
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 20 August 2021
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.716786
 Daniele Zama1
Daniele Zama1 Edoardo Muratore1*
Edoardo Muratore1* Arianna Giannetti1
Arianna Giannetti1 Iria Neri2
Iria Neri2 Francesca Conti1
Francesca Conti1 Pamela Magini3
Pamela Magini3 Simona Ferrari3
Simona Ferrari3 Andrea Pession1
Andrea Pession1Hereditary alpha tryptasemia (HαT) is a recently described autosomal dominant genetic trait caused by an increased copy number of the TPSAB1 gene. It commonly leads to elevated basal serum tryptase levels, and it is associated with heterogeneous clinical manifestations. Some individuals report few to no symptoms, while others present with a spectrum of debilitating features. Most symptoms related to HαT may be explained by mast cell activation and mediator release, namely multiple allergies, anaphylaxis, and skin rash. However, the genotype-phenotype correlation has not yet been clearly understood. In particular, the characterization of the clinical spectrum lacks in children, where differential diagnosis could be challenging. Systemic mastocytosis, HαT, and mast cell activation syndrome are all associated with overlapping pathophysiology and symptoms, making the distinction between these conditions a difficult task. We herein describe two pediatric cases of HαT and their respective families at our tertiary care teaching hospital, highlighting the diagnostic workup and differential diagnosis. We also provide a brief review of the literature to underline the peculiar features of this condition in children.
Hereditary alpha tryptasemia (HαT) is a highly penetrant autosomal dominant genetic trait associated with an increased copy number of the TPSAB1 gene on chromosome 16 (1). The alpha-tryptase-encoding locus presents a high frequency of copy number variations. For example, 29% of people express no alpha-tryptase alleles without clinical manifestations, more commonly in Caucasians (up to 45%) and less frequently in other ethnical background (2). In addition to the canonical genotypes, increased germline TPSAB1 copy number has recently been described in association with elevated basal serum tryptase and clinical features compatible with mast cell activation syndrome (MCAS) (1).
This new genetic condition, HαT, described by Lyons et al. in 2016, potentially explains the majority of cases with slightly elevated basal serum tryptase (3, 4). The upper limit of normal serum tryptase is mainly defined as 11.4 ng/ml, and it is believed to be elevated at baseline in a high percentage of Caucasians, around 5% (5). Most patients with increased TPSAB1 copy number have elevated basal serum tryptase levels (1, 6, 7). The clinical expressivity of the genetic trait is variable and not yet fully characterized. Some individuals report few to no symptoms, while others present a spectrum of debilitating clinical features. Recurrent cutaneous symptoms, like urticaria, angioedema, and skin flushing, are commonly reported (1, 4). Gastrointestinal complaints are also frequent, comprising abdominal pain, gastroesophageal reflux, diarrhea, and food intolerances, sometimes meeting the required criteria for the diagnosis of functional gastrointestinal disorders (1, 4). Systemic allergic reactions and anaphylaxis are key clinical features, most notably triggered by hymenoptera venom (8). Autonomic dysfunction, comprising postural tachycardia syndrome and hypotension, retained primary dentition, behavioral complaints, sleep disturbance, chronic pain, and joint hypermobility, are also reported (1, 4, 6). Moreover, a higher prevalence of increased TPSAB1 germline copy number was observed in a cohort of patients with mastocytosis than the general population (17.2 vs. 4.4%), suggesting a possible correlation between the two conditions (9) (Table 1).
