 Yu'e Chen1†
Yu'e Chen1† Yingjun Xie2,3†Yuying Jiang4Qi Luo5Lijing Shi1Shuhong Zeng4
Yingjun Xie2,3†Yuying Jiang4Qi Luo5Lijing Shi1Shuhong Zeng4 Jianlong Zhuang4*Guorong Lyu6*
Jianlong Zhuang4*Guorong Lyu6*- 1Ultrasonography, Quanzhou Women's and Children's Hospital, Quanzhou, China
- 2Department of Obstetrics and Gynecology, Key Laboratory for Major Obstetric Diseases of Guangdong Province, The Third Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
- 3Key Laboratory of Reproduction and Genetics of Guangdong Higher Education Institutes, The Third Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
- 4Prenatal Diagnosis Center, Quanzhou Women's and Children's Hospital, Quanzhou, China
- 5Department of Public Health for Women and Children, Quanzhou Women's and Children's Hospital, Quanzhou, China
- 6Ultrasonography, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, China
Background: An increase in pathogenic copy number variants (pCNVs) has been recognized to associate with fetal growth restriction (FGR). Here, we aim to explore the application value of chromosomal microarray analysis (CMA) in prenatal diagnosis of FGR.
Methods: Prenatal ultrasound was applied to identify FGR. A total of 149 pregnant women with FGR were enrolled in our study. All subjects underwent karyotype analysis and CMA to reveal the chromosomal abnormalities.
Results: In this study, all subjects were successfully detected by karyotype and CMA analyses. Of these subjects, the chromosomal abnormalities detection rate was 5.37% (8/149) for karyotyping and 13.42% (20/149) for CMA, respectively. Among them, an 8.05% (12/149) incremental yield of CMA over karyotype analysis was observed (p = 0.004). In addition, a significant difference of pCNV detection rate was observed between the groups with different high-risk factors (p = 0.005). The FGR with structural anomalies group showed the highest pCNV detection rate (33.33%), followed by the FGR with non-structural anomalies group (8.77%) and the isolated FGR group (8.06%).
Conclusion: In conclusion, CMA technology showed an effective application value in etiology diagnosis of FGR. We believe that CMA should be recommended as first-line detection technology for prenatal diagnosis in FGR.
Background
Fetal growth restriction (FGR) refers to the fetus that has not reached its growth potential, and the weight or abdominal circumference is below the 10th percentile of its gestational age. Currently, FGR is one of the most common and complex diseases in obstetrics, which is an important factor for perinatal morbidity and death. Moreover, it may also result in long-term adverse outcomes, including childhood cognitive impairment and increased adult diseases (1). Studies have shown that chromosomal abnormalities can explain 15 to 20% of FGR (2). Therefore, it is of great value in the early screening and diagnosis of FGR.
Although the karyotype analysis technology can identify the large structural variants and chromosome aneuploidy in FGR, the resolution is still limited. Chromosomal microarray analysis (CMA) showed great advantages over traditional karyotype analysis, including array-based comparative genomic hybridization (aCGH) technology and single-nucleotide polymorphism array (SNP array) technology. Among them, SNP array technology can not only provide information of copy number variants but also identify loss of heterozygosity (LOH), uniparental disomy (UPD), and triploid (3–5). A large-scale study indicated that CMA can detect an additional 10% of pathogenic copy number variants over karyotype analysis in FGR with structural abnormalities (6). Moreover, present studies revealed that LOH and UPD are also observed in FGR (7, 8).
To date, there are few studies or with limited subjects available on the application of CMA in the genetic etiology diagnosis of FGR. Our initial aim was to explore the application value of CMA technology in the prenatal diagnosis of FGR, which was the first large-scale study in Fujian province to our knowledge.
Materials and Methods
Subjects
A total of 171 singleton pregnant women from January 2017 to December 2019, who were diagnosed with FGR by fetal ultrasound in our hospital, were enrolled. Among them, 149 cases underwent interventional prenatal diagnosis. In the study, multiple pregnancies, cytomegalovirus infection, and chronic diseases related to drug use and abuse were excluded. All subjects signed an informed consent form and this study obtained approval from the Ethics Committee of Quanzhou Women's and Children's Hospital (2020No.31).
Diagnostic Standard
Fetal crown rump length (CRL) was measured by ultrasound during the first trimester of pregnancy to assess the gestation. In the second trimester, two-dimensional obstetric ultrasound is used to evaluate the growth indicators of the fetus, and the weight of the fetus is estimated based on the growth status of different gestational weeks. Hadlock formula is utilized to calculate the estimated fetal weight (EFW) from the biparietal diameter, abdominal circumference, and femur length. FGR is diagnosed when EFW is below the 10th percentile of gestational age. Additionally, according to the standard of the International Society of Ultrasound in Obstetrics & Gynecology, the fetal growth curve was drawn to evaluate the growth potential of the fetuses. In general, fetal ultrasound structural anomalies referred to morphological defects in fetal organs or parts of the body, such as cleft lip and palate, spina bifida, etc., while other anomalies without any structural anomalies were defined as non-structural anomalies, such as dilatation of the lateral ventricles, enhanced intestinal echo, etc.
