 Simona Gatti
Simona Gatti Giulia Gelzoni
Giulia Gelzoni Giulia N. Catassi2
Giulia N. Catassi2 Carlo Catassi
Carlo Catassi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Pediatr. , 26 October 2021
Sec. Pediatric Gastroenterology, Hepatology and Nutrition
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.742830
Background and Aims: Inflammatory bowel disease (IBD) is a typical polygenic disorder and less frequently shows a monogenic origin. Furthermore, IBD can originate in the context of specific genetic syndromes associated with a risk of autoimmune disorders. We aimed to systematically evaluate the prevalence of IBD in specific genetic syndromes and to review the clinical characteristics of the published cases.
Methods: According to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines, studies describing patients with IBD and a genetic syndrome and/or studies indicating the prevalence or incidence of IBD in subjects with a genetic syndrome were included.
Results: Forty-six studies describing a total of 67 cases of IBD in six genetic syndromes and two personally assessed unpublished cases were included in the review. The majority of cases were associated with Turner syndrome (TS) (38 cases), Down syndrome (DS) (18 cases) and neurofibromatosis type 1 (NF1) (8 cases). Sporadic cases were described in DiGeorge syndrome (2), Kabuki syndrome (2), and Williams syndrome (1). The prevalence of IBD ranged from 0.67 to 4% in TS and from 0.2 to 1.57% in DS. The incidence of IBD was increased in TS and DS compared to the general population. Eight cases of IBD in TS had a severe/lethal course, many of which described before the year 2000. Two IBD cases in DS were particularly severe.
Conclusion: Evidence of a greater prevalence of IBD is accumulating in TS, DS, and NF1. Management of IBD in patients with these genetic conditions should consider the presence of comorbidities and possible drug toxicities.
Systematic Review Registration: PROSPERO, identifier: CRD42021249820
Inflammatory bowel disease (IBD) includes a group of chronic intestinal inflammatory conditions with three main clinical subtypes: Crohn's disease (CD), ulcerative colitis (UC), and IBD-unclassified (IBD-U). IBD has a multifactorial pathogenesis, involving genetic susceptibility, environmental factors, intestinal microbiota, and immune system. The contribution of genetic factors has been extensively described; however, the impact of genetics in the development of IBD is variable. The most common form of IBD is a polygenic disorder where the implication of genetic factors can significantly impact on the susceptibility and/or in the development of specific clinical characteristics and complications of the disease (1). So far, more than 230 disease loci related to polygenic IBD have been defined by genome-wide association studies (GWAS) (2). Differences between IBD subtypes, associations with clinical features, and prognosis can be explained, at least partially, by GWAS (3). Conversely, fewer common variants in IBD risk loci have been discovered to have a causative role including NOD2, ATG16L1, IRGM, IL23R, CARD9, RNF186, and PRDM1, with most risk loci containing different candidate genes (4, 5). More recently, monogenic forms of IBD have been identified as a group of diseases characterized by an early onset (generally in children <10 years of age: EO-IBD or very early in children <6 years of age: VEO-IBD) and a severe course, refractory to conventional therapies. More than 70 monogenic defects have been described, with a considerable number of the implicated genes involved in the maintenance of the immune homeostasis (6).
In addition to these groups of IBD (polygenic and monogenic forms), some genetic syndromes, mainly characterized by chromosomal abnormalities (numerical or structural), can present with IBD or are at high risk of development of IBD. These syndromes can be associated with autoimmune comorbidities, with the usual paradigms being represented by Turner syndrome (TS) and trisomy 21 or Down syndrome (DS). IBD has been reported, although sporadically, in other genetic syndromes. Possible mechanisms to explain the risk of autoimmunity in such syndromes include the following: genetic defects of immune regulatory pathways, impaired apoptosis and defense against oxidative stress, and molecular mimicry of viral or bacterial antigens (7). Clinical course of IBD in patients with a genetic syndrome can substantially differ from non-syndromic IBD for several reasons, including the following: the intellectual disability, the difficulties in adherence to treatment, a different response to certain medications, and the presence of comorbidities (Figure 1). So far, no specific protocols or guidelines exist for such vulnerable patients.
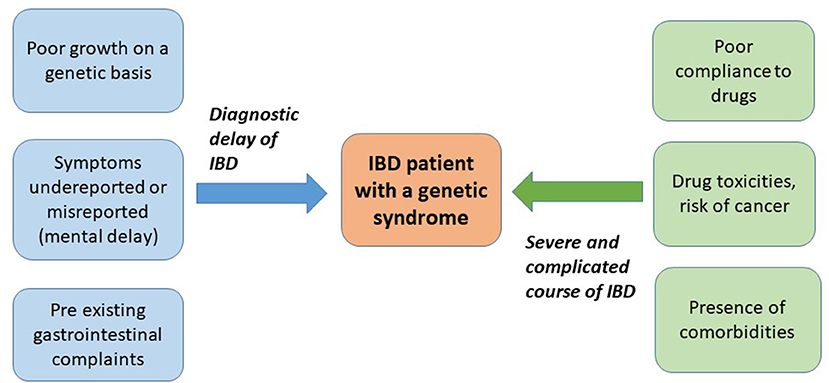
Figure 1. Peculiarities of inflammatory bowel disease (IBD) diagnosis and course in the context of a genetic syndrome.
We aimed to describe this specific scenario starting by illustrating two challenging pediatric cases of IBD in subjects with a genetic syndrome. We further decided to perform a systematic review of literature to identify the frequency of IBD in specific syndromes and to describe the clinical characteristics of IBD in patients with these genetic conditions.
A 3-year-old Italian girl presented to our attention with a 4-week history of bloody diarrhea. She was born from non-consanguineous Italian parents; her grandfather was affected by IBD, and her mother was followed up for mild thrombocytopenia and neutropenia of unknown origin. At the time of admission, she was clinically well and her weight and height were both below the 3rd centile. Blood tests were unremarkable. Stool culture and examination for fecal parasites were negative. Fecal calprotectin was mildly elevated (245 mg/kg) and p-ANCAs (anti-neutrophil cytoplasmic antibodies) were positive. Ileo-colonoscopy revealed pancolitis with edema, signs of inflammation, and tendency to bleeding along the entire colon. Histological findings showed mostly acute inflammation with minimal signs of chronicity. In the suspicion of UC, she was started on oral mesalazine with clinical remission achieved after 2 weeks. At the 3-month follow-up, she was further investigated for her persistent failure to thrive with short stature and retarded growth velocity. Mild dimorphisms were noted at that time. Atrial septal defect was found at echocardiogram. Chromosomal analysis indicated a Turner syndrome (mosaic pattern, 96% 45,X and 4% 46,XX). Five months later, she presented recurrence of gastrointestinal symptoms (bloody diarrhea) and raised fecal calprotectin of 350 mg/kg. She was endoscopically re-evaluated, and the examination showed hyperemia and edema of colic mucosa with mild ulcers in the sigmoid and transverse colon and normal appearance of the terminal ileum. Histology showed signs of acute and chronic colitis with infiltration of lymphocytes, eosinophilic, and neutrophil granulocytes; aphthous erosions; and focal alteration of glandular architecture along the ascending, transverse, and descending colon. The data confirmed a diagnosis of UC. She was started on oral steroids and azathioprine, and clinical and laboratory remission was achieved. In the following months, she required another course of steroids. A year later, the patient was admitted for an endoscopic re-evaluation, and the colonoscopy showed mild hyperemia and edema (Mayo score 1). Treatment with azathioprine was continued. At the age of 4, she started growth hormone (GH) therapy. Currently, she is 6 years old and she maintains clinical remission with azathioprine alone (2.6 mg/kg/day).