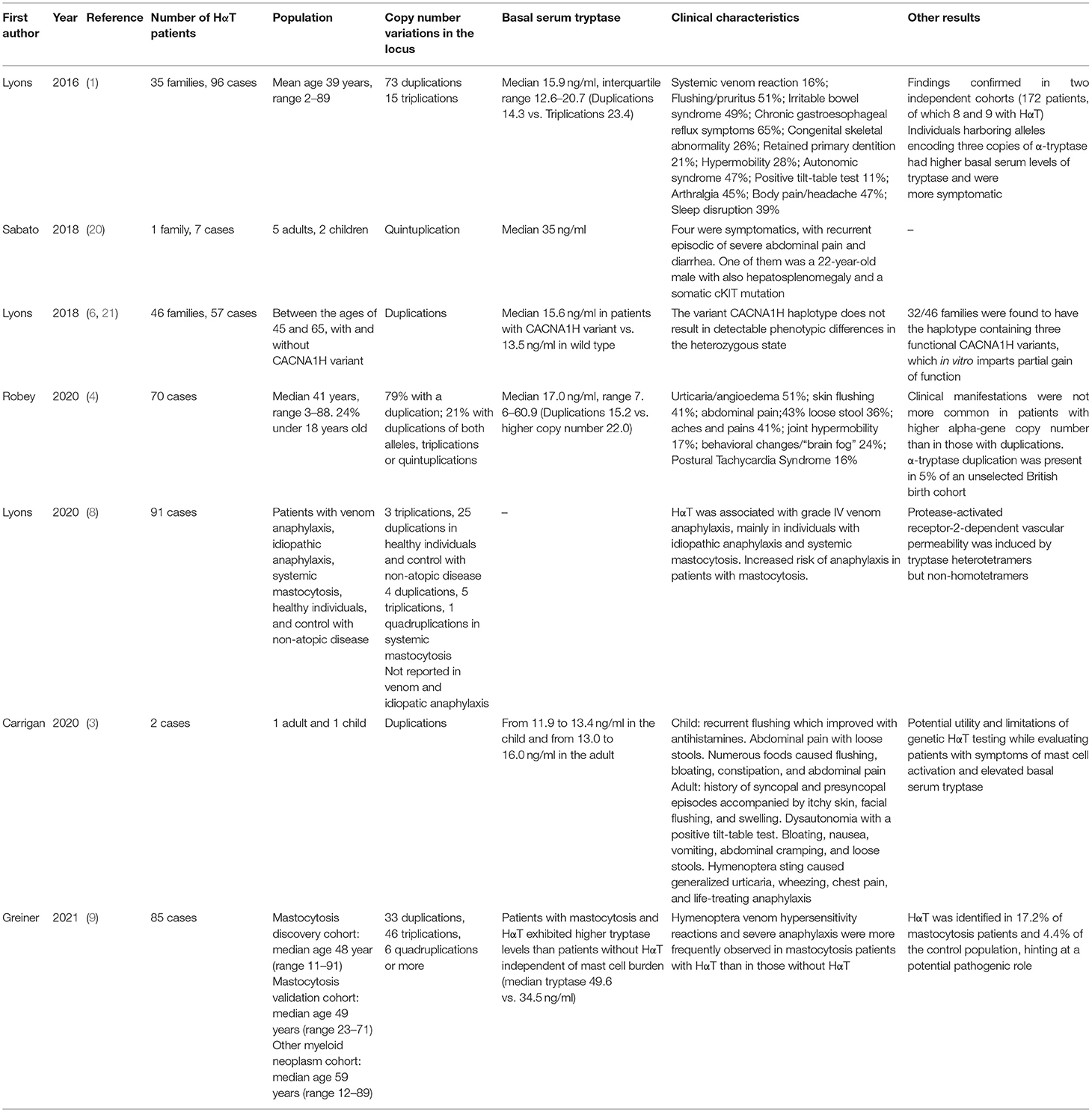
Table 1. Summary of studies regarding HαT.
The characterization of the clinical spectrum of this condition lacks in children, where differential diagnosis could be challenging. We herein describe two pediatric cases of HαT and their respective families that occurred at our tertiary care teaching hospital, in the pediatric unit of the “IRCCS Azienda Ospedaliero-Universitaria di Bologna.”
We describe a two-generation family of Russian origins. The proband is a 4-year old boy referred to our center for suspect systemic mastocytosis (Figure 1A).
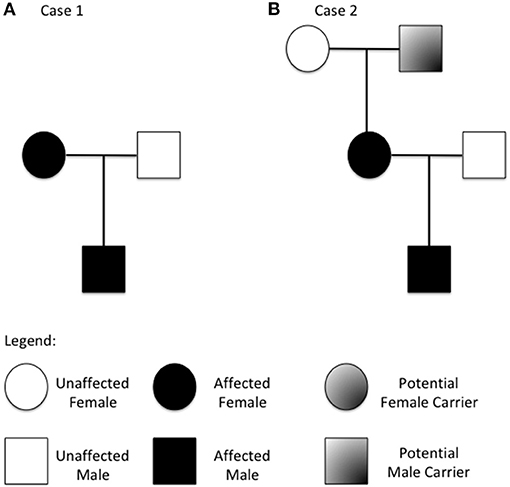
Figure 1. Family tree from the two described families (A refers to case 1 and B to case 2). Patients with increased copy number of the TPSAB1 gene are highlighted.
The proband's mother is a 28 years old woman, previously diagnosed with allergic asthma, Hashimoto thyroiditis, and type 1 diabetes mellitus complicated by necrobiosis lipoidica. She also had numerous allergic reactions to drugs and food in addition to multiple episodes of anaphylaxis after hymenoptera stings and others not specified triggers. The patient also experienced chronic diarrhea (2–20 episodes per day), autonomic dysfunction (recurrent syncope and hyperhidrosis), recurrent papular skin lesions and flushing, juvenile osteoporosis with T8 fracture, multiple lymphadenomegaly, and multiple miscarriage in the first trimester of pregnancy. The diagnostic work-up revealed increased levels of bee venom-specific IgE and elevated basal serum tryptase.
