 Ping Wang1,2†
Ping Wang1,2† Chunhua Zhang
Chunhua Zhang Chunquan Cai
Chunquan Cai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 25 November 2021
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.751895
This article is part of the Research Topic Inherited Metabolic Diseases in Pediatrics: Clinical and Molecular Features View all 25 articles
Introduction: Combined malonic and methylmalonic aciduria (CMAMMA) is a rare metabolic disease caused by biallelic variants in ACSF3 gene. The clinical phenotype is highly heterogeneous in this disorder, ranging from asymptomatic to severe symptoms. No cases with CMAMMA were reported in China.
Materials and Methods: In this study, three Chinese pediatric patients were diagnosed with CMAMMA unexpectedly while being treated for other ailments. To better characterize CMAMMA in a Chinese population, we made a multidimensional analysis with detailed clinical phenotype, semi-quantitative detection of urine organic acid, and analysis of ACSF3 gene variants.
Results: The clinical presentation of these patients is quite different; their main complaints were anemia, jaundice, or abnormal urine test, respectively. They showed no symptoms of the classic methylmalonic academia, but urine organic acid analysis showed elevated malonic acid and methylmalonic acid in all the patients repeatedly. Variants were found at four sites in ACSF3 gene. Patient 1 carried the compound heterogeneous variant c.689G> A (p.Trp230*)/c.1456G> A (p.Ala486Thr). A compound heterozygous variant c.473C> T (p.Pro158Leu)/c.1456G> A (p.Ala486Thr) was identified in patient 2. Patient 3 harbored a novel homozygous variant c.1447A> G (p.Lys483Glu).
Conclusions: Three Chinese patients were diagnosed with CMAMMA caused by ACSF3 variants. Their clinical course revealed that CMAMMA can be a benign condition that does not affect individual growth and development, but severe clinical phenotype may appear when other triggers exist. This study systematically elaborates CMAMMA in a Chinese population for the first time, broadens the spectrum of gene variant, and provides a strong basis for the etiological study of this disorder.
Methylmalonic academia (MMAemia) is a common inborn error of metabolism characterized by abnormal accumulation of methylmalonic acid in body fluids, resulting in many serious clinical manifestations. MMAemia is mainly caused by the defect of methylmalonyl-CoA mutase (MUT) or its coenzyme cobalamin (Cbl) (1). Combined malonic and methylmalonic aciduria (CMAMMA) is a rare atypical form of MMAemia featured with increased concentrations of malonic acid (MA) and methylmalonic acid (MMA) in urine (2). CMAMMA is caused by biallelic variants in the AcylCoA synthetase family member 3 (ACSF3, OMIM:614265) gene. ACSF3 encodes a mitochondrial acyl-CoA synthetase, which is essential for the synthesis of malonyl-CoA as well as methylmalonyl-CoA (3). Patients who carried ACSF3 gene variants excrete more MMA than MA in reported cases, which make them distinct from the patients with classical MMAemia (4).
The gene of ACSF3 is located on chromosome 16q24.3. It consists of 11 exons and encodes a 576-amino-acid protein with the first 83 residues representing the predicted mitochondrial transit peptide (4). To date, more than 50 patients worldwide have been reported to have CMAMMA caused by homozygous or compound heterozygous variants in ACSF3, including missense, nonsense, deletion, frameshift, and splice site variants. The clinical presentation of CMAMMA is quite controversial. Signs and symptoms reported so far are involved in neurological abnormalities in adults and infection induced encephalopathy in pediatric patients. Nevertheless, asymptomatic patients with normal outcomes strongly suggest that CMAMMA can be a benign state (2, 4–9). No case of the disease has been reported in China so far.
In this study, we summarized the clinical course, urine organic acid screening results, and analysis of ACSF3 gene variants in three Chinese patients with CMAMMA diagnosed in our hospital. We identified four variants inherited from the parents separately, among which c.1447A> G (p.K483E) is a novel variant. Based on the present study and literature review, we speculate that the CMAMMA is a benign state when other triggers are absent in Chinese population.