A 10-year-old boy of Arab origin with DS was admitted to our institution for a 3-week history of fever, diarrhea, and erythema nodosum. In the preceding 4 years, he had been complaining of dysphagia for solid foods, feeding on only creamy foods and liquids. At the time of our evaluation, his chemical panel revealed the following: normocytic anemia, low albumin, raised C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR), and raised fecal calprotectin (>500 mg/kg). Anti-Saccharomyces cerevisiae antibodies (ASCA) were present. Stool cultures were negative. An endoscopic examination showed a proximal esophageal stricture (with mild signs of mucosal inflammation), edema, and aphthous ulcers in the rectum, descending, and transverse colon. Histological features of the colic and rectal mucosa included severe infiltration of lymphocytes and eosinophilic granulocytes and rare crypt abscesses; in the sigmoid colon, there was an epithelioid granuloma. From these findings, a diagnosis of CD was made. In consideration of the mild signs of inflammation and the proximal site of the esophageal stricture (not typical for CD), further etiologies, different from CD, were considered, including a previously not reported caustic ingestion and a congenital stenosis, although the appearance at 6 years of age was less likely. Treatment of CD was started with exclusive enteral nutrition (EEN) associated to anti-tumor necrosis factor (TNF) therapy (infliximab 5 mg/kg). Clinical remission was initially achieved. Three months after the initial treatment, he was admitted again for fever; diarrhea; and reappearance of erythema nodosum, asthenia, and weight loss. Abdominal ultrasound showed wall thickening (5–6 mm) of the descending and sigmoid colon and rectum. Azathioprine and steroid therapy was initiated, and the infusion of infliximab was anticipated (every 6 weeks) at a higher dose (7 mg/kg). Five months later, another hospitalization was necessary following an episode of impact of the bolus, dysphagia, and diarrhea with blood. An endoscopic dilation of the esophageal stricture was performed. The abdominal ultrasound revealed a worse wall thickening of the descending and sigmoid colon and rectum (12 mm) and a thickening of the terminal ileum, ileocecal valve, cecum, and ascending colon (7 mm). Another course of EEN was commenced, treatment with infliximab was optimized (every 4 weeks at the dose of 10 mg/kg), and azathioprine was continued. Almost a year after the diagnosis, the patient was endoscopically re-evaluated. A Savary dilation of the esophagus was repeated; the colonoscopy showed hyperemia, edema, ulcers, and fibrin deposition in the sigmoid colon and rectum. Histological findings revealed a chronic inflammation of the esophagus (with signs of fibrosis), stomach, cecum, colon, and rectum. Six months later, he required another esophageal endoscopic dilation, for an episode of impact of bolus. Due to the persistence of clinical activity and raised inflammatory markers (including CRP, ESR, and calprotectin), maintenance therapy with infliximab and azathioprine was switched to adalimumab and azathioprine and another course of oral steroids was required. At the 2-month follow-up, the patient is in clinical remission.
The study was registered in the PROSPERO database registry (PROSPERO registration number CRD 42021249820). The medical literature was searched using MEDLINE, EMBASE, and the Cochrane Central Register of Controlled Trials, until December 2020, according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines (8, 9). A separate systematic research was performed for the following syndromes: Turner syndrome or monosomy X, Down syndrome or trisomy 21, Klinefelter syndrome or XXY syndrome, Edward syndrome or trisomy 18, Patau syndrome or trisomy 13, Williams syndrome, Noonan syndrome or RASopathies, Prader Willi syndrome, Angelman syndrome, DiGeorge syndrome or 22q deletion syndrome, Kabuki syndrome, and neurofibromatosis or von Recklinghausen's disease. The genetic conditions were chosen considering the most frequent chromosomal abnormalities and syndromes with a known risk of autoimmune disorders. A non-specific research was also performed using the term “genetic syndrome” instead of a specific name. For each syndrome, the different names of the condition were combined with “Inflammatory Bowel Disease” OR “IBD” OR “ulcerative colitis” OR “Crohn.” A separate search was performed matching each syndrome name or the generic term “genetic syndrome” AND the terms “autoimmune OR autoimmunity.”
We included all the prospective controlled trials, observational studies, case–control studies, case reports, and series describing cases of IBD (both in children and adults) or studies reporting epidemiological data (prevalence or incidence) of IBD in a population of subjects with a genetic syndrome. Due to the paucity of data, we also included letters, comments, and review articles. We excluded non-English literature. Two independent reviewers (SG and GG) performed the first selection, screened title and abstract of the papers identified by electronic search, and completed an inclusion form for eligible studies. Additional articles were obtained through citation snowballing to locate primary sources. When no abstract was available, the article was always screened by full text. The same two authors read in full all the selected articles, and disagreements were discussed and resolved with the aim of two further authors (GNC and CC). The prevalence or incidence of IBD in a specific syndrome was extracted from the cohort studies, whenever available. Clinical characteristics, medical and surgical treatments, and outcomes of subjects with IBD and a genetic syndrome were summarized. The description of our two cases was performed, following parental consent.
The systematic research identified a total of 986 articles, of which 58 were considered relevant based upon the established inclusion criteria (selection through title and abstract). Furthermore, four papers were identified through reference snowballing. A total of 62 full-text articles were assessed for eligibility and, after accurate reading, 46 were included in the final review. Sixteen articles were excluded, and the reasons for exclusion were as follows: duplicates (reporting cases described elsewhere), not reporting cases of IBD, or not describing either clinical or epidemiological aspects (Figure 2). The 46 articles reported a total of 67 cases of IBD in 6 different syndromes: 37 in TS, 17 in DS, and 13 in other syndromes including 2 cases in DiGeorge syndrome (DGS), 8 cases in neurofibromatosis type 1 (NF1), 2 cases in Kabuki syndrome (KS), and 1 case in Williams syndrome (WS).
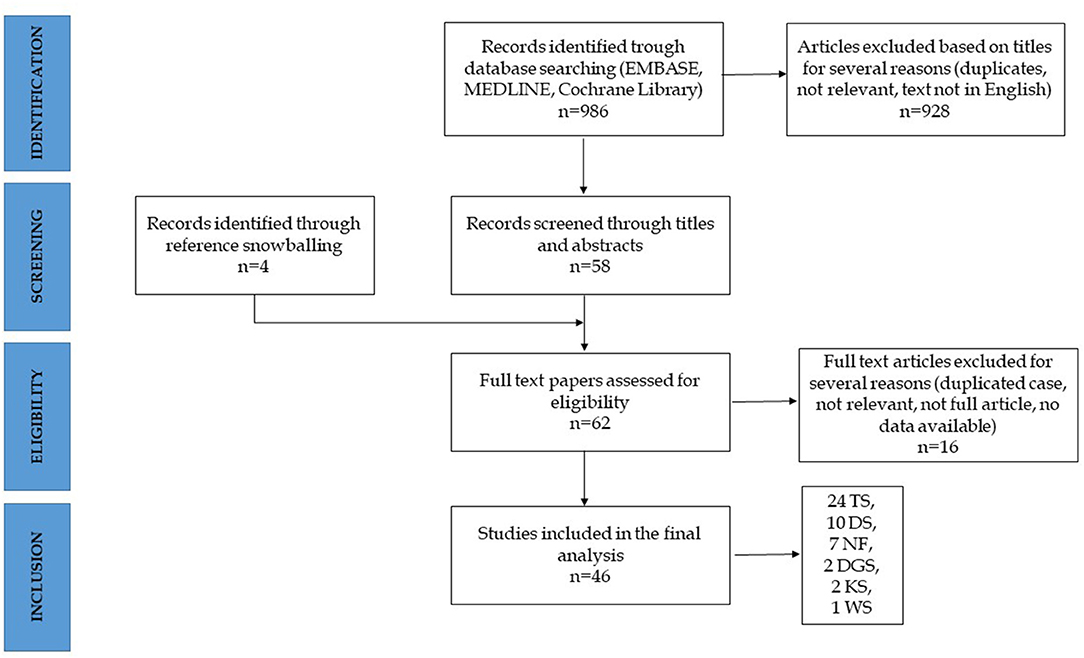
Figure 2. Algorithm of inclusion. TS, Turner's syndrome; DS, Down syndrome; NF, neurofibromatosis; DGS, DiGeorge syndrome; KS, Kabuki syndrome; WS, Williams syndrome.