Karyotype Analysis
A total of 149 pregnant women with FGR received genetic counseling and signed informed consent. Ultrasound-guided amniocentesis was performed at gestational age of 16–24 weeks, and 30 ml of amniotic fluid was drawn. The 20 ml amniotic fluid was analyzed according to the amniotic fluid karyotype operation procedure of the prenatal diagnosis department of our hospital (9).
SNP Array Analysis
The remaining 10 ml amniotic fluid is used for chromosomal microarray analysis. DNA extractions were performed using the QIAamp DNA Blood Kit (QIAGEN, Hilden, Germany) following the kit Handbook (www.qiagen.com). The detection of SNP array is performed in accordance with the standard experimental procedure of the Affymetrix CytoScan 750 K chip kit. The pathogenicity of copy number variations was interpreted with reference to DGV (http://dgv.tcag.ca/dgv), OMIM (https://omim.org/), DECIPHER (https://decipher.sanger.ac.uk/), PubMed (https://www.ncbi.nlm.nih.gov/pubmed/), and other databases.
Statistical Analysis
The SPSS20.0 software was used for data analysis. The chi-square test was used for statistical analysis among the groups, and the Fisher exact probability test was used for statistical analysis when the chi-square test is not satisfied. A value of p < 0.05 was considered as statistical significance.
Results
Cases Information
A total of 149 subjects underwent interventional amniocentesis prenatal diagnosis, with maternal age range of 20–46 years and gestational age range of 16–24 weeks. In the present study, all cases were divided into three groups, including the FGR with ultrasound structural anomalies group (n = 30), FGR with non-structural anomalies group (n = 57), and isolated FGR group (n = 62).
Chromosomal Abnormalities Detected by Karyotyping
In this study, all subjects were successfully detected by the karyotyping and CMA. Among them, three cases of chromosome aneuploidy and five cases of chromosomal structural abnormalities were detected by karyotype analysis (Table 1), with a chromosomal abnormalities detection rate being 5.37% (8/149). In addition, one case of small supernumerary marker chromosome and two cases of chromosomal polymorphism were also detected.
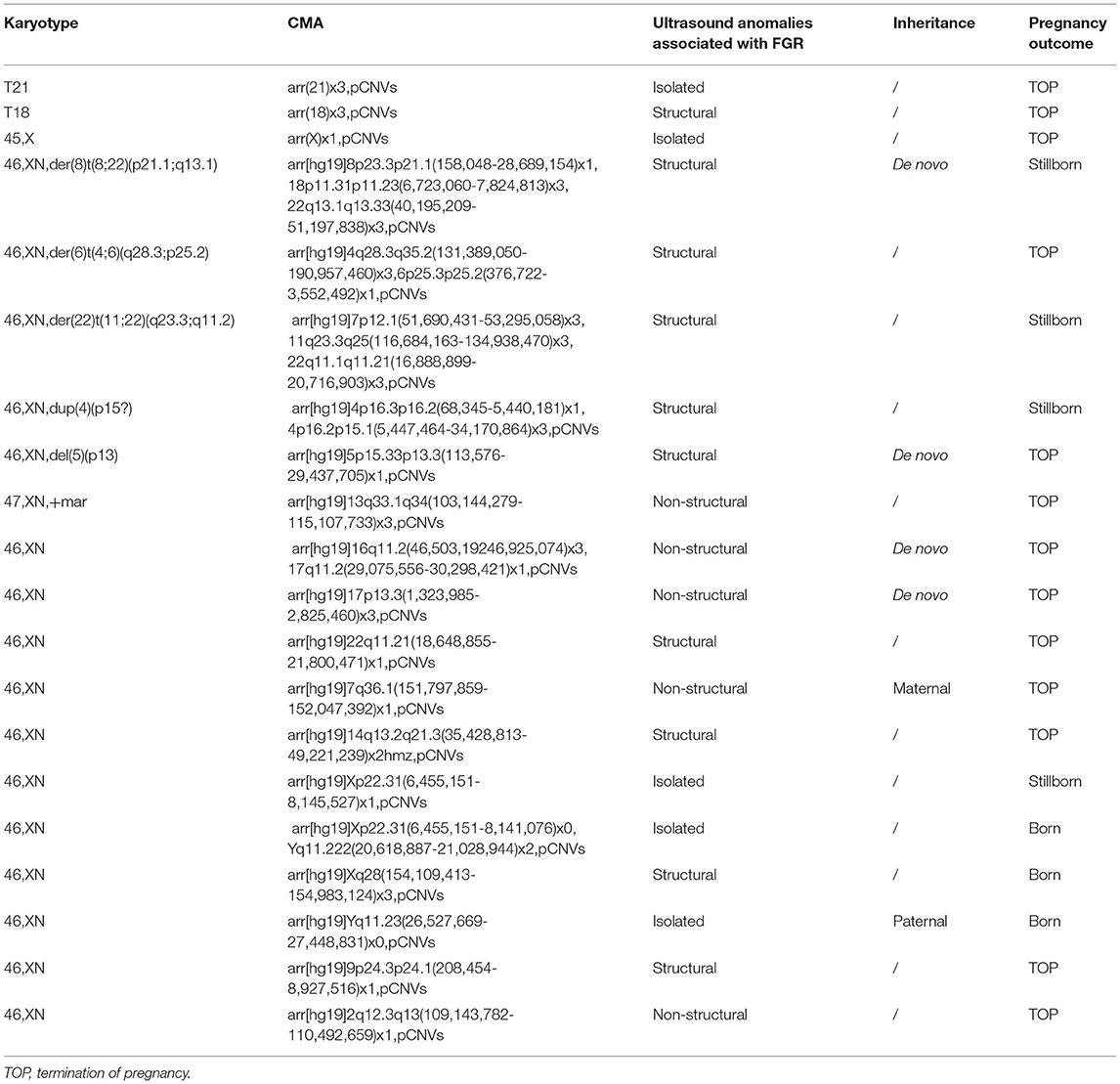
Table 1. Comparison of chromosomal abnormalities detected by karyotyping and CMA.
