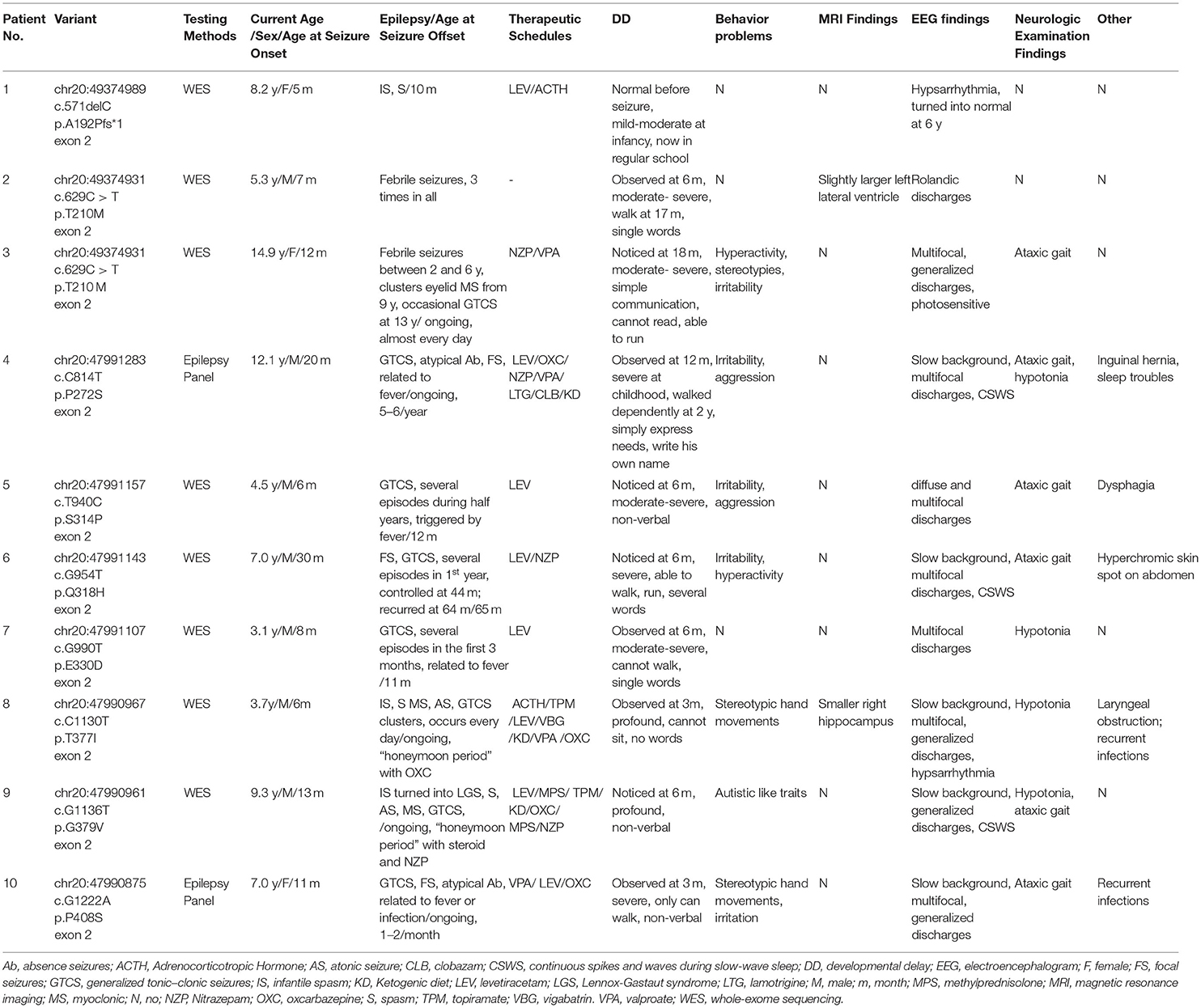
Table 1. Clinical characteristics and genetic information of patients with KCNB1 related-neurodevelopmental disorder reported in our cohort.
According to the manufacturer's instructions, the variant was introduced into the human KCNB1 cDNA (NM_004975) cloned into the pcDNA3.1 vector by site-directed mutagenesis (TOYOBO, Osaka, Japan). A similar transfection protocol was adopted to represent homomeric and heteromeric configurations (11). The total DNA amount was 2ug. Currents from CHO-K1 cells were recorded using an EPC-10 USB patch-clamp amplifier operated by PatchMaster software (HEKA Elektronik, Lambrecht, Germany) room temperature (20–22°C) 24–48 h after transfection. Bath solution contained (in mmol/L) 138 NaCl, 5.4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose and 10 HEPES (pH 7.4 with NaOH); pipette solution contained (in mmol/L): 140 KCl, 2 MgCl2, 10 EGTA, 10 HEPES, 5 Mg-ATP (pH 7.4 with KOH).