A total of 24 papers describing cases or reporting epidemiological data of IBD in TS were included. Table 1 illustrates the characteristics of all the 24 studies (10–33). Thirty-seven cases of TS and IBD (17 with CD, 14 with UC, and 6 cases named as IBD) were reported by 21 articles (10–24, 26–29, 32, 33). A detailed clinical description was available for 25 cases only (10–21, 24, 26–29, 33). Table 2 summarizes the clinical features of the published cases and the unpublished cases presented in this article. Thirteen (50%) were pediatric cases (7 CD, 5 UC, and 1 IBD) (11, 12, 14–16, 18, 21, 27, 28).
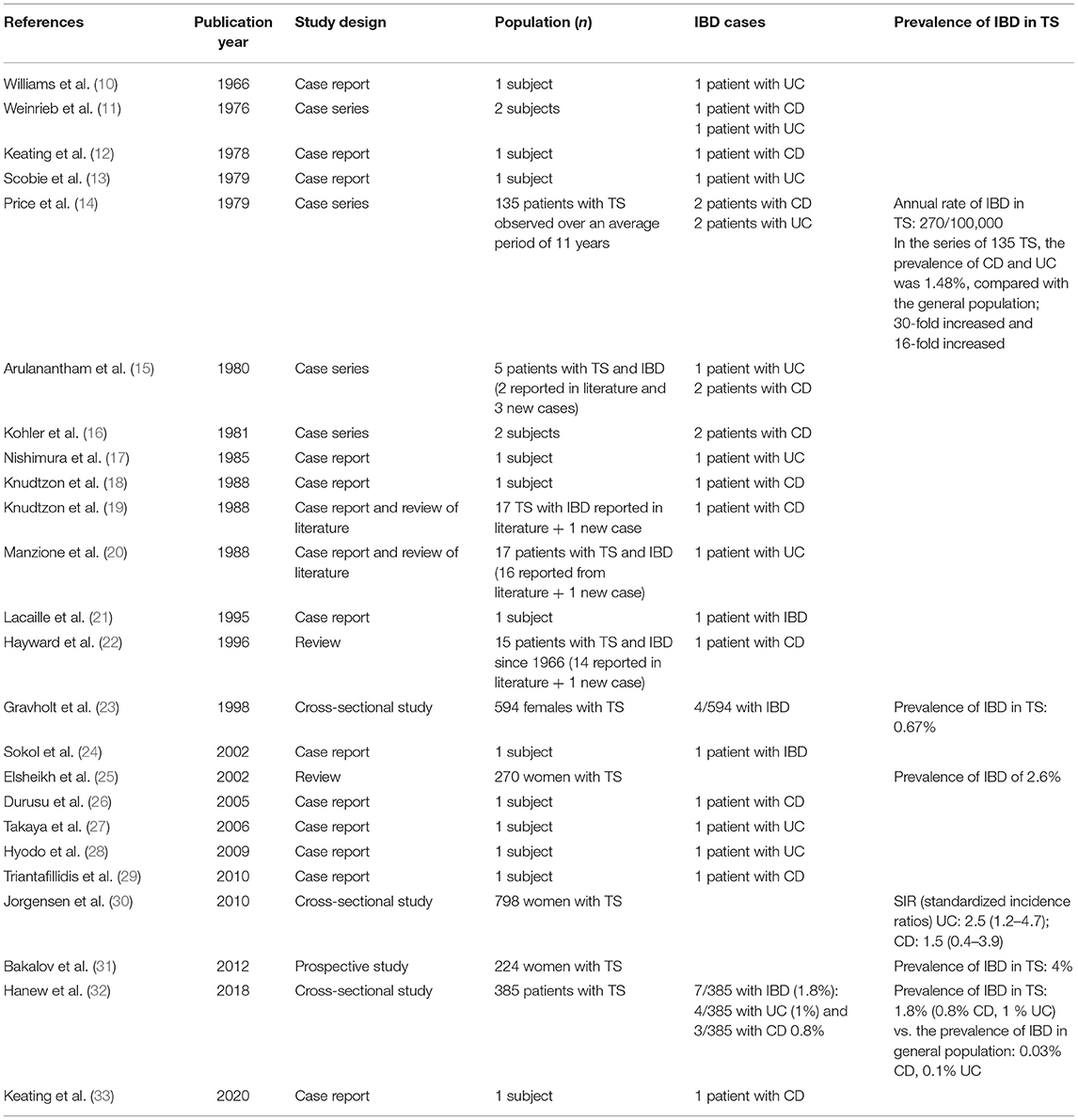
Table 1. Characteristics of published studies reporting cases of IBD in TS subjects.

Table 2. Clinical characteristics of the cases of IBD and TS detailed in literature.
Nine cases (34%) were associated to other comorbidities such as autoimmune diseases (Hashimoto thyroiditis, primary biliary cirrhosis, acquired von Willebrand disease, celiac disease, and primary sclerosing cholangitis) and cardiac defects [bicuspid aortic valve, coarctation of the aorta, and atrial septal defect) (13, 17, 21, 24, 26, 27, 29, 33). Treatment with biologic drugs (infliximab, adalimumab, and ustekinumab) was reported in two cases, and in one case, a drug-induced cardiomyopathy during treatment with adalimumab was described (29, 33). Eight cases (31%) needed surgical treatments (four CD and four UC), and four of them died after surgery because of postoperative complications (cardiorespiratory arrest, hearth attack, and tetanus) (10–12, 14–16, 19).
The karyotype of the included cases was characterized by the absence of an X chromosome (monosomy or mosaic monosomy) in 12 cases (12, 14–16, 18, 21, 24, 26, 27, 29) or by the presence of a structurally abnormal X chromosome (or mosaic pattern with structurally abnormal X chromosome) in the remaining 13 cases (10, 11, 13–17, 19, 20, 28). In one case, the karyotype was not specified (33).
Three articles (a review and two observational studies) reported the prevalence rate of IBD in cohorts of TS without a clinical description of the cases (25, 30, 31). Overall, the prevalence of IBD in TS ranged from 0.67% to 4% (23, 25, 31, 32). One study, performed in Japan, described a 13-fold increase of the prevalence of IBD in women with TS compared to the general population (1.8 vs. 0.13%) with a difference more pronounced in CD (0.8 vs. 0.03%) compared to UC (1 vs. 0.1%) (32). The annual incidence of IBD in women with TS reported by one study was 270/100.000 (14). In another observational study, the standardized incidence ratio (SIR) of IBD in TS compared to the general population was 2.5 (95% CI 1.2–4.7) and 1.5 (95% CI 0.4–3.9) for UC and CD, respectively (30).