A bone marrow biopsy was performed to exclude systemic mastocytosis that revealed no morphologic, immunophenotyping, and cytogenetic abnormalities. The research of c-KIT D816V mutation was negative. Esophagogastroduodenoscopy and colonoscopy with multiple biopsies were also negative. Thus, a diagnosis of MCAS was made. She was initially treated with intravenous corticosteroids and interferon, and she is currently receiving oral antihistaminic before meals and pancreatic enzymes. The father of the proband reported no relevant clinical history.
The proband was born at 31 weeks of gestational age by cesarean section due to maternal pathology. The pregnancy was complicated by intrauterine growth restriction and agenesis of the ductus venosus. At birth, the patient weighed 1,838 g and had moderate respiratory distress, jaundice, hypoglycemia, transitory elevated TSH due to transplacental passage of maternal autoantibody and anemia, and was therefore transferred to neonatal intensive care unit for 1 month. During the first months of life, he presented repeated episodes of vomiting (2–3 episodes/day) without relation with the meal, for which a diagnosis of gastroesophageal reflux was made. Starting from about 1 year of age, he experienced multiple episodes of acute otitis media and tonsillitis treated with antibiotics, and daily episodes of bronchospasm treated initially with inhaled short-acting beta2 agonist and cortisone and afterward with inhaled corticosteroid only. From 15 months of age, he started to present multiple daily episodes of diarrhea (3–11 episodes per day) and abdominal pain exacerbated by meals. Celiac disease screening, stool culture, total Ig, and IgE levels, skin prick test for inhalant allergens, and RAST for alimentary allergens were all negative, whereas an elevated basal serum tryptase was observed (13 ng/ml). Symptoms worsened in the summer and partially responded to a clinical trial of cetirizine and nedocromil but recurred after therapy withdrawal.
Due to recurrent upper airway infection, the patients underwent rhinolaryngoscopy, which revealed adenoid hypertrophy. The proband also presented with different episodes of skin rash associated with hot bath, administration of ibuprofen, and upper airway infection. These symptoms receded after the administration of ketotifen. Moreover, the family reported significant local reactions to insect stings. An increment in the level of basal serum tryptase compared to the previous control was also observed (17 ng/ml). Clinical and laboratory findings, taken together with family history, raised the suspicion of systemic mastocytosis, and therefore the proband was referred to our center at 2 years of age.
At our referral center, the child underwent a complete diagnostic work-up. Basal serum tryptase was found to be 16.4 ng/ml. Levels of Vitamine D 25 OH were reduced (12 ng/ml, n.v. > 20 ng/ml), with slight elevation in alkaline phosphatase and osteocalcin levels and normal parathormone. Fecal calprotectin was also increased (405 mcg/g, n.v. <50 mcg/g). Complete blood count, liver and kidney function total serum Ig, Ig subclasses, and lymphocyte subset were normal, as well as complete abdominal ultrasound.
In the suspicion of systemic mastocytosis, we performed bone marrow biopsy, esophagogastroduodenoscopy, and colonoscopy with multiple biopsies, finding no evidence of mast-cell infiltration. The analysis of the c-KIT D816V mutation was negative on bone marrow specimen. Systemic mastocytosis was therefore excluded, and a diagnosis of MCAS was proposed.
Clinical re-evaluation in the following period highlighted red dermographism, iron deficiency, reduced attention and hyperactivity, recurrent skin rash, and diarrhea generally exacerbated by hot temperatures (e.g., in the summer and after exercise). Multiple episodes of anaphylaxis occurred after hymenoptera stings and others not clearly specified triggers. Moreover, the patient experienced laryngeal edema during laryngitis that required multiple intramuscular adrenaline injections and prompted hospitalization. In consideration of these multiple life-threatening events, an adrenaline autoinjector was prescribed to be held at home.
Basal serum tryptase levels slightly decreased over time, with the highest value being 14.7 ng/ml and the lowest 11 ng/ml. The gastrointestinal complaints were treated first with cromoglycate and then with ketotifen, but in both cases, treatment was suspended due to constipation.
In the suspicion of HαT, genetic testing was performed on the proband and parents. qPCR assay was done with the Universal ProbeLibrary System (Roche). A specific region within the TPSAB1 gene was amplified using the UPL probe 38 and the following flanking primers: forward 5′-aacccctgctgtccaaaac-3′; reverse 5′-taatgaggtccagcactcagg-3′ (chr16:1,292,280–1,292,539, hg19). Copy number variation was calculated with the ΔΔCt method, using HTR7 as the reference gene for normalization and two DNA samples from healthy donors as control. Duplication of the TPSAB1 gene was found in the proband and the mother but not in the father.