Comparison of Chromosomal Abnormalities Detected by Karyotyping and CMA
In the study, 20 cases of pCNVs were detected by CMA, with a pCNV detection rate of 13.42% (20/149) (Table 1). Twenty cases of variants of unknown significance (VOUS) and one case of benign CNVs (bCNVs) were detected as well (Table 2). Among them, all chromosomal abnormalities detected by karyotyping were confirmed by CMA (Figures 1, 2); moreover, an 8.05% (12/149) incremental yield of CMA over karyotyping was observed (χ2 = 8.100, p = 0.004) (Table 2). Among them, three cases were from the isolated FGR group (4.84%, 3/62), five cases were from the FGR with non-structural anomalies group (8.77%, 5/57), and four cases were from the FGR with structural anomalies group (13.33%, 4/30). Additionally, one case of small supernumerary marker chromosome was detected by chromosome karyotype analysis and further confirmed by CMA, which was a pathogenic variant and derived from chromosome 13 (13q33.1q34 microduplication).
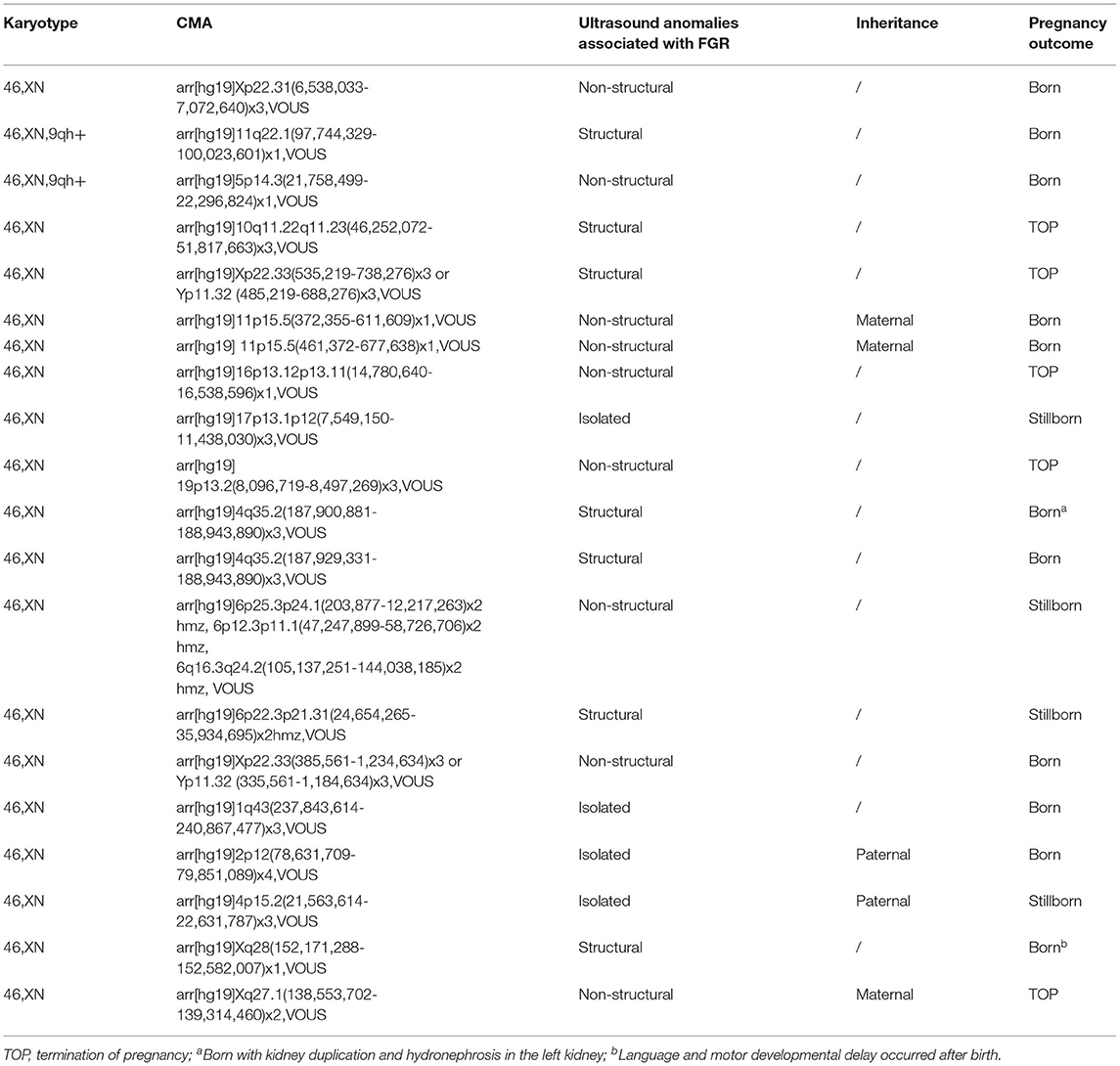
Table 2. The variants of unknown significance detected by CMA.