Ten articles reported cases of IBD in subjects with DS, adding up to a total of 18 cases, including our cases (12 with CD, 5 with UC, and 1 case reported as IBD) (34–43). Five (28%) were pediatric cases (36, 38, 39, 43). The three observational studies indicated a prevalence of IBD in cohorts of adults with DS ranging from 0.2 to 1.57% (35, 40, 41). Only one study compared data with a control population (subjects not affected by DS), describing adjusted rate ratios of 3.3 for UC (95%CI: 0.7–9.6) and 2.6 for CD (95%CI: 05–7.7) (40). Table 3 illustrates the characteristics of the published studies.
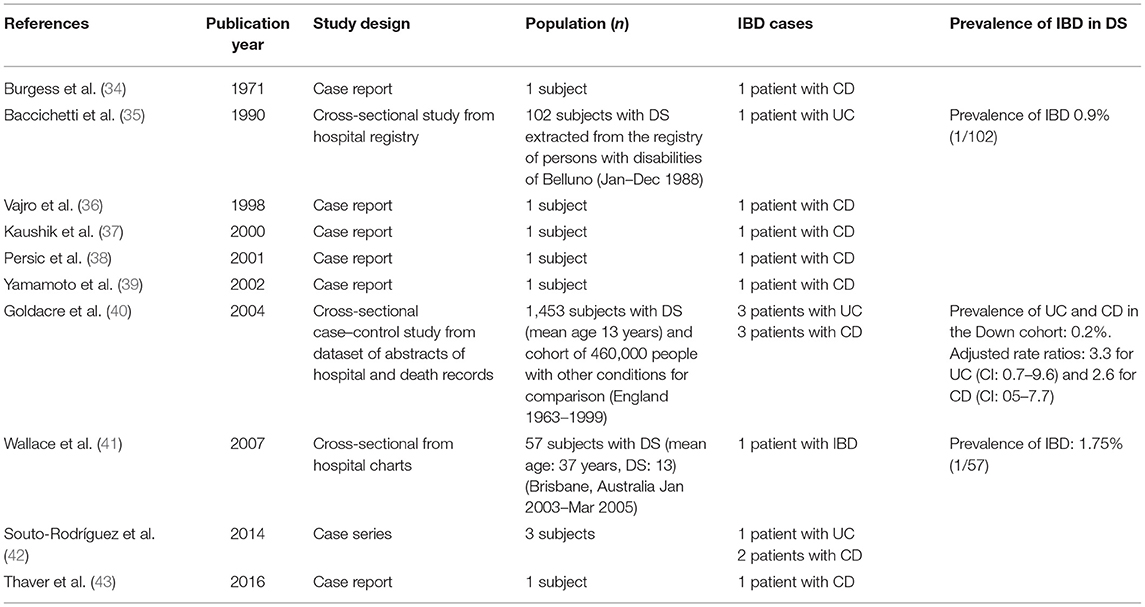
Table 3. Characteristics of published studies reporting cases of IBD in DS subjects.
Detailed clinical descriptions were available for nine cases only (36–39, 42, 43). Table 4 shows the clinical characteristics of the reported cases (including the case presented by our group). Two pediatric cases of CD were also affected by primary sclerosing cholangitis (PSC) (36, 37), one child had CD with pulmonary involvement (requiring infliximab and methotrexate) (43) and one adult woman with UC required colectomy at 34 years of age (42).

Table 4. Clinical characteristics of the cases of IBD and DS detailed in literature.
Twelve articles reported cases of IBD in subjects with other genetic syndromes (44–55). Seven papers reported eight cases of IBD (three CD and five UC) in patients with NF1 (44–50), with two pediatric UC cases (48, 49). Table 5 illustrates the characteristics and results of these studies. Cases of IBD in NF1 were not described as particularly severe, and no lethal cases were reported. Two articles described two cases of IBD in subjects with DGS (an 8-year-old boy and a 17-year-old boy), treated with biological drugs (vedolizumab and infliximab) (54, 55). In literature, two cases with KS and IBD were reported (one child with CD and the other case was not detailed) (52, 53) and one case (a 20-year-old female) with WS and CD, subjected to medical treatment, was also described (51).
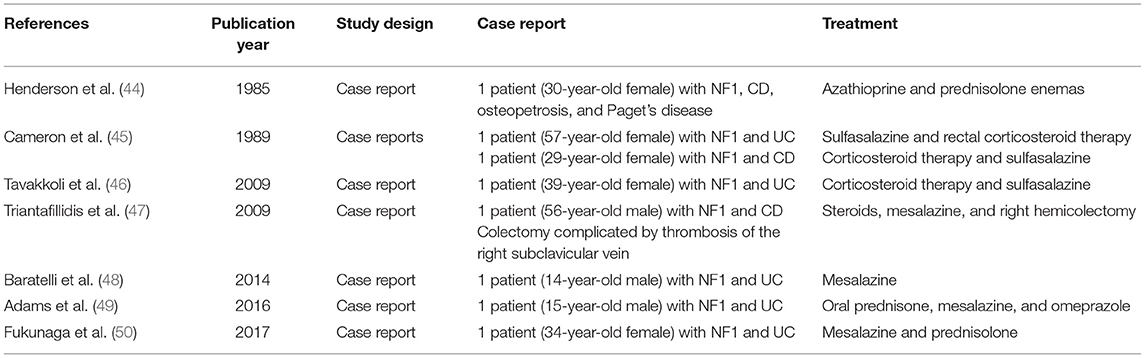
Table 5. Characteristics and main results of studies on NF1 and IBD.
With this systematic review, we aimed to characterize the clinical features and epidemiological aspects of IBD in specific genetic syndromes. Following our inclusion criteria, we identified a total of 69 cases of IBD (considering our two unpublished cases) described in six different genetic syndromes, including DS, TS, NF1, DGS, WS, and KS. DS and TS are conditions related to chromosomal abnormalities (21 and X chromosome, respectively), DGS and WS are chromosomal deletion syndromes (22q and 7q11.23, respectively), while NF1 and KS are related to single-gene mutations (NF1 and MLL2 or KDM6A for NF1 and KS, respectively). All of these are genetic diseases associated with a known risk of autoimmune comorbidities, with the only exception being represented by the NF1, where this association has been rarely described (56–59). The presence of an immunological dysfunction has been widely documented in DS, TS, KS, WS, and DGS. Although the association between these genetic conditions and IBD is known, many confounding factors can delay this diagnosis. In fact, all these syndromes share other relevant features, such as the presence of poor growth or short stature (on a genetic basis) and gastrointestinal symptoms (due to gastrointestinal malformations or comorbidities) that overlap with the main symptoms of IBD. Additionally, a variable degree of intellectual disability is reported in all these syndromes, complicating the symptom reporting and their correct assessment. Therefore, it can be difficult to maintain a high index of suspicion of IBD in this particular scenario, with a possible misdiagnosis or a diagnostic delay. Furthermore, the presence of other comorbidities in all these genetic conditions (e.g., cardiac, renal, hematological, and immunological) can complicate the course of IBD, making the patient more vulnerable to developing drug toxicities, infections, and surgical complications and higher risk of mortality (Figure 1).
The results of our systematic search show that the majority of cases of IBD were described in subjects with TS (38 cases including our novel case). Ten cases were particularly severe, with eight requiring surgery and two treated with biologics. It should be noted that all cases with surgical complications or with a lethal outcome and 50% of cases of TS and IBD were published before the year 2000, when biological treatment was rarely used in IBD. In the biologic era, no mortality for IBD has been recorded in females with TS. No early-onset IBD (before 5 years of age) was reported, apart from our case. We could then speculate that TS is not associated to a particularly severe phenotype of IBD; however, no comparison data with the general IBD population (not affected by TS) is available so far. Interestingly, a case of cardiomyopathy induced by adalimumab in a TS patient with bicuspid aortic valve has been recently presented, suggesting caution when using anti-TNF alpha in this fragile category of patients (33).