Herein, we describe a three-generation family of Italian origins. The proband is a 2 years old boy referred to our center for suspected Food-Protein induced Enterocolitis (Figure 1B).
The mother of the proband is a 31 years old woman. She reported allergic rhinoconjunctivitis and recurrent gastrointestinal complaints from a young age, described as frequent loose stools and abdominal pain, interpreted as food allergies. She also experienced dizzy spells, vertigo, and small skin rash. Laboratory findings included normal Ig levels, normal total IgE levels, and no specific Ig for known allergens, whereas increased basal serum tryptase was found. The maternal grandfather also reported non-specific gastrointestinal symptoms and elevated serum tryptase. The father of the proband had no relevant clinical history.
The proband was born at term via vaginal delivery from an uncomplicated pregnancy. The baby at birth weighed 2,750 g. He received mixed feeding for the first month and only breastfeeding in the following period. He then developed cradle cap and peri-auricular dermatitis. Gastroesophageal reflux was also diagnosed. The child started weaning at 6 months of age, and after the introduction of wheat at 8 months, he developed facial erythematous skin rash and diarrhea (5–6 episodes per day) starting 2 h after the aliment ingestion. After wheat reintroduction, the symptoms recurred, and so it was excluded from the diet. He then also experienced episodes of diarrhea after 3–4 h from the intake of milk derivatives and vomiting linked to the ingestion of egg yolk, which led to the exclusion of these two aliments. Skin prick test for alimentary allergens resulted negative. In the following weeks, reintroducing small quantities of wheat was attempted, but that prompted a severe episode of vomiting that required hospitalization. A diagnosis of Food-Protein induced Enterocolitis was therefore suspected. At 1 year of age, he presented atopic dermatitis and a maculopapular hyperchromic lesion with a positive Darier sign in the right wrist, compatible with solitary cutaneous mastocytoma (Figure 2). Basal serum tryptase was 19.8 ng/ml. Levels of Vitamine D 25 OH, alkaline phosphatase, and parathormone were normal. Total IgE levels were also normal, but positive specific IgE antibodies for dust-mite were found in the absence of related respiratory symptoms. After the first trial of cow-milk reintroduction, the proband experienced upward and rightward gaze deviation followed by generalized hypotonia and lack of responsiveness lasting about 5 s, with normal vital signs. Brief episodes of gaze deviation recurred weekly in the following months, associated with forward movements of the neck and reduced responsiveness. They mainly happened when the child was tired or agitated. No other neurological alteration was found.
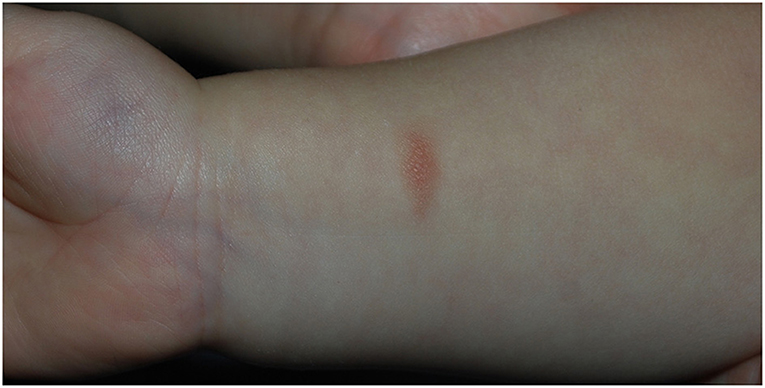
Figure 2. Solitary cutaneous mastocytoma of case 2, presenting as a maculo-papular hypercromic lesion with positive Darier sign in the right wrist.
The patient presented also sleep disturbances, including insomnia, pavor nocturnus, and somnambulism. Polysomnography ruled out the epileptic origin of these signs. Basal serum tryptase levels fluctuated in the following months, from 15.8 to 18.8 ng/ml. In order to better characterize the reason for the persistent rise in serum tryptase, the proband, the mother, and the father underwent genetic testing on the TPSAB1 gene, using the same method as previously described in Case 1. Triplication (four copies) of the TPSAB1 gene was found in the proband and the mother but not in the father.
After genetic testing, progressive reintroduction of food was attempted. Cow milk, wheat, and egg were successfully reintroduced in the diet without additional specific treatments. Neurological symptoms also receded with time.
Hereditary alpha tryptasemia is a recently described, apparently common genetic trait affecting mast cells (MC). Herein, we described four cases from two different families focusing on the two pediatric patients, characterized by remarkably different clinical presentation and severity. The genotype–phenotype correlation in this genetic condition still needs to be clearly understood (1) and, in particular, how increased copy number of the TPSAB1 gene could lead to elevated basal serum tryptase level and the associated heterogeneous clinical manifestations.