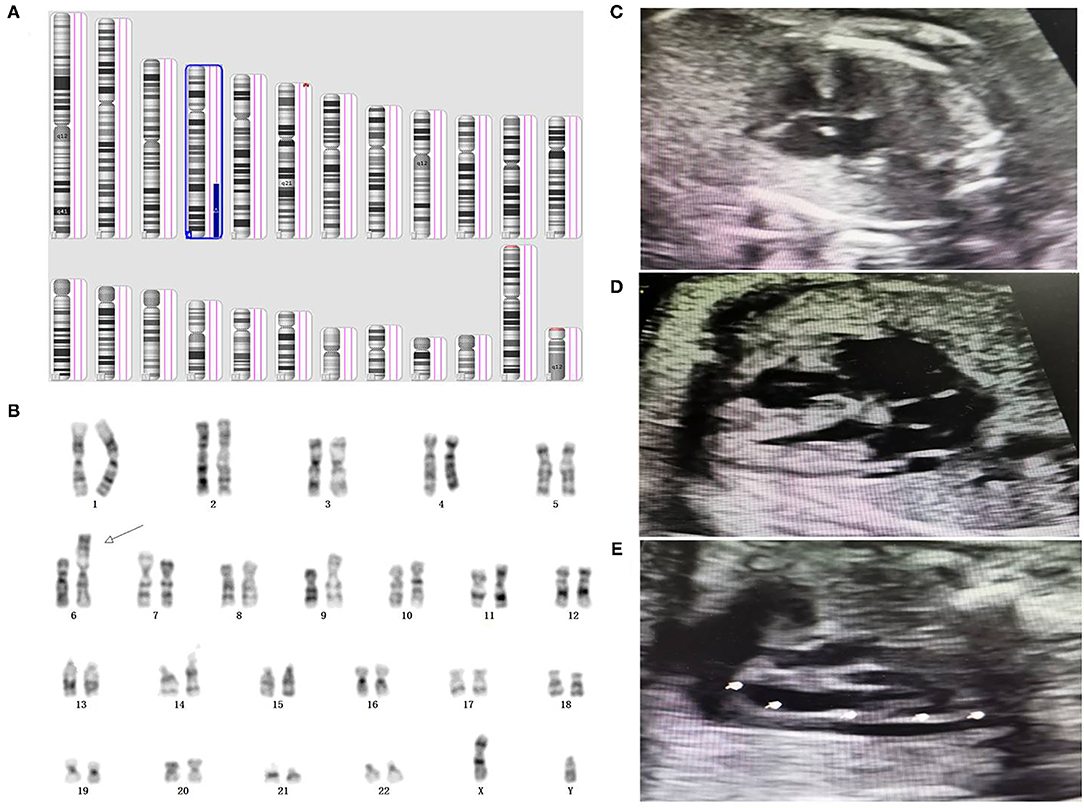
Figure 1. Genetic diagnosis and ultrasound phenotype of derivative chromosome 6. (A) SNP array result indicated a 4q28.3q35.2 duplication associated with 6p25.3p25.2 deletion in the fetus. (B) Karyotype analysis indicated a derivative chromosome 6 in the fetus. (C–E) prenatal ultrasound detection indicated atrioventricular septal defect, coronary sinus dilation, and persistent left superior vena cava in the fetus, respectively.