Epidemiological data for TS were extracted from six studies. Both prevalence (according to five studies) and incidence (based on two reports) indicated a higher risk of IBD in females with TS compared to the general population. These data undoubtedly indicate the need for a gastroenterological follow-up in subjects with TS.
The link between TS and autoimmune or inflammatory disorders could be mediated by genes located on the X-chromosome (e.g., the MHC-paralogs and the Toll-like receptor 8 signaling) and hormonal imbalance (characterized by elevated FSH and LH) that can activate immune pathogenic mechanisms. Based on a previous review of literature including a total of 15 cases of IBD in TS, it has been suggested that females with the isochromosome Xq (found in 60% of the women with IBD and TS vs. 17% of TS women without IBD) are more susceptible to IBD compared to those with the monosomy X (22). Our review, based on 25 genetically confirmed cases, does not reiterate this data, indicating an equal distribution of monosomy and structurally abnormal X chromosome.
Even if cases of IBD and DS were less frequently reported in literature, with a total of 13 adults and 5 children described (including our case), prevalence data (retrospectively collected) from two cross-sectional studies and from one cross-sectional controlled study show a two- to three-fold increase in the prevalence of IBD in DS compared to the general population (35, 40, 41). No longitudinal study was available, with a lack of data on the incidence of IBD in cohorts of subjects with DS. Plausible reasons for an increased risk of autoimmune disorders in DS include the following: involvement of genes on chromosome 21 implicated in the immune response (such as interferon receptor, CD 18, or the gene AIRE), defective T-cell control mechanisms and a reduced expansion of T-cell precursors in the thymus, a higher representation of immature NK cells with lower intrinsic activity, reduced IL-2 expression, defective phagocyte chemotaxis, and oxygen radical production (55).
Besides our pediatric case, one other case described in literature was particularly complicated due to the pulmonary involvement, which is a very rare complication of CD (43). Our case was challenging for several reasons, specifically, 1. the presence of a previously unrecognized esophageal stricture and a long-standing history of dysphagia with solid food refusal and 2. the poor response to standard treatments. Esophageal involvement in CD is uncommon, and its presentation with an esophageal stricture is even less common. The most frequent symptoms are dysphagia and odynophagia, and a large number of patients (up to 1/3) also have oral lesions. Esophageal CD can present as an erosive–ulcerative esophagitis or with an esophageal stricture (generally in the medium or distal esophagus) or fistula. Histology can be unspecific, adding on diagnostic difficulties (60, 61). Differential diagnoses include congenital stenosis or post esophageal atresia repair, gastroesophageal reflux disease, eosinophilic esophagitis, unknown caustic ingestion, and few other rarer causes. All of these conditions should be investigated in patients with DS and esophageal stricture, considering the high incidence of congenital gastrointestinal malformations, gastrointestinal reflux, and intellectual disability (with possible unrecorded foreign bodies or caustic ingestion) (62). In our case, the proximal esophageal stricture was considered compatible with CD, although histology was not specific and the response to anti-inflammatory treatments was poor, requiring endoscopic dilation. Even if there was no history, an origin related to a previous ingestion of caustics of foreign body cannot completely be excluded in our case, considering the severe intellectual disability, equally a congenital origin.
According to the current CD guidelines (63), our patient was considered at high risk at diagnosis; therefore, upfront infliximab was chosen as induction treatment along with EEN. An immunomodulator (azathioprine) was introduced at the first relapse (3 months after diagnosis) and, despite the optimization of infliximab schedule (10 mg/kg every 4 weeks) and the combination with azathioprine, the response was unsatisfactory with several course of steroids required during the follow-up and the necessity to switch to adalimumab. In other cases of IBD in DS reported in literature, response to standard treatments is described as good, except for one case of UC where colectomy was required and the CD case with pulmonary involvement that was treated with infliximab and methotrexate. Patients with DS are generally prone to drug side effects; this is frequently reported with some antiblastic agents, and toxicities (including cardiotoxicity, myelosuppression, infections, and mucositis) have a multifactorial origin (64–67). The greater risk of side effects with high doses of methotrexate in acute leukemia in patients with DS is a typical example; therefore, specific protocols with dose reduction for subjects with DS are currently used (68–71). In the same subjects, a higher accumulation of thioguanine inside erythrocytes in comparison to non-DS children has also been described (72). No specific reports of toxicities related to IBD medications (including thiopurines, methotrexate, and anti-TNF-α) in subjects with DS have been published so far. The risk of malignancy is another concern dealing with DS. Trisomy 21 is associated with a 10- to 20-fold risk for developing acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML), as compared to non-DS children, while the risk of developing solid tumors is not increased (73, 74). Long-term use of some IBD medications (mainly thiopurines with or without anti TNF-α) has been associated to an increased risk of malignancy, particularly a rare form of lymphoma (hepatosplenic T-cell lymphoma) (75–79). In the absence of specific data on toxicity and carcinogenesis of IBD medications in subjects with DS, caution and close clinical follow-up is recommended, particularly with drugs such as azathioprine, methotrexate, and anti-TNF-α.
Lastly, the systematic research identified sporadic cases of IBD in other genetic syndromes. Interestingly, eight cases (six adults and two children), five with UC, have been described in NF1 with five cases reported since 2009. Unfortunately, there are no longitudinal or controlled studies exploring the link between NF1 and IBD, but as more cases accumulate, a correlation beyond coincidence becomes likely. NF1 is one of the RASopathies, a group of genetic syndromes due to germline mutations in genes that encode protein components of the RAS–mitogen-activated protein kinase (MAPK) pathway. RAS has an essential role in adaptive immunity and normal immune cell function, and the MAPK is crucial in the process of inflammation and apoptosis (80). Activated MAPK pathway has been reported previously in acetic acid-induced colitis (81, 82), and MAPK inhibitors were found to reduce inflammation (83) and specifically to ameliorate colitis in animal models of IBD (84, 85). These evidences can explain the basis for a susceptibility to IBD in patients with NF1. Furthermore, an altered mast cell function has been advocated as a possible common mechanism in the pathogenesis of both NF1 and IBD (particularly UC) and could be a link between the two conditions (86–90). From the systematic review, there is no evidence of an enhanced severity of these cases; however, NF1 is another condition associated to the risk of malignancy; therefore, a particular attention to the treatment choice should be reserved to these patients.
Despite the rigorous methodology followed and the extensive review (including several genetic syndromes), this systematic review has some limitations. The major limitations are related to the quality of the studies under review and the small number of available studies. Our results were necessarily based on case reports and series and few cohort studies; as a result, it was not possible to perform a meta-analysis. These limitations further highlight the need for the following: 1. population-based epidemiological studies for specific syndromes focused on the development of IBD and 2. specific studies or registry data sub-analysis investigating clinical characteristics, outcomes, and drug toxicities in subjects with IBD and a genetic syndrome compared to subjects with IBD without an underlying genetic condition.
In conclusion, we found evidence of IBD in several genetic syndromes, with most of the reported cases in TS and DS. Literature on the coexistence of NF1 and IBD has been accumulating in the last years, suggesting a possible link between these two conditions. Both the prevalence and incidence of IBD seem to be increased in subjects with TS and DS compared to the general population, although there is a paucity of high-quality and longitudinal epidemiological data. Patients with IBD and a genetic syndrome require a particular attention during the follow-up, considering the possible association with gastrointestinal and extra-intestinal comorbidities. A careful choice of the immunosuppressive regimens is necessary, particularly in DS, relating to the high risk of malignancy and the enhanced susceptibility to drug side effects, and the development of targeted protocols is required.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
SG and CC designed the study. SG and GG performed literature search and review, wrote the manuscript, and critically revised it. SG, GG, GC, and CC critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Actis GC, Pellicano R. The pathologic galaxy modulating the genotype and phenotype of inflammatory bowel disease: comorbidity, contiguity, and genetic and epigenetic factors. Minerva Med. (2016) 107:401–12.