The multiple tryptase genetic loci in humans are located within the gene-rich region at 16p13.3 and include four tryptase encoding genes (TPSG1, TPSB2, TPSAB1, and TPSD1), of which only two, TPSB2 and TPSAB1, encode the secreted isoforms. The TPSAB1 locus encodes either alpha or beta isoforms, while TPSB2 is considered to encode only beta tryptase (6, 10). These isoforms are 97% similar, making detection of distinct types extremely difficult (6).
Tryptases are trypsin-like serine proteases expressed by MC and basophils (11, 12). The biologic activity of the enzymatically active tetrameric form has not yet been fully understood, but it is speculated to contribute to allergic inflammation (13). Alpha-tryptase tetramers lack protease activity, whereas β-tryptase tetramers are active proteases. Heterotetramers composed of 2α- and 2β-tryptase, but not homotetramer, activate protease-activated receptor-2 (PAR2), expressed on smooth muscle, neurons, and endothelium. Also, only α/β-tryptase tetramers cleave the α subunit of the EGF-like module-containing mucin-like hormone receptor-like 2 (EMR2) mechanosensory receptor, making MC susceptible to vibration-triggered degranulation. The allosteric effect of alpha tryptase is supposed to explain the different biological effects of heterotetramers compared to homotetramers (14). Pro-tryptases, which have not undergone enzymatic conversion into mature tetrameric tryptases, are constitutively secreted into serum in their monomeric form by MC at rest, and provide the vast majority of measured basal serum tryptase in a healthy individual. In contrast, mature tryptases are stored in secretory granules until their release by activated cells (15). Mast cell release tetrameric tryptases during a severe anaphylactic event, and serum tryptase level increases substantially over the individual's baseline. Therefore, serum tryptase is a valuable biomarker for assessing MC burden (as in mastocytosis) and, in some settings, MC activation (16). Increased copy number of the TPSAB1 gene could thus directly explain the increased basal serum tryptase levels observed in HαT patients. Other diseases in which elevated total tryptase levels could be found are acute myeloid leukemia, myelodysplastic syndromes, hypereosinophilic syndrome with the FLP1L1-PDGFRA fusion gene, and end-stage renal disease (16, 17).
Most symptoms related to HαT may be explained by MC mediator release, namely multiple allergies, anaphylaxis, and skin rash (6). Moreover, the portion α/β-tryptase heterotetramers increase with the alpha-tryptase gene dosage, and this could explain the connective tissue abnormalities, such as joint hypermobility, through the activation of PAR2-dependent pathways. The stimulation of the same receptor in the endothelium could also potentiate hypotension and anaphylaxis by increasing vascular permeability (18). Some cases of vibratory urticaria or other allergic manifestations are, instead, probably associated with higher susceptibility of MC to vibration-triggered degranulation due to the increased cleavage of EMR2α (14). Hereditary alpha tryptasemia is also associated with increased small intestinal MC, located throughout the mucosa and submucosa, including the superficial villi, contributing to the observed gastrointestinal manifestations (19).
No generalized conclusions or statements can be derived from the description of our two families. Moreover, the presented cases are influenced by selection bias, given that their tryptase levels would not have been checked if they did not present with their clinical manifestations. Larger case series are needed to understand whether there is a pattern or a clinical effect of HaT in the pediatric population. Moreover, HαT may modify the expression of multifactorial allergic diseases rather than directly cause specific phenotypes (4). Patients with mastocytosis, venom allergies, or idiopathic anaphylaxis have more severe manifestations of anaphylaxis if they also present alpha tryptase encoding TPSAB1 copy number gain (8, 9), possibly indicating that HaT could be a disease-modifying factor rather than a disease-causing mutation. Further studies are needed to address the specific role of HαT in the genesis of the associated symptoms.
Interestingly, in our experience, the first patient had a more severe clinical course, but with lower levels of basal serum tryptase and fewer TPSAB1 gene copies, whereas the second showed a milder clinical course, although he had TPSAB1 triplication and higher levels of tryptase. The described cases seem to contrast with the hypothesis of a linear gene-dosage effect between the number of additional TPSAB1 copies, the basal serum tryptase level, and the severity of clinical symptoms (20). The presented cases are instead in line with the idea that no precise genotype–phenotype correlation could be established between TPSAB1 copy number variation and clinical presentation (4). Thus, other genetic modifiers may contribute to shaping the clinical phenotype. Due to the genetic complexity of the tryptase locus, the germline sequence of this portion of the genome has not been entirely determined. Therefore, coinherited variants may contribute to one or more heterogeneous clinical manifestations observed among individuals affected by HαT. To date, no such variants were linked to a specific phenotype (21).
