
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 16 August 2021
Sec. Pediatric Immunology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.713921
 Amal M. Yahya1†
Amal M. Yahya1† Asia A. AlMulla2Haydar J. AlRufaye2Ahmed Al Dhaheri1Abdulghani S. Elomami3
Asia A. AlMulla2Haydar J. AlRufaye2Ahmed Al Dhaheri1Abdulghani S. Elomami3 Suleiman Al-Hammadi4,5†Lalitha Kailas6
Suleiman Al-Hammadi4,5†Lalitha Kailas6 Ranjit Vijayan7*†
Ranjit Vijayan7*† Abdul-Kader Souid5*†
Abdul-Kader Souid5*†Fermitin family homolog 3 (FERMT3), alternatively kindlin-3 (KIND3), is an integrin binding protein (of 667 residues) encoded by the FERMT3 gene. The molecule is essential for activating integrin αIIbβ3 (the fibrinogen receptor) on platelets and for the integrin-mediated hematopoietic cell (including platelets, T lymphocytes, B lymphocytes, and granulocytes) adhesion. Its defects are associated with impaired primary hemostasis, described as “Glanzmann's thrombasthenia (MIM#273800)-like bleeding problem.” The defects are also associated with infections, designated as “LAD1 (leukocyte adhesion deficiency, type I; MIM#116920)-like immune deficiency.” The entity that joins the impaired primary hemostasis with the leukocyte malfunction has been termed “leukocyte adhesion deficiency, type III” (LAD3, autosomal recessive, MIM#612840), representing a defective activation of the integrins β1, β2, and β3 on leukocytes and platelets. Here, we report a male toddler with novel compound heterozygous variants, NM_178443.2(FERMT3):c.1800G>A, p.Trp600* (a non-sense variant) and NM_178443.2(FERMT3):c.2001del p.*668Glufs*106 (a non-stop variant). His umbilical cord separated at about 3 weeks of age. A skin rash (mainly petechiae and purpura) and recurrent episodes of severe epistaxis required blood transfusions in early infancy. His hemostatic work-up was remarkable for a normal platelet count, but abnormal platelet function screen with markedly prolonged collagen-epinephrine and collagen-ADP closure times. The impaired platelet function was associated with reduced platelet aggregation with all agonists. The expression of platelet receptors was normal. Other remarkable findings were persistent lymphocytosis and granulocytosis, representing defects in diapedesis due to the integrin dysfunction. The natural history of his condition, structure and sequence analysis of the variations, and comparison with other LAD3 cases reported in the literature are presented.
The kindlin family member 3 [kindlin-3, or KIND3; preferred name FERMT3 (Uniprot accession Q86UX7)] is a cytosolic “β-integrin adapter protein” expressed mainly in hematopoietic cells (1, 2). Deficiencies in FERMT3 have been linked to the autosomal recessive “leukocyte adhesion deficiency, type III” (LAD3, MIM#612840), where all the β-integrins (β1, β2, and β3) are defective (3–8). This entity is a joint of LAD1-like leukocyte dysfunction (persistent leukocytosis plus infections, typically without pus formation) and Glanzmann thrombasthenia-like platelet dysfunction (mucocutaneous bleeding) (9). Individuals with this condition present in early infancy with severe bleeding and frequent infections (10).
The FERM central domain (residues 258 to 558; InterPro accession: Q86UX7) of FERMT3 binds to and activates platelet integrin αIIbβ3 (CD41/CD61). This receptor is activated by ADP, epinephrine, collagen, arachidonic acid, and thrombin. Activated receptors then bind fibrinogen, von Willebrand factor (vWF), fibronectin, and vitronectin (11–13). FERM domain is also essential for activating integrins β1 and β2 on the surface of granulocytes and lymphocytes (14–16). FERM defects, thus, demonstrate the essential role of F-actin-rich structures in the podosomes of lymphocytes, granulocytes and platelets (17, 18).
Only very few children with LAD3 have been reported in the literature. Thus, clinical descriptions of new patients are important to define the disease phenotype. We present here a toddler with novel compound heterozygous variants in FERMT3 (MIM#607901). This report features his characteristics and natural history. Its purpose is to enhance the recognition of this disorder and perform a structural bioinformatics analysis of his variations.