Three patients at the age of 11 days to 8 months were diagnosed as CMAMMA in Tianjin Children's Hospital in China. There was no family history in these patients. The informed consents from guardians and the approval of the Medical Ethics Committee of Tianjin Children's Hospital were obtained.
Routine tests were conducted in three patients, such as physical examination, routine blood, urine and stool test, blood gas analysis, biochemical test, imaging examination, and etiological examination for infected patients. Semi-quantitative analysis of organic acid in urine was performed using gas chromatography–mass spectrometry (GC/MS).
The peripheral blood samples were collected to extract the genomic DNA. Peripheral blood from the patients and their parents were collected. A Blood Genomic DNA Mini kit was used to extract the genomic DNA according to the manufacturer's protocol. WES was performed to detect the pathogenic gene variants. Paired-end sequencing was performed on more than 95% of the target regions, with a read length of 150 bp and an average coverage depth of 100-fold, covering all coding regions and exon–intron boundaries. The Burrows-Wheeler Aligner (BWA) software was used to align the raw data with the human reference genome hg19. Insertions, deletions, and single-nucleotide polymorphism sites (SNPs) were analyzed by the Genome Analysis Tool Kit (GATK) software. Annovar software was utilized to add the annotated information of databases such as HGMD, DBSNP, OMIM, and 1000 Genomes. Protein function was predicted by SIFT and Polyphen2 software. The filtering of the variants was performed based on the phenotype, gene frequency, variant type, inherited pattern, and bioinformatics analysis. Suspected variants were validated by Sanger sequencing.
Patient 1 was a 3-month-old girl. She was admitted to the hospital due to pale face for 2 weeks, fever, and cough for 2 days. Detailed physical examination and laboratory tests revealed that the patient had anemia, splenomegaly, hepatomegaly, thrombocytopenia, and bronchitis. Widened interval outside the cerebrum was noted in computed tomography. These clinical features were mainly attributed to suspected Evans syndrome and viral infection. The patient is 5 years old now and shows no sign of physical or psychomotor retardation.
Patient 2 was an 11-day-old boy. He was born at term by normal delivery. He was referred to the hospital due to jaundice for 7 days. Detailed physical examination showed neonatal omphalitis, myocardial damage, testicular hydrocele, and liver function damage. Laboratory tests showed that the direct bilirubin level was 17.1 μmol/L (normal: 0–5 μmol/L), and indirect bilirubin level was 184.4 μmol/L (normal: 3.4–10.3 μmol/L). These clinical manifestations were mainly caused by jaundice and infection. He is 3 years old now and attains a development milestone.
Patient 3 was an 8-month-old boy. He was born at term by cesarean section. He was admitted because of abnormal urine test for 3 months with red urine and urinary frequency. Urinary tract infection, pneumonia, and diarrhea were found in this patient. No obvious abnormality in his growth development was present. Infection factors were the main cause of his clinical presentations. He shows no sign of psychomotor retardation at the age of 5 in our recent follow-up.
GC/MS demonstrated elevated MA and MMA in all the patients repeatedly (Figure 1), which suggested the suspected diagnosis of MMAemia. However, the patients did not present with the clinical symptoms of classic MMAemia.
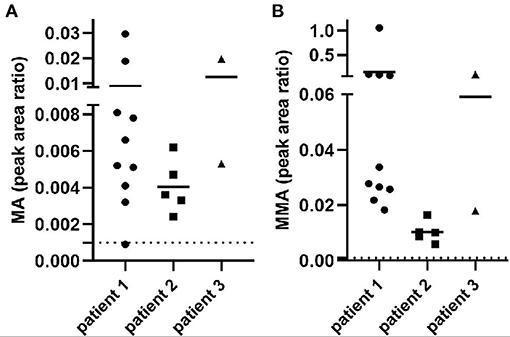
Figure 1. MA and MMA excretion in urine from all the patients. The x-axis represents different patients, and the value of the y-axis means metabolite peak area ratio to creatinine (Cr). Dotted lines represent reference range. (A) Peak area ratio of MA/Cr indicated the MA excretion in patients. (B) Peak area ratio of MMA/Cr indicated the MMA excretion in patients.