2. de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. (2017) 49:256–61. doi: 10.1038/ng.3760
3. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. (2012) 491:119–24. doi: 10.1038/nature11582
4. Liu JZ, van Sommeren S, Huang H, Ng SC, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. (2015) 47:979–86. doi: 10.1038/ng.3359
5. Uniken Venema WT, Voskuil MD, Dijkstra G, Weersma RK, Festen EA. The genetic background of inflammatory bowel disease: from correlation to causality. J Pathol. (2017) 241:146–58. doi: 10.1002/path.4817
6. Uhlig HH, Muise AM. Clinical genomics in inflammatory bowel disease. Trends Genet. (2017) 33:629–41. doi: 10.1016/j.tig.2017.06.008
7. Lee KH, Ahn BS, Cha D, Jang WW, Choi E, Park S, et al. Understanding the immunopathogenesis of autoimmune diseases by animal studies using gene modulation: a comprehensive review. Autoimmun Rev. (2020) 19:102469. doi: 10.1016/j.autrev.2020.102469
8. Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JPA, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. (2009) 339:b2700. doi: 10.1136/bmj.b2700
9. Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. (2009) 151:264–9. doi: 10.7326/0003-4819-151-4-200908180-00135
10. Williams ED, Engel E, Taft PD, Forbes AP. Gonadal dysgenesis and ulcerative colitis. A case report with clinical, cytogenetic, and post-mortem studies. J Med Genet. (1966) 3:51–5. doi: 10.1136/jmg.3.1.51
11. Weinrieb IJ, Fineman RM, Spiro HM. Turner syndrome and inflammatory bowel disease. N Engl J Med. (1976) 294:1221–2. doi: 10.1056/NEJM197605272942207
12. Keating JP, Ternberg JL, Packman R. Association of Crohn disease and turner syndrome. J Pediatr. (1978) 92:160–1. doi: 10.1016/S0022-3476(78)80104-8
13. Scobie BA. Co-existing coeliac and inflammatory bowel disease in a patient with Turner's syndrome. Aust N Z J Med. (1979) 9:316–7. doi: 10.1111/j.1445-5994.1979.tb04148.x
14. Price WH. A high incidence of chronic inflammatory bowel disease in patients with Turner's syndrome. J Med Genet. (1979) 16:263–6. doi: 10.1136/jmg.16.4.263
15. Arulanantham K, Kramer MS, Gryboski JD. The association of inflammatory bowel disease and X chromosomal abnormality. Pediatrics. (1980) 66:63–7.
16. Kohler JA, Grant DB. Crohn's disease in Turner's syndrome. Br Med J. (1981) 282:950. doi: 10.1136/bmj.282.6268.950
17. Nishimura H, Kino M, Kubo S, Kawamura K. Hashimoto's thyroiditis and ulcerative colitis in a patient with Turner's syndrome. JAMA. (1985) 254:357. doi: 10.1001/jama.1985.03360030047008
18. Knudtzon J, Ledaal P, Middelthon-Moe M, Aarskog D. 45,X/47,XY,+13 mosaicism and Crohn's disease. Acta Paediatr Scand. (1988) 77:922–4. doi: 10.1111/j.1651-2227.1988.tb10784.x
19. Knudtzon J, Svane S. Turner's syndrome associated with chronic inflammatory bowel disease. a case report and review of the literature. Acta Med Scand. (1988) 223:375–8. doi: 10.1111/j.0954-6820.1988.tb15887.x
20. Manzione NC, Kram M, Kram E, Das KM. Turner's syndrome and inflammatory bowel disease: a case report with immunologic studies. Am J Gastroenterol. (1988) 83:1294–7.
21. Lacaille F, Canioni D, Bernard O, Fabre M, Brousse N, Schmitz J. Celiac disease, inflammatory colitis, and primary sclerosing cholangitis in a girl with Turner's syndrome. J Pediatr Gastroenterol Nutr. (1995) 21:463–7. doi: 10.1097/00005176-199511000-00017
22. Hayward PA, Satsangi J, Jewell DP. Inflammatory bowel disease and the X chromosome. QJM. (1996) 89:713–8. doi: 10.1093/qjmed/89.9.713
23. Gravholt CH, Juul S, Naeraa RW, Hansen J. Morbidity in turner syndrome. J Clin Epidemiol. (1998) 51:147–58. doi: 10.1016/S0895-4356(97)00237-0
24. Sokol L, Stueben ET, Jaikishen JP, Lamarche MB. Turner syndrome associated with acquired von Willebrand disease, primary biliary cirrhosis, and inflammatory bowel disease. Am J Hematol. (2002) 70:257–9. doi: 10.1002/ajh.10120
25. Elsheikh M, Dunger DB, Conway GS, Wass JA. Turner's syndrome in adulthood. Endocr Rev. (2002) 23:120–40. doi: 10.1210/edrv.23.1.0457
26. Durusu M, Gurlek A, Simsek H, Balaban Y, Tatar G. Coincidence or causality: celiac and Crohn diseases in a case of Turner syndrome. Am J Med Sci. (2005) 329:214–6. doi: 10.1097/00000441-200504000-00010
27. Takaya J, Teraguchi M, Ikemoto Y, Yoshimura K, Yamato F, Higashino H, et al. Turner syndrome associated with ulcerative colitis. Clin Pediatr Endocrinol. (2006) 15:97–100. doi: 10.1297/cpe.15.97
28. Hyodo H, Tomita Y, Hirai K, Hirakawa H, Ueno S, Ishiguro H. Turner syndrome with ulcerative colitis. Clin Pediatr Endocrinol. (2009) 18:101–5. doi: 10.1297/cpe.18.101
29. Triantafillidis JK, Nadia el F, Fostira F, Terzoudi G, Fouskas J, Pinis S. Turner's syndrome, autoimmune thyroiditis, and Crohn's disease in the same patient: a combination emphasizing the role of X-chromosome in inflammatory bowel disease patients. Inflamm Bowel Dis. (2010) 16:1088–9. doi: 10.1002/ibd.21125
30. Jorgensen KT, Rostgaard K, Bache I, Biggar RJ, Nielsen NM, Tommerup N, et al. Autoimmune diseases in women with Turner's syndrome. Arthritis Rheum. (2010) 62:658–66. doi: 10.1002/art.27270
31. Bakalov VK, Gutin L, Cheng CM, Biggar RJ, Nielsen NM, Tommerup N, et al. Autoimmune disorders in women with turner syndrome and women with karyotypically normal primary ovarian insufficiency. J Autoimmun. (2012) 38:315–21. doi: 10.1016/j.jaut.2012.01.015
32. Hanew K, Tanaka T, Horikawa R, Hasegawa T, Yokoya S. Prevalence of diverse complications and its association with karyotypes in Japanese adult women with Turner syndrome-a questionnaire survey by the Foundation for Growth Science. Endocr J. (2018) 65:509–19. doi: 10.1507/endocrj.EJ17-0401
33. Keating E, Kelleher TB, Lahiff C. De novo Anti-TNF-alpha-induced congestive heart failure in a patient with turner syndrome and Crohn's Disease. Inflamm Bowel Dis. (2020) 26:e161–e2. doi: 10.1093/ibd/izaa176
35. Baccichetti C, Lenzini E, Pegoraro R. Down syndrome in the Belluno district (Veneto region, northeast Italy): age distribution and morbidity. Am J Med Genet Suppl. (1990) 7:84–6. doi: 10.1002/ajmg.1320370716
36. Vajro P, Cucchiara S, Vegnente A, Iorio R, de Silva C, Cipolletta L, et al. Primary sclerosing cholangitis preceding Crohn's disease in a child with Down's syndrome. Dig Dis Sci. (1998) 43:166–9. doi: 10.1023/A:1018800826608
37. Kaushik SP, Kaye G, Clarke AC. Autoimmune hepatobiliary disease in trisomy 21. J Clin Gastroenterol. (2000) 30:330–2. doi: 10.1097/00004836-200004000-00029
38. Persic M, Dessardo S, Subat-Dezulovic M, Ahel V, Rozmanic V. Down syndrome and Crohn's disease: an extremely rare association. Pediatr Int. (2001) 43:519–21. doi: 10.1046/j.1442-200X.2001.01431.x
39. Yamamoto M, Abo W, Hori T, et al. Crohn's disease in a child with Down syndrome. Pediatr Int. (2002) 44:537–9. doi: 10.1046/j.1442-200X.2002.01598.x
40. Goldacre MJ, Wotton CJ, Seagroatt V, Yeates D. Cancers and immune related diseases associated with Down's syndrome: a record linkage study. Arch Dis Child. (2004) 89:1014–7. doi: 10.1136/adc.2003.046219
41. Wallace RA. Clinical audit of gastrointestinal conditions occurring among adults with Down syndrome attending a specialist clinic. J Intellect Dev Disabil. (2007) 32:45–50. doi: 10.1080/13668250601146761
42. Souto-Rodriguez R, Barreiro-de-Acosta M, Dominguez-Munoz JE. Down's syndrome and inflammatory bowel disease: is there a real link? Rev Esp Enferm Dig. (2014) 106:220–2.