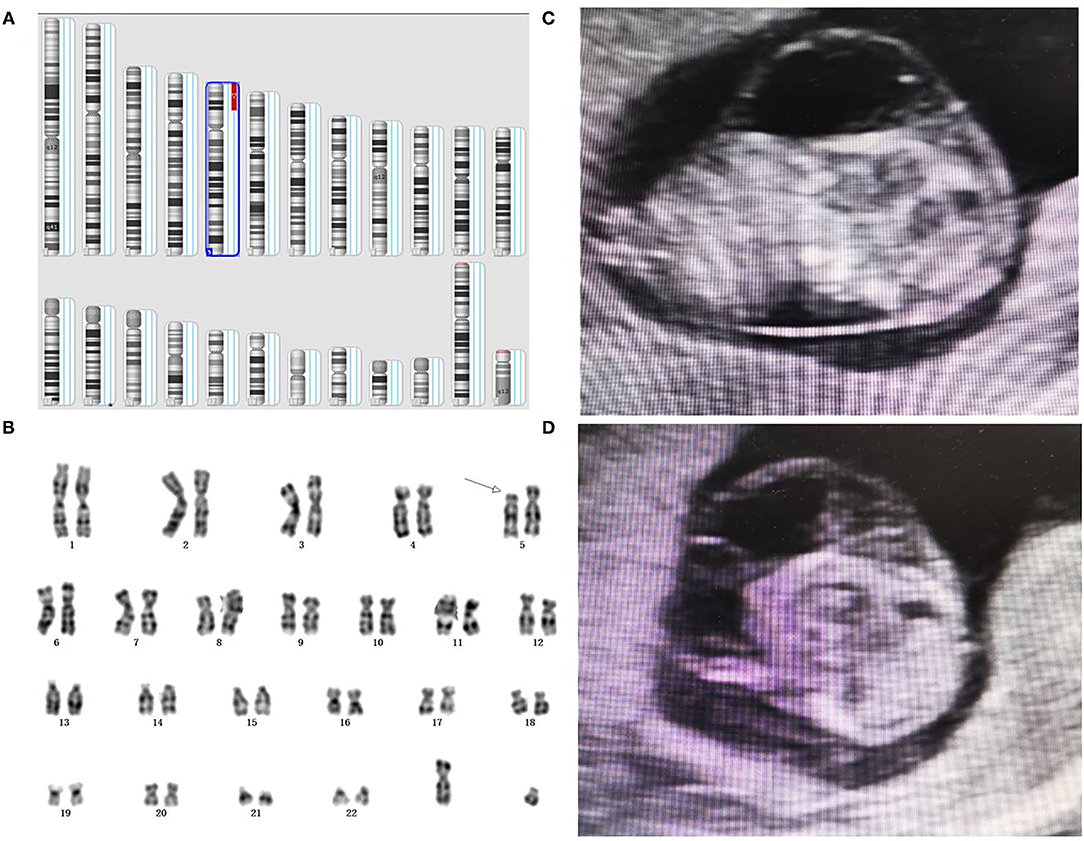
Figure 2. Genetic diagnosis and ultrasound phenotype of partial 5p deletion. (A) A detailed view from an SNP array analysis report for chromosomes involved. (B) Karyotype analysis revealed a partial 5p deletion in the fetus. (C, D) Prenatal ultrasound elicited cystic hygroma colli in the fetus.
Comparison of the Detection Rates of CNVs Among the Groups
In this study, we further analyzed the detection rate of pCNVs in groups of FGR with different ultrasound phenotypes (Table 3). A significant difference of pCNV detection rates was observed among the groups (χ2 = 10.678, p = 0.005). Among them, the FGR with structural anomalies group showed a higher pCNV detection rate than that in the FGR with non-structural anomalies group and isolated FGR group (χ2 = 8.310, p = 0.004; χ2 = 9.460, p = 0.002). In addition, no significant difference of pCNV detection rates was observed between the FGR with non-structural anomalies group and isolated FGR group (χ2 = 0.019, p = 0.889).

Table 3. Detection rates of CNVs among FGR groups with different ultrasound anomalies.
Follow-Ups of Pregnancy Outcome
In this study, 90.60% (135/149) were successfully followed up. Among the cases with pCNVs, 13 of them chose to terminate their pregnancy, four fetuses died in utero, and three cases continued their pregnancy without obvious abnormality observed after birth (Table 1). Of the cases with VOUS, five cases chose to terminate their pregnancy (two cases with structural anomalies and three cases with soft index anomalies), four cases encountered intrauterine death (three cases with structural anomalies and 1 cases was isolated FGR), and 11 cases continued their pregnancy. The follow-up results showed that one case (4q35.2 microduplication) exhibited kidney duplication and hydronephrosis in the left kidney, and one case (Xq28 microdeletion) exhibited language and motor developmental delay. No obvious abnormalities were observed in the remaining cases.