All the patients were detected to carry variants in ACSF3 gene (NM_174917.4) by WES. Patient 1 carried a compound heterozygous variant. The variant c.689G> A (p.Trp230*) was inherited from her mother and the other variant c.1456G> A (p.Ala486Thr) was inherited from her father. Besides, the compound heterozygous variant, c.473C> T (p.Pro158Leu)/c.1456G> A (p.Ala486Thr), was identified in the ACSF3 gene in patient 2. The two heterozygous variants were inherited from the mother and father, respectively. Patient 3 harbored a homozygous variant of c.1447A> G (p.Lys483Glu) in ACSF3 gene, which was inherited from his parents (Table 1). Sanger sequencing was used to confirm the variants (Figures 2–4). The homozygous variant c.1447A> G in ACSF3 gene is a novel discovery.

Table 1. Genetic and phenotypic finding of patients with CMAMMA.
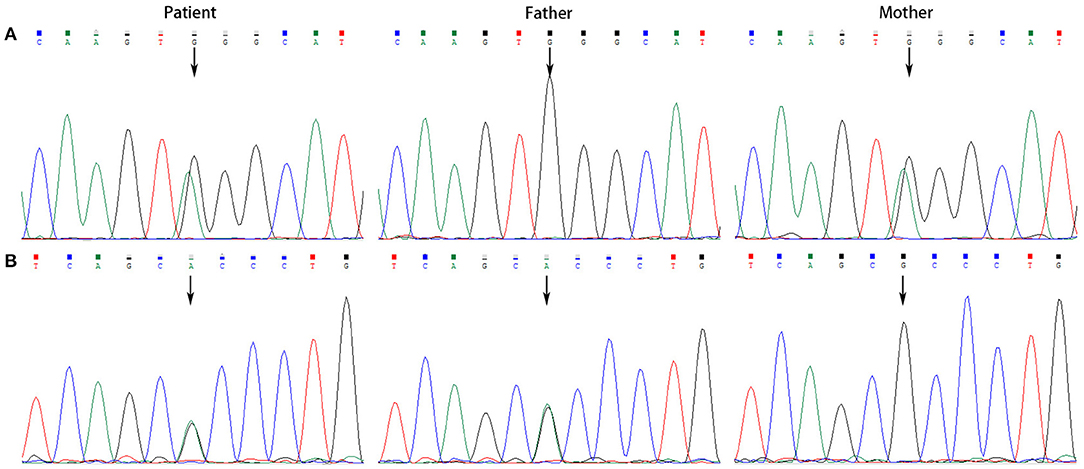
Figure 2. Sanger sequencing of the genomic DNA from patient 1 and her parents. (A) Patient 1 carried with a heterozygous variant of c.689G> A, her father with a normal genotype, and mother with a heterozygous variant of c.689G> A. (B) Patient 1 carried with a heterozygous variant of c.1456G> A, her father with a heterozygous variant of c.1456G> A, and mother with a normal genotype.
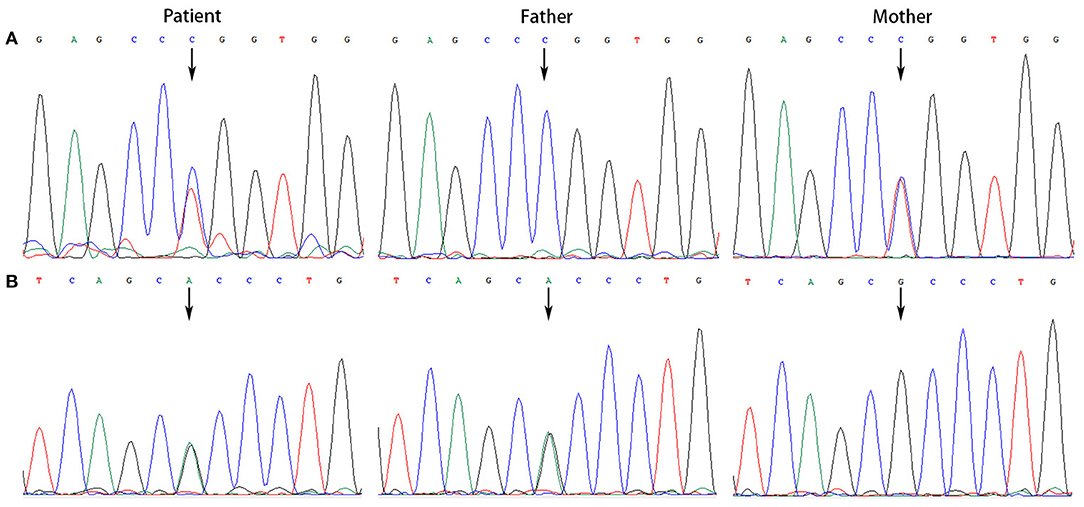
Figure 3. Sanger sequencing of the genomic DNA from patient 2 and his parents. (A) Patient 2 with a heterozygous variant of c.473C> T, his father with a normal genotype, and mother with a heterozygous variant of c.473C> T. (B) Patient 2 with a heterozygous variant of c.1456G> A, his father with a heterozygous variant of c.1456G> A, and mother with a normal genotype.