The first patient eventually received a diagnosis of MCAS after the exclusion of systemic mastocytosis. Mast cell activation syndrome is defined as excessive MC activity without clonal proliferation or obvious secondary triggers (7). The key feature for the diagnosis is recurrent episodes of systemic anaphylaxis with concurrent involvement of at least two among cardiovascular, respiratory, dermatologic, and gastrointestinal systems. The clinical symptoms have to be associated with an acute increase in specific biologic mediator levels secreted by MC (e.g., histamine, tryptase, prostaglandin D2, Leukotriene C4) (22). Clinical heterogeneity in MCAS patients likely is due to MC mutational complexity and heterogeneity. Few studies have investigated the genetic basis of MCAS, reporting associations with somatic mutations in KIT and other MC regulatory genes, but with the possibility of germline mutations occurring (23).
The second proband also presented a solitary cutaneous mastocytoma. A higher prevalence of HαT in patients with mastocytosis compared to matched-control subjects has been recently reported, even if with lower frequency in cutaneous mastocytosis than systemic forms (9). The pathogenic role of germline α-tryptase encoding TPSAB1 copy number gains in the development of clonal MC infiltration of the skin or other organs still needs to be understood. It is speculated that increased α-tryptase load could exert direct mitogenic activity on MC or influence the microenvironment with autocrine or paracrine mechanism, facilitating clonal proliferation (24–26).
Mastocytosis is defined by clonal MC expansion and accumulation in various tissues, mainly the bone marrow and skin (27). Clinical presentations reflect organ damage due to MC infiltration and symptoms of MC activation, similar to MCAS (11). The diagnosis of systemic mastocytosis is based on the criteria established by the WHO (28). Total serum tryptase levels should be measured in all patients with symptoms compatible with mastocytosis at least 24 h after the symptomatic event. If the basal tryptase level is ≥20.0 ng/ml, the likelihood of systemic mastocytosis is high, and a bone marrow biopsy is warranted. Normal or slightly elevated basal serum tryptase levels (11.5–20.0 ng/ml) do not rule out the diagnosis, and highly sensitive polymerase-chain-reaction techniques for KIT D816V transcripts in peripheral-blood leukocytes could be used to confirm or exclude the clinical suspicion (29). For patients in whom the KIT mutation is detected in peripheral blood, a bone marrow evaluation is indicated (11). Due to high clinical suspicion, bone marrow and gastrointestinal biopsies were performed directly in the first patient, even with levels of tryptase <20.0 ng/ml. The exclusion of MC clonal proliferation in the bone marrow and the gut prompted the diagnosis of MCAS.
The most common form of mastocytosis in children is cutaneous mastocytosis. Disease-onset is usually within the first 2 years of life and commonly experience spontaneous regression of skin lesions at puberty (7). Three major variants are defined: monomorphic and polymorphic maculopapular cutaneous mastocytosis, diffuse cutaneous mastocytosis, and solitary mastocytoma of skin (30). The hallmark of cutaneous mastocytosis is Darier's sign, which is the lesions that become urticarial when rubbed or scratched (11). Children with typical cutaneous lesions usually do not require a bone marrow biopsy if hepatosplenomegaly, lymphadenopathy, or peripheral-blood abnormalities are absent (11). Patients with cutaneous mastocytosis do not fulfill the criteria for the diagnosis of systemic mastocytosis. However, these patients could also present systemic symptoms such as anaphylaxis or gastrointestinal complaints, similar to those documented in MCAS. In this regard, the second proband presented both a solitary mastocytoma of the skin and Food-Protein induced Enterocolitis, but bone marrow examination was not performed because no indication of systemic MC proliferation was found. Nevertheless, elevation in basal serum tryptase levels prompted the genetic testing on the TPSAB1 gene.
The recent association of increased TPSAB1 copy number with HαT, with few supporting scientific publications and the complex structure of the genomic region, likely contribute to a delay of the diagnostic offer by public institutions. Since the increased copy number cannot be easily identified through routine genetic testing, no public laboratories routinely offer this test in Europe. A commercial one is offered by at least one company in the USA (Gene by Gene, Houston, Texas). However, specialists, including medical geneticists, should evaluate the access to a targeted genetic test for TPSAB1 copy number and its results. Moreover, the clinician must consider the differential diagnostic possibility of other genetic diseases, such as congenital autoinflammatory syndromes or congenital malabsorption syndromes, mainly inborn disaccharidase deficiency, that were excluded in our patients.
Systemic mastocytosis, HαT, and MCAS are associated with overlapping symptoms and pathophysiology, and distinguishing them only on clinical grounds could be challenging (3). Pediatric patients with increased basal serum tryptase should be all screened for increased copy number variations in the TPSAB1 locus, the genetic hallmark of HαT. If compatible symptoms and slightly elevated tryptase levels are present, HαT could be diagnosed, and further diagnostic testing is not needed. However, if clinical and laboratory findings suggest mastocytosis, the diagnosis of HαT must not rule out the need for an invasive investigation because the two conditions could coexist. More extensive studies are therefore needed to establish the role of tryptase genotyping in the diagnostic algorithm when considering the necessity for bone marrow biopsy.