Discussion
CMA technology can detect unbalanced CNVs effectively, which has been recommended as a first-line detection technology for the genetic etiology diagnosis in fetus with structural anomalies (10). However, there are few studies available in the application of CMA for FGR genetic etiology diagnosis. Research on the application of CMA technology in FGR with normal karyotypes showed a 10% incremental yield of CMA over karyotyping in FGR with structural anomalies; and 4% pathogenic copy number variation can also be detected in FGR with non-structural anomalies (6). In our study, 12 cases with pCNVs were detected by CMA, with an additional detection rate of 8.05% (12/149) over karyotyping. This study is similar to that reported in the literature (2, 6, 11, 12), which showed a great advantage of CMA technology in the prenatal diagnosis of FGR over karyotyping.
In the present study, based on the ultrasound phenotypes, three groups were divided; a significant difference of pCNV detection rate was observed among the groups. Among them, the detection rate of pCNVs in the FGR with structural anomalies group showed higher pCNV detection rate. This further strengthens the application value of CMA in fetuses with structural anomalies. Moreover, we believe that FGR combined with ultrasound soft indicator anomalies may not increase the risk of chromosomal abnormality.
Chromosome aneuploidy can explain approximately 7% of FGR, of which trisomy 18 was the most common etiologic factor (13). Additionally, studies have shown that 90% of trisomy 18 can express FGR, while it exists in 30% of trisomy 21 (14). Three cases of chromosome aneuploidy were identified in this study, including one case of trisomy 21, one case of trisomy 18, and one case of Turner syndrome. Among them, two cases exhibited isolated FGR. Thus, the genetic diagnosis and clinical consultation of isolated FGR should also be given equal attention. In addition, five cases of common pCNVs associated with FGR were detected (6), including two cases of Xp22.31 microdeletion, one case of 22q11.21 microdeletion syndrome, one case of 22q11.1q11.21 microduplication syndrome, and one case 4p16.3 microdeletion syndrome. Previous studies have shown that Xp22.31 microdeletion in fetuses only exhibited isolated FGR (12, 15). In this study, two cases of Xp22.31 microdeletion were detected with isolated FGR, which is consistent with the previous reports. Studies have shown that 22q11.21 microdeletion was detected in FGR with structural malformations (16). In this study, a 22q11.21 microdeletion in FGR without cardiac abnormality was observed, which may relate to the non-penetrance in 22q11.21 microdeletion. Moreover, researchers indicated that the 22q11.21 microduplication can also be observed in FGR with oligohydramnios or isolated FGR (6, 17). The 4p16.3 microdeletion covering the “Wolf–Hirschhorn syndrome” region would exhibit intellectual disability, developmental delay, and hypotonia, and has been identified in FGR previously (16, 18). In this study, the fetus with 4p16.3 microdeletion referred to as Wolf–Hirschhorn syndrome exhibited FGR with multiple structural anomalies.
VOUS is a great challenge in clinical consultation; because of low ratio of parent verification and the incomplete localized database, it is difficult to interpret the pathogenicity of the subjects with VOUS. Thus, it is very important to apply fetal ultrasound to further monitor the growth and development of the fetuses. In the current study, 20 cases of VOUS were detected in this study, which may be the etiology for FGR. Among them, one case of Xq28 region involving 410.7kb deletion was detected, including PNMA3, PNMA6A, and MAGEA1 genes. As shown in the database, there are cases with similar and smaller deletions than the fragment exhibiting developmental delay and ventricular septal defect. Moreover, a previous study showed that a patient who harbored a deletion of chromosome Xq28 exhibited growth delay (19). Ultrasound in this case showed ventricular septal defect and FGR, which was similar to the clinical phenotypes reported in the database and literature. In addition, an unknown significance of Xp22.33 microduplication was detected, containing SHOX/SHOXY gene, which was related to autosomal dominant diseases of Leri–Weill cartilage osteogenesis disorder due to gene mutation or deletion. Previous studies reported that duplication involving the SHOX gene would lead to short stature, intellectual disability, and developmental delay (20, 21). Thus, in the present study, we believe that Xq28 deletion and Xp22.33 duplication may be the genetic etiology for FGR. It can be seen that prenatal ultrasound combined with CMA detection technology would be helpful for the clinical consultation of VOUS and the exploration of genotype–phenotype relationship.
To date, more studies have shown that UPD will result in FGR (22–24). Our study identified a loss of heterozygosity in the 14q13.2q21.3 region who exhibit FGR combined with ventricular septal defect, auricle abnormality and hypoxia. Studies have shown that maternal UPD of chromosome 14 will result in Temple syndrome, which have been reported to be associate with short stature, intrauterine growth retardation, abnormal facial appearance, and low birth weight (25), while paternal UPD of chromosome 14 will leading to Kagami-Ogata syndrome, with the clinical phenotypes including abnormal bone development, joint contractures, abnormal facial features, developmental delay, intellectual disability, etc (26, 27). According to the phenotype, the maternal 14q13.2q21.3 loss of heterozygosity may harbor. However, more work should be done to determine the source of loss of heterozygosity in this case.