This 18-month-old male toddler was born at term to asymptomatic, non-consanguineous parents (from Kerala, India). The pregnancy and delivery were uneventful. His birth weight was 2.9 kg. He had no siblings; the mother was gravida 2, para 1, abortion (spontaneous) 1. His paternal grandmother had severe menorrhagia, requiring multiple transfusions. Otherwise, the family history was negative.
He was born in the United Arab Emirates and received the BCG (Bacillus Calmette–Guérin) vaccine [0.05 mL, intradermal injection in the upper left arm (manufactured by Serum Institute of India Pvt. Ltd., India)] at birth (Figure 1, left upper panel). His postnatal period was complicated by a delayed umbilical cord separation for about 3 weeks. He presented at 2 months of age with petechial rash over his arms and seborrheic dermatitis over his scalp. Starting at 8 months of age, he had recurrent episodes of severe nose bleeding; consequently, several blood counts were requested. He had 10 admissions since birth; six for severe epistaxis, two for work-up of marked leukocytosis, one for the bone marrow procedure, and one for a significant skin rash (eczema-like). His overall management included two red cell transfusions (over a 4 month period), three platelet transfusions, and tranexamic acid (used only for a short time period).
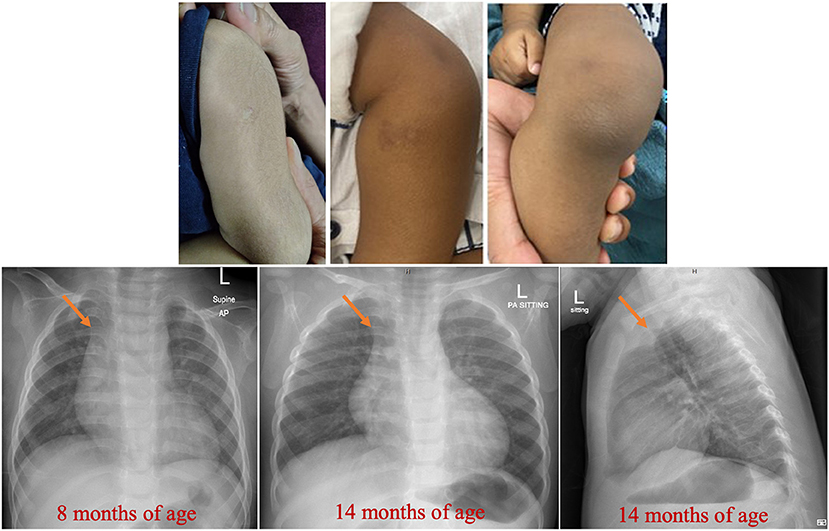
Figure 1. Findings on the skin and chest radiographs. Healed BCG site and dry skin with bruises and scars are evident. Prominent thymus (arrowed) at 8 mo of age is evident. Please also note the normal skeletal radiographs.
He was never febrile; his temperatures were ≤37.2°C. On two occasions, his blood cultures (requested because of the marked leukocytosis) grew Micrococcus luteus and Staphylococcus haemolyticus, which were considered skin flora contaminants. His physical examination was remarkable for the skin findings of dryness, bruises (petechiae and purpura), and a few scars, as shown in Figure 1 (middle and right upper panel). He was uncircumcised. His growth and development were appropriate for age.
Investigations were requested because of the unexplained persistent leukocytosis. His white blood cell differential counts are shown in Figures 2A,B. Persistent marked leukocytosis (mainly lymphocytosis, monocytosis, and eosinophilia) were evident. In addition, “left shift” (neutrophil bands, metamyelocytes, and myelocytes) was a frequent finding (Figures 3A,B). His chest radiograph was remarkable for a notably prominent thymus (reported as a “round opacity at the mediastinum”), Figure 1 (lower panels). Findings on the abdominal ultrasound and echocardiogram were normal. Serum ferritin was ≤20 μg/L and C-reactive protein (CRP) ≤ 1.8 mg/L. Serum immunoglobulins (IgA, IgG, and IgM) were normal. At 9 months of age, his lymphocyte subset was: CD3 T cells [11,002/μL (41%), reference values (10th−90th centiles) = 1,900–5,900], CD4 T cells [6,423/μL (24%), reference values = 1,400–4,300], CD8 T cells [3,785/μL (14%), reference values = 500–1,700], CD19 B cells [14,206/μL (53%), reference values = 610–2,600], and NK cells (426/μL, 2%). Bone marrow examination was normal; the megakaryocytes were adequate in number and had normal morphology (Figures 3C,D). Leukocyte RNA was negative for leukemia fusion gene transcripts (MDx Leukemia Fusion Gene Q30 screen; QuanDx, Menlo Park, CA, USA).