43. Thaver D, Beg M. Pulmonary Crohn's disease in down syndrome: a link or linkage problem. Case Rep Gastroenterol. (2016) 10:206–11. doi: 10.1159/000445975
44. Henderson RG, Long PJ, Al-Nafussi AI, Coombs RR, Gibson RN. Paget's disease with benign osteopetrosis and Crohn's disease. J R Soc Med. (1985) 78:766–8. doi: 10.1177/014107688507800913
45. Cameron EM, Raeburn A, Ford MJ. Neurofibromatosis and inflammatory bowel disease. Scott Med J. (1989) 34:500–1. doi: 10.1177/003693308903400407
46. Tavakkoli H, Asadi M, Mahzouni P, Foroozmehr A. Ulcerative colitis and neurofibromatosis type 1 with bilateral psoas muscle neurofibromas: a case report. J Res Med Sci. (2009) 14:261-5.
47. Triantafillidis JK, Peros G, Merikas E, Malgarinos G, Gikas A. Crohn's disease in a patient with von recklinghausen's disease: A rare combination of two disorders with strong genetic background. Annal. Gastroenterol. (2009) 22:119–22.
48. Baratelli F, Le M, Gershman GB, French SW. Do mast cells play a pathogenetic role in neurofibromatosis type 1 and ulcerative colitis? Exp Mol Pathol. (2014) 96:230–4. doi: 10.1016/j.yexmp.2014.02.006
49. Adams W, Mitchell L, Candelaria-Santiago R, Hefner J, Gramling J. Concurrent ulcerative colitis and neurofibromatosis type 1: the question of a common pathway. Pediatrics. (2016) 137:e20150973. doi: 10.1542/peds.2015-0973
50. Fukunaga S, Takedatsu H, Mitsuyama K, Torimura T. A rare case of ulcerative colitis with neurofibromatosis type 1. Kurume Med J. (2018) 64:25–7. doi: 10.2739/kurumemedj.MS00014
51. Gilbert-Barness E, Fox T, Morrow G, Luquette M, Pomerance HH. Williams syndrome associated with Crohn disease, multiple infections, and chronic granulomatous disease. Fetal Pediatr Pathol. (2004) 23:29–37. doi: 10.1080/15227950490423016
52. Ho J, Fox D, Innes AM, McLeod R, Butzner D, Johnson N, et al. Kabuki syndrome and Crohn disease in a child with familial hypocalciuric hypercalcemia. J Pediatr Endocrinol Metab. (2010) 23:975–9. doi: 10.1515/jpem.2010.156
53. Margot H, Boursier G, Duflos C. Immunopathological manifestations in Kabuki syndrome: a registry study of 177 individuals. Genet Med. (2020) 22:181-8. doi: 10.1038/s41436-019-0623-x
54. Thau E, Moore HA, Stein P, Chen K. Efficacy of Vedolizumab for inflammatory bowel disease in the setting of DiGeorge syndrome. J Pediatr Gastroenterol Nutr. (2019) 68:e88. doi: 10.1097/MPG.0000000000002322
55. Uy R, Jacobs N, Mziray-Andrew C. Inflammatory bowel disease and diverticulosis in an adolescent with digeorge syndrome. J Pediatr Gastroenterol Nutr. (2016) 62:e43–5. doi: 10.1097/MPG.0000000000000497
56. Ferrari M, Stagi S. Autoimmunity and genetic syndromes: a focus on down syndrome. Genes. (2021) 12:20268. doi: 10.3390/genes12020268
57. Kanakatti Shankar R. Immunological profile and autoimmunity in turner syndrome. Horm Res Paediatr. (2020) 93:415–22. doi: 10.1159/000512904
58. Stagi S, Gulino AV, Lapi E, Rigante D. Epigenetic control of the immune system: a lesson from Kabuki syndrome. Immunol Res. (2016) 64:345–59. doi: 10.1007/s12026-015-8707-4
59. Casto C, Pepe G, Li Pomi A, Corica D, Aversa T, Wasniewska M. Hashimoto's thyroiditis and graves' disease in genetic syndromes in pediatric age. Genes. (2021) 12:222. doi: 10.3390/genes12020222
60. Vale Rodrigues R, Sladek M, Katsanos K, van der Woude CJ, Wei J, Vavricka SR, et al. Diagnosis and outcome of oesophageal crohn's disease. J Crohns Colitis. (2020) 14:624–9. doi: 10.1093/ecco-jcc/jjz201
61. Decker GA, Loftus EV, Pasha TM, Tremaine WJ, Sandborn WJ. Crohn's disease of the esophagus: clinical features and outcomes. Inflamm Bowel Dis. (2001) 7:113–9. doi: 10.1097/00054725-200105000-00006
62. Ravel A, Mircher C, Rebillat AS, Cieuta-Walti C, Megarbane A. Feeding problems and gastrointestinal diseases in down syndrome. Arch Pediatr. (2020) 27:53–60. doi: 10.1016/j.arcped.2019.11.008
63. van Rheenen PF, Aloi M, Assa A, Bronsky J, Escher JC, Fagerberg UL, et al. The medical management of paediatric Crohn's disease: an ECCO-ESPGHAN guideline update. J Crohns Colitis. (2021) 15:171–94. doi: 10.1093/ecco-jcc/jjaa161
64. O'Brien MM, Taub JW, Chang MN, Massey GV, Stine KC, RaimondiDavid Becton SC, et al. Cardiomyopathy in children with down syndrome treated for acute myeloid leukemia: a report from the children's oncology group study POG 9421. J Clin Oncol. (2008) 26:414–20. doi: 10.1200/JCO.2007.13.2209
65. Blatt J, Albo V, Prin W, Orlando S, Wollman M. Excessive chemotherapy-related myelotoxicity in children with Down syndrome and acute lymphoblastic leukaemia. Lancet. (1986) 2:914. doi: 10.1016/S0140-6736(86)90429-0
66. Palle J, Frost BM, Peterson C, Gustafsson G, Hellebostad M, Kanerva J, et al. Doxorubicin pharmacokinetics is correlated to the effect of induction therapy in children with acute myeloid leukemia. Anticancer Drugs. (2006) 17:385–92. doi: 10.1097/01.cad.0000198911.98442.16
67. Hefti E, Blanco JG. Anthracycline-related cardiotoxicity in patients with acute myeloid leukemia and down syndrome: a literature review. Cardiovasc Toxicol. (2016) 16:5–13. doi: 10.1007/s12012-015-9307-1
68. Peeters M, Poon A. Down syndrome and leukemia: unusual clinical aspects and unexpected methotrexate sensitivity. Eur J Pediatr. (1987) 146:416–22. doi: 10.1007/BF00444952
69. Buitenkamp TD, Izraeli S, Zimmermann M, Forestier E, Heerema NA, van den Heuvel-Eibrink MM, et al. Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood. (2014) 123:70–7. doi: 10.1182/blood-2013-06-509463