An inadequate diagnostic pathway in children could cause late diagnosis of these conditions, leading to a reduction in the quality of life and several unnecessary medical consultations and therapies, as highlighted by the description of our families. Clinical overlap with common diseases, such as functional gastrointestinal disorders and allergies, makes the diagnosis of HαT in adulthood a problematic task. Therefore, suspecting this condition in children based on clinical phenotype and family history should prompt genetic testing to perform early diagnosis.
The observed heterogeneity in HαT clinical presentation requires further studies to better characterize clinical phenotype in children. The exact correlation between clinical findings and genetic alteration is unclear and should be a research focus in the next future. A better understanding of the associated symptoms could improve diagnosis and genetic counseling, helping physicians manage patients with MCAS-related symptoms previously considered of idiopathic origin.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
DZ and EM: conceptualization. AG, IN, SF, PM, and EM: data collection. EM and DZ: writing—original draft preparation. EM: image and table design. AG, IN, FC, PM, SF, and AP: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lyons JJ, Yu X, Hughes JD, Le QT, Jamil A, Bai Y et al. Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat Genet. (2016) 48:1564–9. doi: 10.1038/ng.3696
2. Soto D, Malmsten C, Blount JL, Muilenburg DJ, Caughey GH. Genetic deficiency of human mast cell alpha-tryptase. Clin Exp Allergy. (2002) 32:1000–6. doi: 10.1046/j.1365-2222.2002.01416.x
3. Carrigan C, Milner JD, Lyons JJ, Vadas P. Usefulness of testing for hereditary alpha tryptasemia in symptomatic patients with elevated tryptase. J allergy Clin Immunol Pract. (2020) 8:2066–7. doi: 10.1016/j.jaip.2020.01.012
4. Robey RC, Wilcock A, Bonin H, Beaman G, Myers B, Grattan C et al. Hereditary alpha-tryptasemia: UK prevalence and variability in disease expression. J allergy Clin Immunol Pract. (2020) 8:3549–56. doi: 10.1016/j.jaip.2020.05.057
5. Fellinger C, Hemmer W, Wöhrl S, Sesztak-Greinecker G, Jarisch R, Wantke F. Clinical characteristics and risk profile of patients with elevated baseline serum tryptase. Allergol Immunopathol (Madr). (2014) 42:544–52. doi: 10.1016/j.aller.2014.05.002
6. Lyons JJ. Hereditary alpha tryptasemia: genotyping and associated clinical features. Immunol Allergy Clin North Am. (2018) 38:483–95. doi: 10.1016/j.iac.2018.04.003
7. Wilcock A, Bahri R, Bulfone-Paus S, Arkwright PD. Mast cell disorders: from infancy to maturity. Allergy. (2019) 74:53–63. doi: 10.1111/all.13657
8. Lyons JJ, Chovanec J, O'Connell MP, Liu Y, Šelb J, Zanotti R et al. Heritable risk for severe anaphylaxis associated with increased α-tryptase-encoding germline copy number at TPSAB1. J Allergy Clin Immunol. (2020) 147:622–32. doi: 10.1016/j.jaci.2020.06.035
9. Greiner G, Sprinzl B, Górska A, Ratzinger F, Gurbisz M, Witzeneder N et al. Hereditary α tryptasemia is a valid genetic biomarker for severe mediator-related symptoms in mastocytosis. Blood. (2021) 137:238–47. doi: 10.1182/blood.2020006157
10. Akin C, Soto D, Brittain E, Chhabra A, Schwartz LB, Caughey GH et al. Tryptase haplotype in mastocytosis: relationship to disease variant and diagnostic utility of total tryptase levels. Clin Immunol. (2007) 123:268–71. doi: 10.1016/j.clim.2007.02.007
11. Theoharides TC, Valent P, Akin C. mast cells, mastocytosis, and related disorders. N Engl J Med. (2015) 373:163–72. doi: 10.1056/NEJMra1409760
12. Foster B, Schwartz LB, Devouassoux G, Metcalfe DD, Prussin C. Characterization of mast-cell tryptase-expressing peripheral blood cells as basophils. J Allergy Clin Immunol. (2002) 109:287–93. doi: 10.1067/mai.2002.121454
13. Caughey GH. Mast cell tryptases and chymases in inflammation and host defense. Immunol Rev. (2007) 217:141–54. doi: 10.1111/j.1600-065X.2007.00509.x