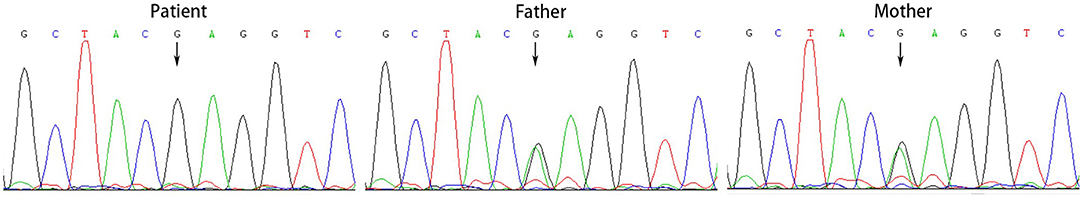
Figure 4. Sanger sequencing of the genomic DNA from patient 3 and his parents. Patient 3 harbored a homozygous variant of c.1447A> G in ACSF3 gene, both his father and mother carried this variant.
CMAMMA is a rare inborn error of metabolism characterized by high excretion of MMA than MA in urine. The pathogenic gene ACSF3 catalyzes the initial reaction in intramitochondrial fatty acid synthesis by activating MA and MMA into their respective CoA thioesters. To date, about 52 patients with 31 homozygous or compound heterozygous variants of ACSF3 have been reported worldwide, including missense, nonsense, deletion, frameshift, and splice site variants. The locations of these variants in ACSF3 are graphically displayed in Figure 5. Most of the variants are missense, mainly located at the carboxyl terminal of the protein (Table 1) (2, 4–9). The most common ACSF3 variants reported so far are c.1075G> A (p.E359K) and c.1672C> T (p.R558W), which are also the most common variants in asymptomatic patients. It indicates that the two variants are more likely to have a mild effect on gene function. Patients who carried the heterozygous variant c.1470G> C (p.E490D) have mild clinical symptoms, while the homozygous variant can result in significant developmental and speech delays. In the present work, we identified four variants of the ACSF3 gene (one nonsense and three missense mutations) in three Chinese patients from three unrelated families, among which the variant c.1447A> G (p.K483E) is novel. The heterozygous variant c.473C> T (p.P158L) has also been reported in another patient with benign clinical presentation (8).
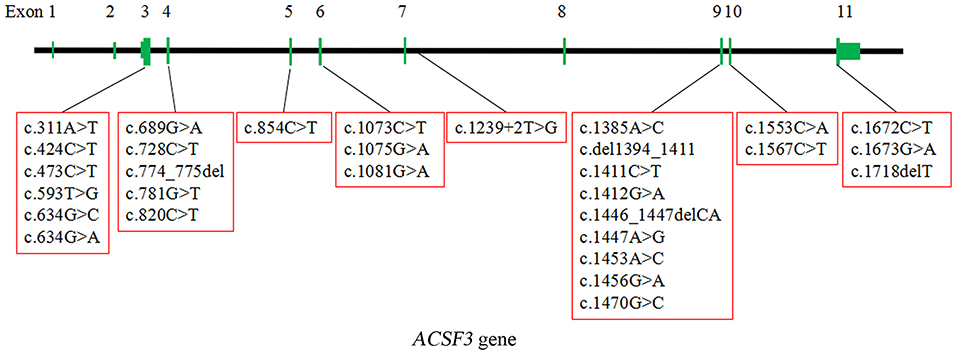
Figure 5. The localization of 30 homozygous or compound heterozygous variants in ACSF3 reported worldwide (except for a Chr16:87441993:89171912 deletion spanning ACSF3). The types of the variants include missense, nonsense, deletion, frameshift, and splice site variants. The most common type is missense.