70. Buitenkamp TD, Mathot RA, de Haas V, Pieters R, Zwaan CM. Methotrexate-induced side effects are not due to differences in pharmacokinetics in children with Down syndrome and acute lymphoblastic leukemia. Haematologica. (2010) 95:1106–13. doi: 10.3324/haematol.2009.019778
71. Kroll M, Kaupat-Bleckmann K, Morickel A, Altenl J, Schewel DM, Stanullal M, et al. Methotrexate-associated toxicity in children with Down syndrome and acute lymphoblastic leukemia during consolidation therapy with high dose methotrexate according to ALL-BFM treatment regimen. Haematologica. (2020) 105:1013–20. doi: 10.3324/haematol.2019.224774
72. Palle J, Frost BM, Petersson C, Hasle H, Hellebostad M, Kanerva J, et al. Thioguanine pharmacokinetics in induction therapy of children with acute myeloid leukemia. Anticancer Drugs. (2009) 20:7–14. doi: 10.1097/CAD.0b013e32831bc086
73. Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down's syndrome. Lancet. (2000) 355:165–9. doi: 10.1016/S0140-6736(99)05264-2
74. Rowley JD. Down Syndrome and acute leukaemia: increased risk may be due to trisomy 21. Lancet. (1981) 2:1020–2. doi: 10.1016/S0140-6736(81)91218-6
75. Herrinton LJ, Liu L, Weng X, Lewis JD, Hutfless S, Allison JE. Role of thiopurine and anti-TNF therapy in lymphoma in inflammatory bowel disease. Am J Gastroenterol. (2011) 106:2146–53. doi: 10.1038/ajg.2011.283
76. Beaugerie L, Brousse N, Bouvier AM, Frédéric Colombel J, Lémann M, Cosnes J, et al. Lymphoproliferative disorders in patients receiving thiopurines for inflammatory bowel disease: a prospective observational cohort study. Lancet. (2009) 374:1617–25. doi: 10.1016/S0140-6736(09)61302-7
77. Afif W, Sandborn WJ, Faubion WA, Rahman M, Harmsen SW, Zinsmeister AR, et al. Risk factors for lymphoma in patients with inflammatory bowel disease: a case-control study. Inflamm Bowel Dis. (2013) 19:1384–9. doi: 10.1097/MIB.0b013e318281325e
78. Lemaitre M, Kirchgesner J, Rudnichi A, et al. Association between use of thiopurines or tumor necrosis factor antagonists alone or in combination and risk of lymphoma in patients with inflammatory bowel disease. JAMA. (2017) 318:1679–86. doi: 10.1001/jama.2017.16071
79. Annese V, Beaugerie L, Egan L, Biancone L, Bolling C, Brandts C, et al. European evidence-based consensus: inflammatory bowel disease and malignancies. J Crohns Colitis. (2015) 9:945–65. doi: 10.1093/ecco-jcc/jjv141
80. Genot E, Cantrell DA. Ras regulation and function in lymphocytes. Curr Opin Immunol. (2000) 12:289–94. doi: 10.1016/S0952-7915(00)00089-3
81. Topcu-Tarladacalisir Y, Akpolat M, Uz YH, Kizilay G, Sapmaz-Metin M, Cerkezkayabekir A, et al. Effects of curcumin on apoptosis and oxidoinflammatory regulation in a rat model of acetic acid-induced colitis: the roles of c-Jun N-terminal kinase and p38 mitogen-activated protein kinase. J Med Food. (2013) 16:296–305. doi: 10.1089/jmf.2012.2550
82. Almeer RS, Mahmoud SM, Amin HK, Abdel Moneim AE. Ziziphus spina-christi fruit extract suppresses oxidative stress and p38 MAPK expression in ulcerative colitis in rats via induction of Nrf2 and HO-1 expression. Food Chem Toxicol. (2018) 115:49–62. doi: 10.1016/j.fct.2018.03.002
83. Terajima M, Inoue T, Magari K, Yamazaki H, Higashi Y, Mizuhara H. Anti-inflammatory effect and selectivity profile of AS1940477, a novel and potent p38 mitogen-activated protein kinase inhibitor. Eur J Pharmacol. (2013) 698:455-62. doi: 10.1016/j.ejphar.2012.11.021
84. Arafa E-S A, Mohamed WR, Zaher DM, Omar HA. Gliclazide attenuates acetic acid-induced colitis via the modulation of PPARγ, NF-κB and MAPK signaling pathways. Toxicol Appl Pharmacol. (2020) 391:114919. doi: 10.1016/j.taap.2020.114919
85. Bai X-S, Bai G, Tang L-D, Li Y, Huan Y, Wang H. MiR-195 alleviates ulcerative colitis in rats via MAPK signaling pathway. Eur Rev Med Pharmacol Sci. (2020) 24:2640–686. doi: 10.26355/eurrev_202003_20533
86. He SH. Key role of mast cells and their major secretory products in inflammatory bowel disease. World J Gastroenterol. (2004) 10:309–18. doi: 10.3748/wjg.v10.i3.309
87. Chen S, Burgin S, McDaniel A, Li X, Yuan J, Chen M, et al. Nf1-/- Schwann cell-conditioned medium modulates mast cell degranulation by c-Kit-mediated hyperactivation of phosphatidylinositol 3-kinase. Am J Pathol. (2010) 177:3125–32. doi: 10.2353/ajpath.2010.100369
88. Staser K, Yang FC, Clapp DW. Mast cells and the neurofibroma microenvironment. Blood. (2010) 116:157–64. doi: 10.1182/blood-2009-09-242875
89. Yang FC, Staser K, Clapp DW. The plexiform neurofibroma microenvironment. Cancer Microenviron. (2012) 5:307–10. doi: 10.1007/s12307-012-0115-x
Keywords: inflammatory bowel disease, Crohn disease, ulcerative colitis, Down syndrome, Turner syndrome, genetic syndromes, neurofibromatosis, DiGeorge syndrome
Citation: Gatti S, Gelzoni G, Catassi GN and Catassi C (2021) The Clinical Spectrum of Inflammatory Bowel Disease Associated With Specific Genetic Syndromes: Two Novel Pediatric Cases and a Systematic Review. Front. Pediatr. 9:742830. doi: 10.3389/fped.2021.742830
Received: 16 July 2021; Accepted: 22 September 2021;
Published: 26 October 2021.
Edited by:
Pietro Vajro, University of Salerno, ItalyReviewed by:
Rinaldo Pellicano, Molinette Hospital, ItalyCopyright © 2021 Gatti, Gelzoni, Catassi and Catassi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simona Gatti, c2ltb25hLmdhdHRpQGhvdG1haWwuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.