Conclusions
In conclusion, the genetic etiology of FGR is complicated, including chromosome aneuploidy, UPD, LOH, and copy number variants. In this study, a 4.84% incremental yield of CMA over karyotyping was observed in isolated FGR. Thus, we believe that CMA should be recommended as a first-line detection technology for prenatal diagnosis in FGR, in the presence of other ultrasound anomalies or not. Moreover, our research indicated that Xp28 microdeletion and Xp22.33 duplication may be the genetic etiology for FGR.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Ethics Committee of Quanzhou women's and children's hospital (2020No.31). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
GL designed the study. YC and JZ wrote the article. JZ, QL, and SZ performed routine chromosome analysis, SNP array detection, and data analysis. GL, LS, YX, and YJ modified and proofread the paper. All authors approved the final article.
Funding
This research was supported by the Fujian Provincial Health Commission Youth Science and Technology Project (2020QNB045); Quanzhou Science and Technology Bureau (2020C026R).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We wish to express our appreciation to Fujian Provincial Health Commission and Quanzhou Science and Technology Bureau for funding this work. We also express our appreciation to the subjects who participated in this study.
References
1. American College of Obstetricians and Gynecologists' Committee on Practice Bulletins—Obstetrics and the Society for Maternal-Fetal Medicine. ACOG practice bulletin No. 204: fetal growth restriction. Obstet Gynecol. (2019) 133:e97–109. doi: 10.1097/AOG.0000000000003070
2. Zhu H, Lin S, Huang L, He Z, Huang X, Zhou Y, et al. Application of chromosomal microarray analysis in prenatal diagnosis of fetal growth restriction. Prenat Diag. (2016) 36:686–92. doi: 10.1002/pd.4844
3. Dugoff L, Norton ME, Kuller JA. The use of chromosomal microarray for prenatal diagnosis. Am J Obstet Gynecol. (2016) 215:B2–9. doi: 10.1016/j.ajog.2016.07.016
4. Breman A, Pursley AN, Hixson P, Bi W, Ward P, Bacino CA, et al. Prenatal chromosomal microarray analysis in a diagnostic laboratory; experience with >1000 cases and review of the literature. Prenat Diagn. (2012) 32:351–61. doi: 10.1002/pd.3861
5. Ganapathi M, Nahum O, Levy B. Prenatal diagnosis using chromosomal SNP microarrays. Methods Mol Biol. (2019) 1885:187–205. doi: 10.1007/978-1-4939-8889-1_13
6. Borrell A, Grande M, Pauta M, Rodriguez-Revenga L, Figueras F. Chromosomal microarray analysis in fetuses with growth restriction and normal karyotype: a systematic review and meta-analysis. Fetal Diagn Ther. (2018) 44:1–9. doi: 10.1159/000479506
7. Yingjun X, Zhiyang H, Linhua L, Fangming S, Linhuan H, Jinfeng T, et al. Chromosomal uniparental disomy 16 and fetal intrauterine growth restriction. Eur J Obstet Gynecol Reprod Biol. (2017) 211:1–7. doi: 10.1016/j.ejogrb.2016.12.019
8. Leung WC, Lau WL, Lo TK, Lau TK, Lam YY, Kan A, et al. Two IUGR foetuses with maternal uniparental disomy of chromosome 6 or UPD(6)mat. J Obstet Gynaecol. (2017) 37:113–5. doi: 10.1080/01443615.2016.1242558
9. Zhuang J, Wang Y, Zeng S, Lv C, Lin Y. Jiang Y. A prenatal diagnosis and genetics study of five pedigrees in the Chinese population with Xp2231 microduplication. Mol Cytogenet. (2019) 12:50. doi: 10.1186/s13039-019-0461-1
10. American College of Obstetricians and Gynecologists Committee on Genetics. Committee opinion No. 581: the use of chromosomal microarray analysis in prenatal diagnosis. Obstet Gynecol. (2013) 122:1374–7. doi: 10.1097/01.AOG.0000438962.16108.d1
11. Gruchy N, Decamp M, Richard N, Jeanne-Pasquier C, Benoist G, Mittre H, et al. Array CGH analysis in high-risk pregnancies: comparing DNA from cultured cells and cell-free fetal DNA. Prenat Diagn. (2012) 32:383–8. doi: 10.1002/pd.2861
12. Borrell A, Grande M, Meler E, Sabrià J, Mazarico E, Muñoz A, et al. Genomic microarray in fetuses with early growth restriction: a multicenter study. Fetal Diagn Ther. (2017) 42:174–80. doi: 10.1159/000452217