Patients who carried ACSF3 variants exhibit controversial clinical phenotypes. Based on the literature review we did, severe clinical manifestations are mainly reported in adults, with the most common symptoms of neurological problems and psychiatric features (2). Because of the long-term damage accumulation, older people are at high risk for neurological illness even without ACSF3 variants. The phenotype of pediatric patients is relatively mild or even asymptomatic. Three pediatric patients diagnosed with CMAMMA due to ACSF3 mutation were reported to be clinically asymptomatic, and they had age-appropriate development (4). A retrospective study described the course of 25 CMAMMA individuals and suggested that CMAMMA is probably a benign condition (8). Another study showed that infection factors could provoke metabolic dysregulation in pediatric patients with CMAMMA, and normal levels of development could be obtained at follow-up (5). Therefore, CMAMMA may be considered as a risk factor instead of a disease, which could lead to clinical symptoms when other influencing factors exist.
ACSF3 variants that were only reported in patients with benign manifestations include c.689G> A, c.1456G> A, c.473C> T, c.1447A> G, c.311A> T, c.1239+2T> G, c.1446_1447delCA, c.424C> T, c.820C> T, c.1553C> A, c.774_775del, and c.1081G> A. Patients carrying these variants were asymptomatic or had infection-induced symptoms that disappeared after treatment (2, 4–9). Besides, although the variant c.1456G> A has a high frequency in population, our present study showed that patients carrying this variant had repeatedly elevated biochemical phenotype without obvious clinical phenotype, which could also be observed in other inherited metabolic disease. The β-ureidopropionase deficiency patients carrying the homozygous or compound heterozygous c.977G> A variant (high prevalence in normal Japanese population) in UPB1 gene could be asymptomatic as well (10). We speculate that the c.1456G> A variant in ACSF3 may act in a similar way. High frequency of c.1456G> A in population indicates that CMAMMA may not be as rare as generally considered, and this kind of variant may contribute to the benign condition in patients. Accumulations of free MA and MMA were neurotoxic in vitro, while the concentrations might be insufficient to produce disease in CMAMMA patients (8). Actually, we cannot completely exclude the possibility that the accumulation of metabolites is associated with the occurrence of neurological symptoms as the patients grow up, or that the people with the ACSF3 gene variants are more genetic susceptible to neurological problems later in life. Increased dependency on β-oxidation for energy production were observed in fibroblasts from CMAMMA patients. As a result, the subsequent increased risk for hypoxia and oxidative stress may be crucial for the onset of neurological symptoms in the long run (11).
In conclusion, our study included three Chinese pediatric patients under the age of 4 years. Their clinical features were mainly caused by infection or other reasons. They showed no sign of physical or psychomotor retardation in our follow-up. Combined with reported cases, our study strongly suggested that the CMAMMA is a benign clinical course, especially for patients carrying certain variants. In addition, the present study had some limitations. Firstly, this study only involved the correlation analysis between genotype and phenotype. Functional verification of variants in vitro will help to elucidate the pathogenesis of CMAMMA, especially for the novel variant c.1447A> G (p.K483E), while functional studies were not available in this study due to the limitations of experimental condition. Secondly, a longer follow-up period is needed to better assess the growth and development of the patients.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of Tianjin Children's Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
PW and JS conceived the concept and wrote the manuscript. CG contributed to literature review and revised the manuscript. XY provided clinical diagnosis. JZ contributed to interpretation of the results. CZ performed the analysis. CC participated in supervision of the project. All authors read and approved the final manuscript.