The holding potential was−80 mV. A family of square voltage steps consisting of 500 ms depolarizing pulses from−80 mV to +120 mV (10 mV steps) followed by a 100-ms step to−80 mV (tail-currents) was applied. The peak current was measured from−80 mV to +60 mV, normalized for cell capacitance, then plotted against voltage to generate current density–voltage relationships. Voltage-dependence of activation was built-up by plotting tail currents normalized to the largest tail current amplitude against the corresponding test potential and fit with the Boltzmann equation. The cells were depolarized from−80 to +50 mV for 2.0 s, followed by an isopotential pulse at +60 mV of 500 ms duration to generate voltage-dependence of inactivation curves. The current values recorded at the beginning of the +60 mV pulse were measured, normalized, and fit to a Boltzmann distribution.
MinuteTM Plasma Membrane Protein Isolation and Cell Fractionation Kit (Invent, Plymouth, USA) were adopted to separate the total and plasma membrane KCNB1 proteins. We collected the total protein fractions before cellular component separation and isolated plasma membrane proteins according to the manufacturer's protocol. The proteins of CHO-K1 co-expressing variants and wild-type were extracted using ice-cold RIPA buffer with a ratio of 100:1 protease inhibitor cocktails (Beyotime, Shanghai, China). The total amount of plasmid DNA here maintained 4 ug.
20 ug of samples of proteins were separated by 10% sodium dodecyl sulfate– polyacrylamide (SDS-PAGE), transferred to PVDF membranes, blocked in protein free rapid blocking buffer (Epizyme, Shanghai, China), and incubated with mouse anti-Kv2.1 (1:1000; Abcam, ab192761), mouse anti-transferrin receptor (1:500; Invitrogen, #13-6800), rabbit anti-GAPDH antibody (1:2000; Servicebio, GB11002) overnight at 4 °C, then incubated with peroxidase-conjugated goat anti-rabbit IgG (1:10,000 Jackson ImmunoResearch) and goat anti-mouse IgG (1:10,000, Jackson ImmunoResearch) (1h at room temperature). Protein quantitation was completed using ImageJ software (NIH, Bethesda, USA).
PubMed literature search was conducted by combining “KCNB1” or “Kv2.1 channel” and “variants” or “mutations” until 16 July 2021. Articles with functional studies of KCNB1 variants were reviewed carefully. A similar method in previous research was used to classify partial LoF and complete LoF variant (11, 12). In vitro heteromeric expression models, variants that exhibited adverse effects on current amplitudes, or channel kinetics, or protein expression of Kv2.1 wild-type subunit were classified as DN effect variants, otherwise grouped as non-DN variants. Variants with controversial conclusions or unclear classifications were excluded. We analyzed published patients carrying enrolled variants with sufficient clinical information and grouped them to compare their clinical features.
The analysis and display of patch-clamp and western blotting data was performed using GraphPad Prism 8. A one-sided Fisher's exact test with p < 0.05 determining statistical significance in analyzing clinical features including seizure and intellectual disabilities in different subgroups (“complete LoF” subgroup vs. “partial LoF” subgroup, “DN effect” subgroup vs. “non-DN effect” subgroup).
Ten unrelated neurodevelopmental disorder patients with KCNB1 variants, including three females, and seven males, were recruited in the study (Table 1). Except for patient 2 with febrile seizures, the rest were diagnosed with epilepsy. The seizure onset age ranged from 5 to 30 months (median age, 11.5 months), and 6/9 of the patients' epileptic events were related to fever. Generalized tonic-clonic seizures (8), spasms (3), myoclonic seizures (3), focal seizures (3), atonic seizures (2), and atypical absence (2) were observed in our patients. Six cases had two or more seizure types. Three cases were diagnosed as infantile spasms, and one turned into Lennox-Gastaut syndrome. The age at the final follow-up ranged from 3.1 to 14.7 years old. Out of those 9 cases, 4 achieved seizure freedom from 10 to 65 months (median age, 11.5 months). The remaining 5 cases had drug-resistant epilepsy (3 cases with daily seizures and 2 with monthly seizures). Five of the 9 cases were treated with levetiracetam as the initial antiepileptic drug, which was effective for 4 cases. Three patients tried nitrazepam as add-on therapy and obtained seizure reduction. One of the two cases with infantile spasms was responsive to ACTH. Two of the three cases that tried ketogenic diet had significant seizure reductions and cognitive improvements, with more verbal expression in case 4, and more flexible eye contact in case 8.
All patients presented with global developmental delay; Eight displayed before seizures onset while patient one experienced developmental regression after epilepsy. The first noticeable symptom was stunted motor skill development. At the last follow-up, only patient one showed average intelligence level. Eight of the nine cases could walk dependently, but with ataxic gait, patient 7 could walk with support at the age of 2.4 years, and patient 8 could not raise his head at the age of 3.0 years. Patient 1 had a mild speech impediment in her infancy. She could not say “Mam/Dad” until 18 month-old age but now communicate with others normally. Of the rest nine patients, four had no words, two could speak single word, one could say several words, and two could communicate simply with short sentences. Of the 10 cases, seven individuals had behavioral problems, including autistic features in Four, hyperactivity in three, and aggression in three.