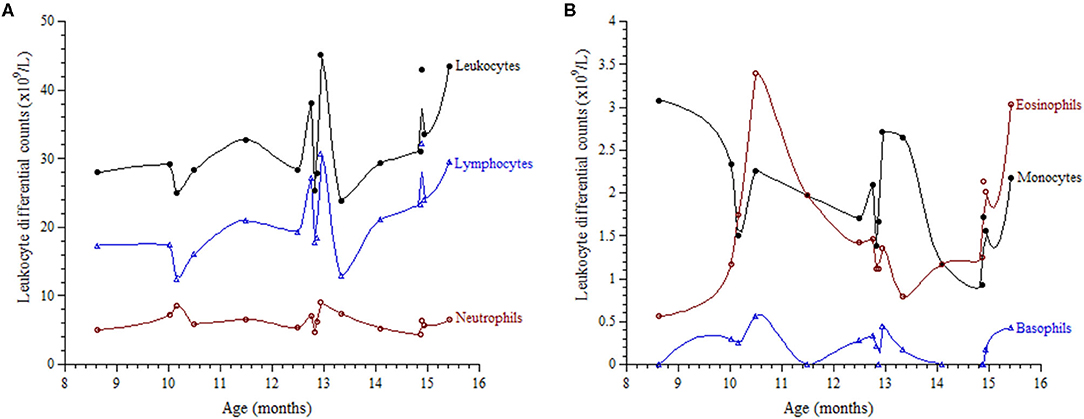
Figure 2. Leukocyte differential counts as function of age.
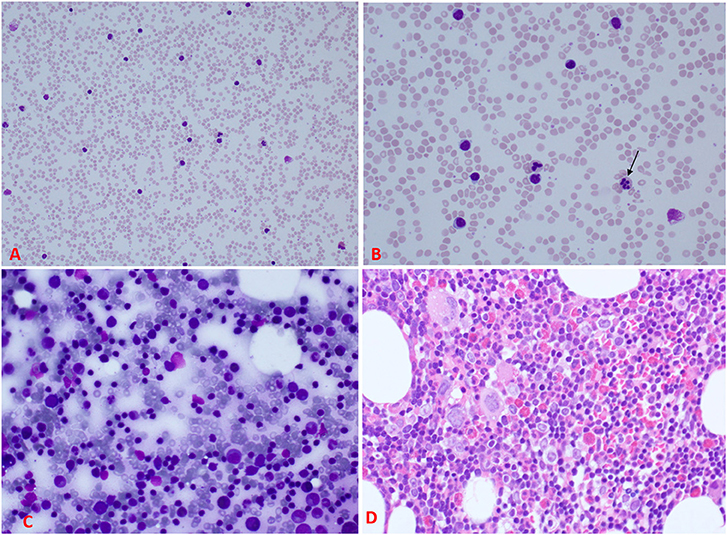
Figure 3. Peripheral blood and bone marrow morphology. (A) A low power view of the peripheral blood smear demonstrating significant leukocytosis. (B) A higher magnification of the smear showing the leukocytosis predominantly lymphocytes mixed with neutrophils; some of the neutrophils reveal abnormal nuclear lobation, like hypersegmentation (arrow). (C) Bone marrow aspirate showing normal maturation of erythroid and myeloid cells and increased lymphocytes with some immaturity suggestive of hematogones. (D) Section from the bone marrow biopsy showing about 70% cellularity (normal for age) of trilineage hematopoiesis with increased eosinophils, lymphocyte aggregates, and interstitial lymphocytes. Megakaryocytes were adequate with normal morphology [For comparison, please see (19) for normal peripheral blood and bone marrow smears].