14. Le QT, Lyons JJ, Naranjo AN, Olivera A, Lazarus RA, Metcalfe DD et al. Impact of naturally forming human α/β-tryptase heterotetramers in the pathogenesis of hereditary α-tryptasemia. J Exp Med. (2019) 216:2348–61. doi: 10.1084/jem.20190701
15. Schwartz LB, Min H-K, Ren S, Xia H-Z, Hu J, Zhao W et al. Tryptase precursors are preferentially and spontaneously released, whereas mature tryptase is retained by HMC-1 cells, Mono-Mac-6 cells, and human skin-derived mast cells. J Immunol. (2003) 170:5667–73. doi: 10.4049/jimmunol.170.11.5667
16. Schwartz LB. Diagnostic value of tryptase in anaphylaxis and mastocytosis. Immunol Allergy Clin North Am. (2006) 26:451–63. doi: 10.1016/j.iac.2006.05.010
17. Khoury P, Lyons JJ. Mast cell activation in the context of elevated basal serum tryptase: genetics and presentations. Curr Allergy Asthma Rep. (2019) 19:55. doi: 10.1007/s11882-019-0887-x
19. Hamilton MJ, Zhao M, Giannetti MP, Weller E, Hufdhi R, Novak P et al. Distinct small intestine mast cell histologic changes in patients with hereditary alpha-tryptasemia and mast cell activation syndrome. Am J Surg Pathol. (2021) 45:997–1004. doi: 10.1097/PAS.0000000000001676
20. Sabato V, Chovanec J, Faber M, Milner JD, Ebo D, Lyons JJ. First identification of an inherited TPSAB1 quintuplication in a patient with clonal mast cell disease. J Clin Immunol. (2018) 38:457–9. doi: 10.1007/s10875-018-0506-y
21. Lyons JJ, Stotz SC, Chovanec J, Liu Y, Lewis KL, Nelson C et al. A common haplotype containing functional CACNA1H variants is frequently coinherited with increased TPSAB1 copy number. Genet Med. (2018) 20:503–12. doi: 10.1038/gim.2017.136
22. Weiler CR, Austen KF, Akin C, Barkoff MS, Bernstein JA, Bonadonna P et al. AAAAI Mast Cell Disorders Committee Work Group Report: mast cell activation syndrome (MCAS) diagnosis and management. J Allergy Clin Immunol. (2019) 144:883–96. doi: 10.1016/j.jaci.2019.08.023
23. Molderings GJ. The genetic basis of mast cell activation disease - looking through a glass darkly. Crit Rev Oncol Hematol. (2015) 93:75–89. doi: 10.1016/j.critrevonc.2014.09.001
24. Mirza H, Schmidt VA, Derian CK, Jesty J, Bahou WF. Mitogenic responses mediated through the proteinase-activated receptor-2 are induced by expressed forms of mast cell α- or β-tryptases. Blood. (1997) 90:3914–22. doi: 10.1182/blood.V90.10.3914
25. Gruber BL, Kew RR, Jelaska A, Marchese MJ, Garlick J, Ren S, et al. Human mast cells activate fibroblasts: tryptase is a fibrogenic factor stimulating collagen messenger ribonucleic acid synthesis and fibroblast chemotaxis. J Immunol. (1997) 158:2310–7.
26. Zhang J, Gruber BL, Marchese MJ, Zucker S, Schwartz LB, Kew RR. Mast cell tryptase does not alter matrix metalloproteinase expression in human dermal fibroblasts: further evidence that proteolytically-active tryptase is a potent fibrogenic factor. J Cell Physiol. (1999) 181:312–8. doi: 10.1002/(SICI)1097-4652(199911)181:2<312::AID-JCP13>3.0.CO;2-1
27. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
28. Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. (2017) 129:1420–7. doi: 10.1182/blood-2016-09-731893
29. Giannetti A, Filice E, Caffarelli C, Ricci G, Pession A. Mast cell activation disorders. Medicina (Kaunas). (2021). 57:124. doi: 10.3390/medicina57020124
Keywords: hereditary alpha tryptasemia, mastocytosis, mast cell activation syndrome, pediatric allergology, genetic disorder
Citation: Zama D, Muratore E, Giannetti A, Neri I, Conti F, Magini P, Ferrari S and Pession A (2021) Case Report: Hereditary Alpha Tryptasemia in Children: A Pediatric Case Series and a Brief Overview of Literature. Front. Pediatr. 9:716786. doi: 10.3389/fped.2021.716786
Received: 29 May 2021; Accepted: 02 August 2021;
Published: 20 August 2021.
Edited by:
Gregor Hoermann, Medical University of Vienna, AustriaReviewed by:
Georg Greiner, Medical University of Vienna, AustriaCopyright © 2021 Zama, Muratore, Giannetti, Neri, Conti, Magini, Ferrari and Pession. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edoardo Muratore, ZWRvYXJkby5tdXJhdG9yZUBzdHVkaW8udW5pYm8uaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.