Video EEG of all patients showed abnormal profiles. Slow background activity was noticed in 5 out of 10 patients. Continuous spikes and waves during slow-wave sleep (CSWS) was detected in three patients, and hypsarrhythmia was found in two. Diffuse or generalized spike waves and polyspike waves were observed in five patients. Multifocal epileptiform discharges were recorded in seven patients.
Nine KCNB1 variants in our cohort were found, including 8 missense variants (p.T210M, p.P272S, p.S314P, p.Q318H, p.E330D, p.T377I, p.G379V, and p.P408S), and 1 in-frame deletion (p.A192Pfs*1) (Table 1). Eight of them were novel. It is worth noting that variant p.E330D detected here is caused by the 990th base substituted by T, instead of the previously reported C (11.20). p.T210M has been identified in four cases except our two patients, which was a hotspot variant8, 10, 11, 21. According to ACMG guidelines, three variants (p.A192Pfs*1, p.T210M and p.E330D) were classified as pathogenic, and five (p.P272S, p.S314P, p.Q318H, p.G379V, and p.P408S) were likely pathogenic variants and one (p.T377I) was uncertain significance variant. These variants were distributed sporadically in KCNB1 protein, two of which were in S4–S5 linker (p.Q318H and p.E330D), two in S5-S6 linker (p.T377I and p.G379V), and one respectively in domain S1 (p.A192Pfs*1), S1–S2 linker (p.T210M), S3 (p.P272S) and S6 (p.P408S).
Except for p.T201M, whose functional alteration had been disclosed (11), vitro experiments on eight identified novel variants were undertaken in the study. Three partial LoF variants were identified, while one of them, variant p.E330D, was classified as complete LoF in Kang's study; the DN effects of 5 variants were confirmed, including variant p.E330D, whose result here was similar to that in Kang's study (11).
The display of representative whole-cell current density traces of CHO-K1 cells that are transiently expressing mock, wild-type, or variants in Figure 1. The voltage protocols used here for electrophysiology studies are depicted in Figure 2A. The current density comparisons of homomeric and heteromeric channels of all variants are shown in Figure 2B. Variants p.P272S, p.Q318H, and p.E330D were identified as partial LoF variants, while the other five were complete LoF here, for no detectable outward potassium current were recorded. When co-expression with the wild-type subunits, the peak current density of variants p.A192Pfs*1, p.P272S, p.E330D, p. T377I, and p.P408S, significantly increased to >75% of which in cells of wild-type. In contrast, other variants amounted to 50%-75% relative to wild-type, and variant p.G379V seemed to have a dominant-negative effect on the magnitude of wild-type current. The volume and number of CHO-K1 cells included for analyzing the current density of each group are listed in Supplementary Table 1.
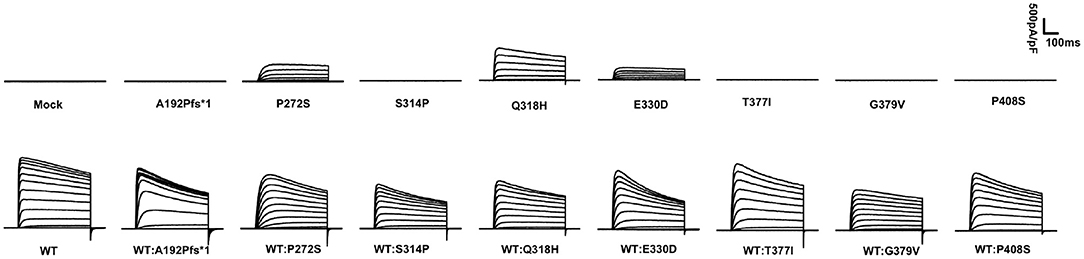
Figure 1. Averaged whole-cell current density traces of CHO-K1 transiently expressing MOCK or KCNB1 subunits.
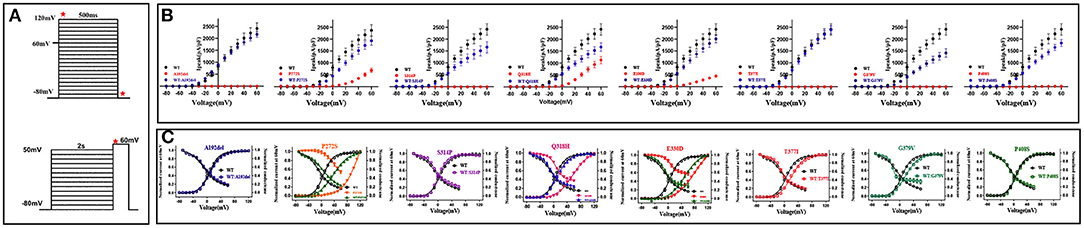
Figure 2. Biophysical properties of KCNB1 variants detected in the study. (A) The voltage protocols used for electrophysiological studies are depicted. In the above image, Ipeak densities of KCNB1 variants were recorded at a voltage from−80 to +60 mV, activation curves were generated at a voltage from−80 to +120 mV. Inactivation curves were recorded as the below figure. Red stars indicate time points of the current measurement. (B) Comparison of Ipeak density of KCNB1 variants in homomeric and heteromeric configurations. (C) Voltage-dependence of activation/inactivation curves of KCNB1 variants in homomeric and (or) heteromeric models.