13. Vanlieferinghen S, Bernard JP, Salomon LJ, Chalouhi GE, Russell NE, Ville Y. Retard de croissance intra-utérin du deuxième trimestre: malformations associées et syndromes sous-jacents [Second trimester growth restriction and underlying fetal anomalies]. Gynecol Obstet Fertil. (2014) 42:567–71. doi: 10.1016/j.gyobfe.2014.07.002
14. Maulik D. Fetal growth restriction:the etiology. Clin Obstet Gynecol. (2006) 49:228–35. doi: 10.1097/00003081-200606000-00006
15. Oneda B, Baldinger R, Reissmann R, Reshetnikova I, Krejci P, Masood R, et al. High-resolution chromosomal microarrays in prenatal diagnosis significantly increase diagnostic power. Prenat Diagn. (2014) 34:525–33. doi: 10.1002/pd.4342
16. Srebniak MI, Diderich KE, Joosten M, Govaerts LC, Knijnenburg J, de Vries FA, et al. Prenatal SNP array testing in 1000 fetuses with ultrasound anomalies: causative, unexpected and susceptibility CNVs. Eur J Hum Genet. (2016) 24:645–51. doi: 10.1038/ejhg.2015.193
17. De Wit MC, Srebniak MI, Joosten M, Govaerts LC, Kornelisse RF, Papatsonis DN, et al. Prenatal and postnatal findings in small-for-gestational-age fetuses without structural ultrasound anomalies at 18–24 weeks. Ultrasound Obstet Gynecol. (2017) 49:342–8. doi: 10.1002/uog.15949
18. Zheng W, Chen B, Yin Z, Huang X. Liang Y. [Identification of a critical region on chromosome 4p163 for wolf-hirschhorn syndrome-associated fetal growth retardation]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2020) 37:731–5. doi: 10.3760/cma.j.issn.1003-9406.2020.07.007
19. Deng X, Fang H, Pathak A, Zou AM, Neufeld-Kaiser W, Malouf EA, et al. Deletion of FUNDC2 and CMC4 on chromosome Xq28 is sufficient to cause hypergonadotropic hypogonadism in men. Front Genet. (2020) 11:557341. doi: 10.3389/fgene.2020.557341
20. Tropeano M, Howley D, Gazzellone MJ, Wilson CE, Ahn JW, Stavropoulos DJ, et al. Microduplications at the pseudoautosomal SHOX locus in autism spectrum disorders and related neurodevelopmental conditions. J Med Genet. (2016) 53:536–47. doi: 10.1136/jmedgenet-2015-103621
21. Jee YH, Baron J, Nilsson O. New developments in the genetic diagnosis of short stature. Curr Opin Pediatr. (2018) 30:541–7. doi: 10.1097/MOP.0000000000000653
22. Kunwar F, Pabst R, Bakshi S. Intrauterine growth restriction associated with paternal isodisomy of chromosome 5: a clinical report and literature survey. J Matern Fetal Neonatal Med. (2020) 33:1027–9. doi: 10.1080/14767058.2018.1506443
23. Lazier J, Martin N, Stavropoulos JD, Chitayat D. Maternal uniparental disomy for chromosome 6 in a patient with IUGR, ambiguous genitalia, and persistent mullerian structures. Am J Med Genet A. (2016) 170:3227–30. doi: 10.1002/ajmg.a.37876
24. Eggermann T, Zerres K, Eggermann K, Moore G, Wollmann HA. Uniparental disomy: clinical indications for testing in growth retardation. Eur J Pediatr. (2002) 161:305–12. doi: 10.1007/s00431-002-0916-x
25. Ioannides Y, Lokulo-Sodipe K, Mackay DJ, Davies JH, Temple IK. Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J Med Genet. (2014) 51:495–501. doi: 10.1136/jmedgenet-2014-102396
26. Sutton VR, McAlister WH, Bertin TK, Kaffe S, Wang JC, Yano S, et al. Skeletal defects in paternal uniparental disomy for chromosome 14 are re-capitulated in the mouse model (paternal uniparental disomy 12). Hum Genet. (2003) 113:447–51. doi: 10.1007/s00439-003-0981-x
Keywords: fetal growth restriction, chromosomal microarray analysis, karyotype analysis, prenatal diagnosis, copy number variants (CNVs)
Citation: Chen Y, Xie Y, Jiang Y, Luo Q, Shi L, Zeng S, Zhuang J and Lyu G (2021) The Genetic Etiology Diagnosis of Fetal Growth Restriction Using Single-Nucleotide Polymorphism-Based Chromosomal Microarray Analysis. Front. Pediatr. 9:743639. doi: 10.3389/fped.2021.743639
Received: 19 July 2021; Accepted: 06 September 2021;
Published: 15 October 2021.
Edited by:
Fan Jin, Zhejiang University, ChinaReviewed by:
Sofia Douzgou Houge, Haukeland University Hospital, NorwayFeodora Stipoljev, University Hospital Sveti Duh, Croatia
Copyright © 2021 Chen, Xie, Jiang, Luo, Shi, Zeng, Zhuang and Lyu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianlong Zhuang, NDE1OTEzMjYxQHFxLmNvbQ==; Guorong Lyu, bGdyLWZldXNAc2luYS5jb20=
†These authors have contributed equally to this work and share first authorship