This research was supported by the National Natural Science Foundation of China (81771589), the Program of Tianjin Science and Technology Plan (18ZXDBSY00170), and the Public Health and Technology project of Tianjin (KJ20166, ZC20120).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Baumgartner MR, Hörster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis. (2014) 9:130. doi: 10.1186/s13023-014-0130-8
2. Sloan JL, Johnston JJ, Manoli I, Chandler RJ, Krause C, Carrillo-Carrasco N, et al. Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria. Nat Genet. (2011) 43:883–6. doi: 10.1038/ng.908
3. Bowman CE, Wolfgang MJ. Role of the malonyl-CoA synthetase ACSF3 in mitochondrial metabolism. Adv Biol Regul. (2019) 71:34–40. doi: 10.1016/j.jbior.2018.09.002
4. Alfares A, Nunez LD, Al-Thihli K, Mitchell J, Melançon S, Anastasio N, et al. Combined malonic and methylmalonic aciduria: exome sequencing reveals mutations in the ACSF3 gene in patients with a non-classic phenotype. J Med Genet. (2011) 48:602–5. doi: 10.1136/jmedgenet-2011-100230
5. de Sain-van der Velden MG, van der Ham M, Jans JJ, Visser G, Prinsen HC, Verhoeven-Duif NM, et al. A new approach for fast metabolic diagnostics in CMAMMA. JIMD Rep. (2016) 30:15–22. doi: 10.1007/8904_2016_531
6. Pupavac M, Tian X, Chu J, Wang G, Feng Y, Chen S, et al. Added value of next generation gene panel analysis for patients with elevated methylmalonic acid and no clinical diagnosis following functional studies of vitamin B12 metabolism. Mol Genet Metab. (2016) 117:363–8. doi: 10.1016/j.ymgme.2016.01.008
7. Brasil S, Leal F, Vega A, Navarrete R, Ecay MJ, Desviat LR, et al. Improving the diagnosis of cobalamin and related defects by genomic analysis, plus functional and structural assessment of novel variants. Orphanet J Rare Dis. (2018) 13:125. doi: 10.1186/s13023-018-0862-y
8. Levtova A, Waters PJ, Buhas D, Lévesque S, Auray-Blais C, Clarke JTR, et al. Combined malonic and methylmalonic aciduria due to ACSF3 mutations: Benign clinical course in an unselected cohort. J Inherit Metab Dis. (2019) 42:107–16. doi: 10.1002/jimd.12032
9. Reid ES, Papandreou A, Drury S, Boustred C, Yue WW, Wedatilake Y, et al. Advantages and pitfalls of an extended gene panel for investigating complex neurometabolic phenotypes. Brain. (2016) 139:2844–54. doi: 10.1093/brain/aww221
10. Nakajima Y, Meijer J, Dobritzsch D, Ito T, Meinsma R, Abeling NG, et al. Clinical, biochemical and molecular analysis of 13 Japanese patients with β-ureidopropionase deficiency demonstrates high prevalence of the c.977G > A (p.R326Q) mutation [corrected]. J Inherit Metab Dis. (2014) 37:801–12. doi: 10.1007/s10545-014-9682-y
Keywords: combined malonic and methylmalonic aciduria, ACSF3 gene, benign condition, Chinese population, novel variant
Citation: Wang P, Shu J, Gu C, Yu X, Zheng J, Zhang C and Cai C (2021) Combined Malonic and Methylmalonic Aciduria Due to ACSF3 Variants Results in Benign Clinical Course in Three Chinese Patients. Front. Pediatr. 9:751895. doi: 10.3389/fped.2021.751895
Received: 02 August 2021; Accepted: 19 October 2021;
Published: 25 November 2021.
Edited by:
Hui Xiong, Peking University First Hospital, ChinaReviewed by:
Chunhua Zeng, Guangzhou Women and Children's Medical Center, ChinaCopyright © 2021 Wang, Shu, Gu, Yu, Zheng, Zhang and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunquan Cai, Y3FjbnM2QDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.