Voltage-dependence of channel activation and inactivation of these variants were also conducted (Figure 2C). The parameters of voltage dependence curves are summarized in Supplementary Table 2. The three partial LoF variants displayed apparent depolarizing shifts in both V50 of channel activation and channel inactivation in homomeric models; but these changes were ameliorated when co-expressed with the wild-type subunit (Supplementary Table 2). Compared with the wild-type, complete LoF variants p.A192Pfs*1, p.S314P, and p.P408S in heteromeric configurations exhibited similar channel activation and inactivation kinetics. Conversely, the other two variants, p.T377I and p.G379V with complete LoF showed depolarizing shifts in the V50 of channel activation, and the latter also caused a hyperpolarizing shift in the V50 of voltage dependence of channel inactivation, indicating that the two variants have dominant-negative effects on the channel dynamics of the wild-type subunit.
The KCNB1 protein expression of all variants was lower than the wild-type subunit in homomeric models. The complete LoF variants, expect for variant p.P408S, in the study exhibited less protein expression than partial LoF variants in homomeric configurations (Figures 3A,D). Unlike other variants, p.P408S had higher protein expression in the homologous model than in the heterologous model, indicating a dominant-negative effect on wild-type protein expression (Figures 3B,D). Noticeably, variant p.A192Pfs*1 led to a truncated protein due to premature stop codon but showed nearly regular KCNB1 protein expression in heteromeric channels. Variant p.A192Pfs*1, p.S314P, p.Q318H, p.T377I, and p.G379V displayed decreased membrane expression than wild-type, suggesting defective efficient cell-surface trafficking of these variants; while the remaining had similar surface trafficking efficiencies (Figures 3C,E).
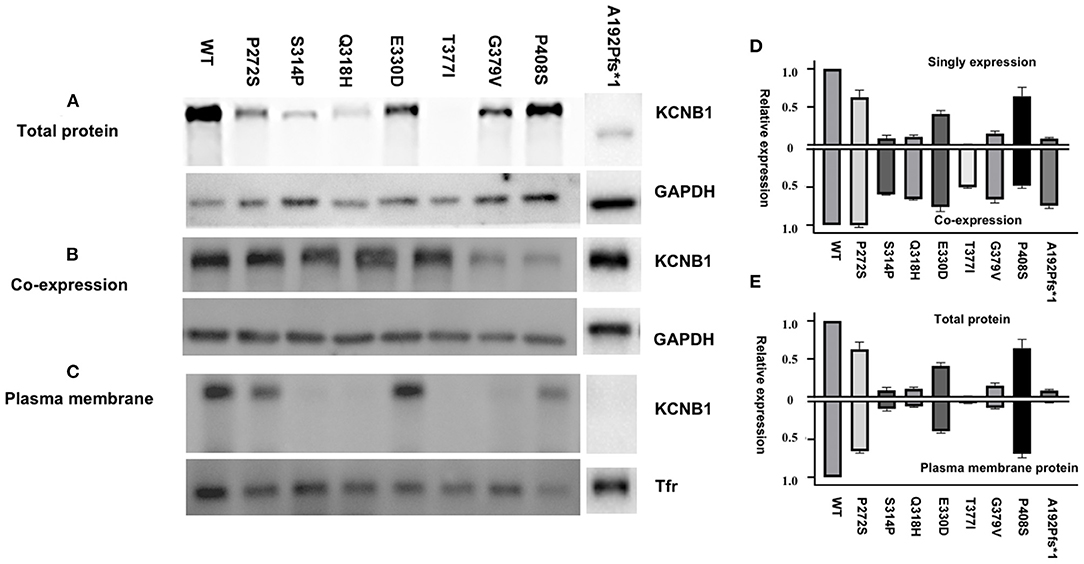
Figure 3. Total and cell-surface protein expression levels of KCNB1 variants. (A,C) Mutant Kv2.1 proteins were expressed and trafficked to the cell surface. (B) Representative western blot of total protein expression of KCNB1 variants in heteromeric models. (D) Comparison of KCNB1 protein expression levels of detected variants and wild-type in both configurations. (E) Plasma membrane trafficking efficiency of Kv2.1 channels in CHO-K1 transfected with KCNB1 variants compared to the wild-type.
Thirty-one KCNB1 variants (23 from the published literature and eight newly reported from this study) with functional results were found (Supplementary Table 3) (7–13, 20–28). After excluding controversial variants and individuals with insufficient clinical descriptions, 33 patients with 19 complete LoF variants (Complete LoF group) and 13 patients with 6 partial LoF variants (partial LoF group), 30 patients with 16 DN effect KCNB1 variants (DN effect group) and 18 patients with 10 non-DN effect KCNB1 variants (non-DN effect group) were included for further analyses (Supplementary Table 3). In all, patients in complete LoF subgroup had earlier seizure onset age (64.3 vs. 16.7%, p = 0.007), worse seizure outcomes (18.8 vs. 66.7%, p = 0.025), and more kinds of seizure types (58.7 vs. 25.0%, p = 0.052) than those in partial LoF subgroup. Furthermore, patients older than 2-year-old in partial LoF group were more likely to have the ability to walk (90.9 vs. 60.0%, p = 0.077) (Table 2). Compared to non-DN effect group, more individuals in DN effect group had epilepsy (90.0 vs. 77.8%, p = 0.227), more than 3 kinds of seizure types (65.5 vs. 30.8%, p=0.039), and less achieved seizure freedom (17.6 vs. 30.0%, p = 0.387). Similarly, patients older than 2-year-old in the non-DN effect group were prone to walk (81.8 vs. 64.7%, p = 0.296) (Table 3). Patients from different subgroups exhibited indistinguishable language development levels and incidence rates of behavioral or psychiatric characteristics.
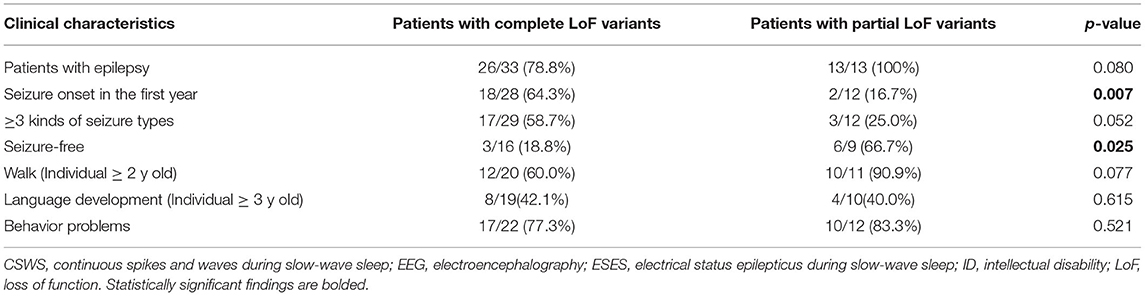
Table 2. Comparisons of patients with partial and complete LoF KCNB1 variants in clinical features.

Table 3. Comparisons of patients with DN and non-DN effect KCNB1 variants in clinical features.
To the best of our knowledge, this is the first Asian cohort study of KCNB1-related neurodevelopmental disorder. We reached similar conclusions from the previous reports; its clinical manifestations include variable epilepsy, global developmental delay, and behavior issues (8–10), indicating no ethnic differences. Ninety percent of patients in the study had epilepsy, 55.6% were recorded to have more than three seizure types, and 44.4% achieved seizure free, as per the last follow-up. The seizure outcomes in disorder were quite variable. It was worth noting that 5/9 cases had a history of fever-induced or fever-related epileptic events, incredibly patient 2 who was diagnosed with febrile convulsion rather than epilepsy. Unlike sodium channel genes such as SCN1A (29), little attention has been paid to the relationship between hyperthermia and seizure occurrence in the disease. Our findings suggest that seizures associated with fever in the early stages of the disease might be an alarm for this condition.
Based on our clinical observations and previous literature, the global developmental delay seems to be more prominent and shared in the disease, which often occurred in the first year of life (8–10). Marini et al. (21) put forward that affected patients would still have moderate or severe cognitive impairment whether the seizure is controlled or not in the long-term follow-up. Strikingly, we identified a patient with a truncated variant who had a similar developmental level as her peers (further discussion on this case can be found in the next part). About 80% of the patients in previous series had behavioral problems including autistic traits, aggression, and hyperactivity (8–10), which is consistent with our results. The typical EEG pattern in the research was slow background activity with multifocal, diffuse, or generalized epileptiform discharges. Malignant EEG patterns, including hypsarrhythmia and CSWS/ESES, were also detected in previously reported patients (8).
Functional analyses of 8 novel KCNB1 variants by manual electrophysiology were conducted in the study, while19 KCNB1 variants using automated patch clamp recording in Kang's study (11). We compared their results with ours and found some controversies. Firstly, in our study, partial LoF variant p.E330D was classified into complete LoF variant in Kang's study, so did p.R306C and p.V378A (11, 13, 26). However, the recorded Kv2.1 currents by the traditional way is much larger than that of the new method. After careful comparison, we found that the current expression of these three partial LoF variants is <25% of the Kv2.1 wild-type. Thus this tiny current might be neglected in automated electrophysiology.
Secondly, Yu et al. proposed that the variant p.R312H has a DN effect on the voltage gate of activation, contrary to Kang's findings (11, 22). Despite the classification divergence, the variant p.E330D in our study and Kang's study had a similar assumption: DN effect (11). Based on the above analyses, these results are beyond our ability to explain why such differences exist in traditional and new methods. Further research is needed to unravel this mystery.
Researchers and clinicians are inquisitive about the possible link between the phenotype and genotype in single-gene inherited diseases. Therefore, introducing the molecular phenotype of genetic variants into analysis might be helpful to understand the relationship better. So far, some correlations have been unveiled. For example, the mixed dysfunctional of LoF/GoF KCNA2 variants are associated to more severe phenotypes than simple LoF or GoF variants (30). According to our and previous researches, mutated Kv2.1 channels often result in loss-of-function of the protein. Calhoun et al. (12) reported the first partial LoF KCNB1 variant p.I199F and suggested that the degree of KCNB1 protein dysfunction is associated with disease severity. However, this view has not been well proved. Here, we concluded and compared clinical characteristics of patients with complete and partial LoF variants, DN and non-DN effect variants, and got some hallmarks about clinical manifestations among different variant effect subgroups.
Firstly, compared with that in complete LoF subgroup, patient in the partial LoF subgroup seemed to have a less severe epilepsy phenotype, with later seizure onset age, fewer seizure types, and better seizure outcomes. Moreover, patients in the DN effect group had a higher rate of epilepsy and multiple seizure types and a lower probability of obtaining seizure-free. From this perspective, our finding was in line with previous assumption that the epilepsy phenotype might be linked to the extent of Kv2.1 channel impairments caused by variants (8). However, we have not found the correlations between intellectual development or behavior characteristics and variant effects.
Even though we got some links between phenotype and genotype by analyzing the molecular phenotype of KCNB1 variants in disorder, larger sample cohort studies and more functional research are invited to confirm these relationships.
DN effect is a classical molecular mechanism in genetic diseases, which is defined as the variant that will adversely affect the co-expressed wild-type activity. Many KCNB1 variants have disease effects in this way, such as the well-validated variant p.G379R, which interferes with wild-type functions in both potassium conductance and non-conductance aspects (7, 15). We found that not all variants could be considered to have DN effects according to their functional results. On the one hand, previous work has been mainly focused on unveiling their deficits in potassium conductance rather than their non-conductance roles (15). Here, we added an experiment on the KCNB1 protein expression in the heteromeric models, which aided us in identifying p.V408S as a DN effect variant. Thus, some KCNB1 variants with DN effects may be masked due to lacking of sufficient experimental evidence.
On the other hand, haploinsufficiency could be another possible mechanism in the disorder. In our cohort, patient 1 with a truncated variant p.A192Pfs*1 almost got remission from the diseases—no epileptic seizure and normal school performances, which is the best prognosis so far. As almost all reported truncated KCNB1 variants, including ours, located in the last exon of the gene, they were presumed to generate truncated proteins rather than undergoing nonsense-mediated mRNA decay. Functional data of variant p.A192Pfs*1 in the study supported the assumption. Truncated KCNB1 variants have been reported to relate to the milder phenotype than missense variants, especially in Bar et al. and colleagues' study, they disclosed that there are statistical differences in epilepsy between patients with missense variants and truncated variants (8–10). The possible underlying pathophysiological explanation for such discrepancy could be that the two mutant types do not share the same molecular mechanism. Researchers uncovered that KCNQ2 variants leading to haploinsufficiency often relate to a milder phenotype—benign familial neonatal seizures, while variants with DN effect are inclined to a severer phenotype—epileptic encephalopathy (31). A similar phenomenon might also exist in patients with KCNB1 variants. Meanwhile, the current functional results of all truncated variants (p.A192fs*1, Y274fs*36, and Y533X) tended to support the mechanism of haploinsufficiency rather than the negative-dominance phenomenon (11).
Although we successfully applied functional data to establish phenotype and genotype correlations in the present study, there are some limitations. The biggest problem is that the functional data we used here is partly from literature, and some of them vary among different laboratories. However, those controversial data were excluded before the analyses. Secondly, all experiments in the study were performed in the CHO-K1 cell line, which could not well reflect functional alterations caused by KCNB1 variants in vivo. Future studies should be taken on patient-derived induced pluripotent stem cells (iPSC) or in vivo to better understand the disorder's pathogenesis (32, 33).
In the study, we found that patients with KCNB1 variants in the Asian cohort have similar clinical manifestations as that of other regions: variable seizures, psychomotor developmental delay, and behavior problems. Molecular phenotypes of KCNB1 variants through some functional analyses aid in establishing genotype and phenotype linkage in the diseases. Truncated KCNB1 variants leading to haploinsufficiency are related to milder phenotypes. KCNB1 variants with complete LoF and DN effect have more severe epilepsy.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of Xiangya Hospital, Central South University, Changsha, Hunan (201605585). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
FY and JX: conception of the study. JX, JP, and ZL: experimental design and study designation. SC, HD, XD, LY, and BC: patient's evaluation. JX, SC, and BC: experiments. JX, HD, SC, and ZL: data acquisition and analysis. JX, MK, and FY: first version of the manuscript. JX, MK, LY, and FY: revised version of the manuscript. All authors read and approved the final version of the manuscript.
This research was funded by the National Natural Science Foundation of China [NO. 81771408], the Graduate Innovation Program of Central South University [NO. 2019zzts338] and Hunan Province Key Field Research and Development Plan [NO. 2019SK2081].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to express our gratitude to the patients and families for participating in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.755344/full#supplementary-material
1. Jedrychowska J, Korzh V. Kv21 voltage-gated potassium channels in developmental perspective. Dev Dyn. (2019) 248:1180–94. doi: 10.1002/dvdy.114
2. Mohapatra DP, Misonou H, Pan SJ, Held JE, Surmeier DJ, Trimmer JS. Regulation of intrinsic excitability in hippocampal neurons by activity-dependent modulation of the Kv21 potassium channel. Channels. (2009) 3:46–56. doi: 10.4161/chan.3.1.7655
3. Du J, Phillips-Tansey E, Russell JT, Russell JT, McBain CJ. Frequency-dependent regulation of rat hippocampal somato-dendritic excitability by the K+ channel subunit Kv21. J Physiol. (2000) 522:19–31. doi: 10.1111/j.1469-7793.2000.t01-2-00019.xm
4. David JP, Stas JI, Schmitt N, Bocksteins E. Auxiliary KCNE subunits modulate both homotetrameric Kv21 and heterotetrameric Kv21/Kv64 channels. Sci Rep. (2015) 5:12813. doi: 10.1038/srep12813
5. Bocksteins E, Snyders DJ. Electrically silent Kv subunits: their molecular and functional characteristics. Physiology. (2012) 27:73–84. doi: 10.1152/physiol.00023.2011
6. Bocksteins E, Raes AL, Van de Vijver G, Bruyns T, Van Bogaert PP, Snyders DJ. Kv21 and silent Kv subunits underlie the delayed rectifier K+ current in cultured small mouse DRG neurons. Am J Physiol Cell Physiol. (2009) 296:C1271–8. doi: 10.1152/ajpcell.00088.2009
7. Cherkasov A, Muratov EN, Fourches D, McKeown SE, Pendziwiat M, Helbig KL, et al. De Novo KCNB1 mutations in epileptic encephalopathy. Ann Neurol. (2014) 76:529–40.
8. Bar C, Kuchenbuch M, Barcia G, Schneider A, Jennesson M, Le Guyader G, et al. Developmental and epilepsy spectrum of KCNB1 encephalopathy with long-term outcome. Epilepsia. (2020) 61:2461–73. doi: 10.1111/epi.16679
9. Bar C, Barcia G, Jennesson M, Le Guyader G, Schneider A, Mignot C, et al. Expanding the genetic and phenotypic relevance of KCNB1 variants in developmental and epileptic encephalopathies: 27 new patients and overview of the literature. Hum Mutat. (2020) 41:69–80. doi: 10.1002/humu.23915
10. De Kovel CGF, Syrbe S, Brilstra EH, Verbeek N, Kerr B, Dubbs H, et al. Neurodevelopmental disorders caused by de novo variants in KCNB1 genotypes and phenotypes. JAMA Neurol. (2017) 74:1228–36. doi: 10.1001/jamaneurol.2017.1714
11. Kang SK, Vanoye CG, Misra SN, Echevarria DM, Calhoun JD, O'Connor JB, et al. Spectrum of Kv21 dysfunction in KCNB1-associated neurodevelopmental disorders. Ann Neurol. (2019) 86:899–912. doi: 10.1002/ana.25607
12. Calhoun JD, Vanoye CG, Kok F, George AL, Kearney JA. Characterization of a KCNB1 variant associated with autism, intellectual disability, and epilepsy. Neurol Genet. (2017) 3:e198. doi: 10.1212/NXG.0000000000000198
13. Saitsu H, Akita T, Tohyama J, Goldberg-Stern H, Kobayashi Y, Cohen R, et al. De novo KCNB1 mutations in infantile epilepsy inhibit repetitive neuronal firing. Sci Rep. (2015) 5:15199. doi: 10.1038/srep15199
14. Sottas V, Abriel H. Negative-dominance phenomenon with genetic variants of the cardiac sodium channel Nav15. Biochim Biophys Acta. (2016) 1863:1791–8. doi: 10.1016/j.bbamcr.2016.02.013
15. Hawkins NA, Misra SN, Jurado M, Kang SK, Vierra NC, Nguyen K, et al. Epilepsy and neurobehavioral abnormalities in mice with a dominant-negative KCNB1 pathogenic variant. Neurobiol Dis. (2021) 147:105141. doi: 10.1016/j.nbd.2020.105141
16. Miao P, Peng J, Chen C, Gai N, Yin F. A novel mutation in KCNB1 gene in a child with neuropsychiatric comorbidities with both intellectual disability and epilepsy and review of literature. Zhonghua Er Ke Za Zhi. (2017) 55:115–9. doi: 10.3760/cma.j.issn.0578-1310.2017.02.012
17. Peng J, Pang N, Wang Y, Wang XL, Chen J, Xiong J, et al. Next-generation sequencing improves treatment efficacy and reduces hospitalization in children with drug-resistant epilepsy. CNS Neurosci Ther. (2019) 25:14–20. doi: 10.1111/cns.12869
18. Peng J, Wang Y, He F, Chen C, Wu LW, Yang LF, et al. Novel West syndrome candidate genes in a Chinese cohort. CNS Neurosci Ther. (2018) 24:1196–206. doi: 10.1111/cns.12860
19. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
20. Miao P, Feng J, Guo Y, Wang J, Xu X, Wang Y, et al. Genotype and phenotype analysis using an epilepsy-associated gene panel in Chinese pediatric epilepsy patients. Clin Genet. (2018) 94:512–20. doi: 10.1111/cge.13441
21. Marini C, Romoli M, Parrini E, Costa C, Mei D, Mari F, et al. Clinical features and outcome of 6 new patients carrying de novo KCNB1 gene mutations. Neurol Genet. (2017) 3:e206. doi: 10.1212/NXG.0000000000000206
22. Yu W, Shin MR, Sesti F. Complexes formed with integrin-α5 and KCNB1 potassium channel wild type or epilepsy-susceptibility variants modulate cellular plasticity via Ras and Akt signaling. FASEB J. (2019) 33:14680–89. doi: 10.1096/fj.201901792R
23. de Kovel CGF, Brilstra EH, van Kempen MJA, Van't Slot R, Nijman IJ, Afawi Z, et al. Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients. Mol Genet Genomic Med. (2016) 4:568–80. doi: 10.1002/mgg3.235
24. Samanta D. Epileptic spasm and de novo KCNB1 mutation: if it is not one potassium channel, it is another! Acta Neurol Belg (2020) 120:417–20. doi: 10.1007/s13760-018-0950-8
25. Allen NM, Conroy J, Shahwan A, Lynch B, Correa RG, Pena SD, et al. Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion. Epilepsia. (2016) 57:e12–7. doi: 10.1111/epi.13250
26. Thiffault I, Speca DJ, Austin DC, Cobb MM, Eum KS, Safina NP, et al. A novel epileptic encephalopathy mutation in KCNB1 disrupts Kv21 ion selectivity, expression, and localization. J Gen Physiol. (2015) 146:399–410. doi: 10.1085/jgp.201511444
27. Latypova X, Matsumoto N, Vinceslas-Muller C, Bézieau S, Isidor B, Miyake N. Novel KCNB1 mutation associated with non-syndromic intellectual disability. J Hum Genet. (2017) 62:569–73. doi: 10.1038/jhg.2016.154
28. Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature. (2015) 519:223–8. doi: 10.1038/nature14135
29. Catterall WA. Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu Rev Pharmacol Toxicol. (2014) 54:317–38. doi: 10.1146/annurev-pharmtox-011112-140232
30. Döring JH, Schröter J, Jüngling J, Biskup S, Klotz KA, Bast T, et al. Refining genotypes and phenotypes in KCNA2-related neurological disorders. Int J Mol Sci. (2021) 22:2824. doi: 10.3390/ijms22062824
31. Orhan G, Bock M, Schepers D, Ilina EI, Reichel SN, Löffler H, et al. Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann Neurol. (2014) 75:382–94. doi: 10.1002/ana.24080
32. Sabitha KR, Shetty AK, Upadhya D. Patient-derived iPSC modeling of rare neurodevelopmental disorders: Molecular pathophysiology and prospective therapies. Neurosci Biobehav Rev. (2021) 121:201–19. doi: 10.1016/j.neubiorev.2020.12.025
Keywords: KCNB1, neurodevelopmental disorder, loss of function, dominant-negative effect, haploinsufficiency
Citation: Xiong J, Liu Z, Chen S, Kessi M, Chen B, Duan H, Deng X, Yang L, Peng J and Yin F (2022) Correlation Analyses of Clinical Manifestations and Variant Effects in KCNB1-Related Neurodevelopmental Disorder. Front. Pediatr. 9:755344. doi: 10.3389/fped.2021.755344
Received: 08 August 2021; Accepted: 14 December 2021;
Published: 05 January 2022.
Edited by:
Alberto Spalice, Sapienza University of Rome, ItalyReviewed by:
Dinesh Upadhya, Manipal Academy of Higher Education, IndiaCopyright © 2022 Xiong, Liu, Chen, Kessi, Chen, Duan, Deng, Yang, Peng and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei Yin, eWYyMzIzQGhvdG1haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.