Hemostatic evaluation was also requested because of the severe epistaxis. His platelet counts and secondary hemostasis [activated partial thromboplastin time (aPTT), prothrombin time (PT), international normalized ratio (INR), thrombin time (TT), and serum fibrinogen] were normal. Platelet function screen (performed using Dade® PFA Collagen/EPI and Collagen/ADP Test Cartridges, Siemens Healthcare Diagnostics GmbH; Marburg, Germany) showed prolonged collagen-epinephrine and collagen-ADP closure times > 300 s (normal, ≤ 164 s). Von Willebrand factor (vWF) antigen was 59.6% (normal, 50–200) and activity 46.7% (normal, 50–200); these tests were performed using Innovance® VWF Ac and VWF Ag (Siemens Healthcare Diagnostics GmbH; Marburg, Germany). The results of vWF were normal as his blood group was “O”; even though, the red blood cell phenotype may have partially contributed to his bleeding tendency. Platelet aggregation studies (performed using the Bio/Data Corporation product; Horsham, PA, USA) revealed severely impaired aggregations in response to all agonists, suggesting a defect in the inside-out activation of integrin αIIbβ3 (Table 1). Spontaneous platelet aggregation, a pathologic finding, was not present.
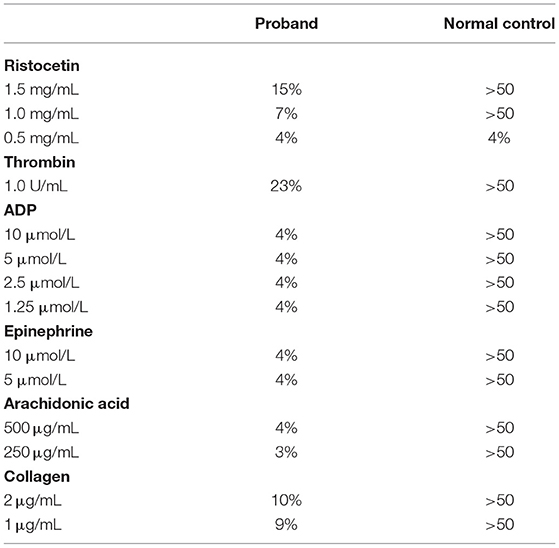
Table 1. Results of his platelet aggregation studies.
Flow cytometry revealed a normal expression of the leukocyte receptors CD11a (integrin, alpha L), CD11b (integrin, alpha M), CD11c (integrin, alpha X), and CD18 (integrin β2). It also revealed a normal expression (99%) of the platelet receptors CD41 (integrin αIIb), CD61 (integrin β3), CD42a (GPIX), and CD42b (GPlb-α). These tests were performed using monoclonal antibodies from Becton, Dickinson and Company, BD Biosciences; San Jose, California, USA (see Supplementary Material).
In view of his abnormal platelet function profile and marked leukocytosis, “diagnostic exome sequencing test” was requested (mainly considering the probability of LAD3). Two novel compound heterozygous variants were found: (1) Non-sense (stop-gain, resulting in a truncated product) NM_178443.2(FERMT3):c.1800G>A, p.Trp600* (inherited from the mother), and (2) Non-stop (stop-loss, resulting in an elongated polypeptide) NM_178443.2(FERMT3):c.2001del p.*668Glufs*106 (inherited from the father).
The non-sense variant FERMT3:p.Trp600* leads to a premature truncation of the protein and the loss of 68 residues from the C-terminal (Figure 4A). This truncation results in the loss of two α-helices and a β-sheet, comprising of three β-strands (Figure 4B). Computational prediction scores from multiple predictors [combined annotation dependent depletion (CADD): 46; likelihood ratio test (LRT): 0.8433; MutationTaster: 0.81] obtained from Ensembl Variant Effect Predictor (VEP; https://www.ensembl.org/Tools/VEP) (20) suggest that this variant is “deleterious,” while Varsome (21) flags it as “pathogenic.” The Trp600* variation shortens the F3 subdomain of FERMT3. The F3 subdomain is a part of the integrin-binding pocket of FERMT3 and this variation is, therefore, expected to inhibit the ability of FERMT3 to bind to integrins.
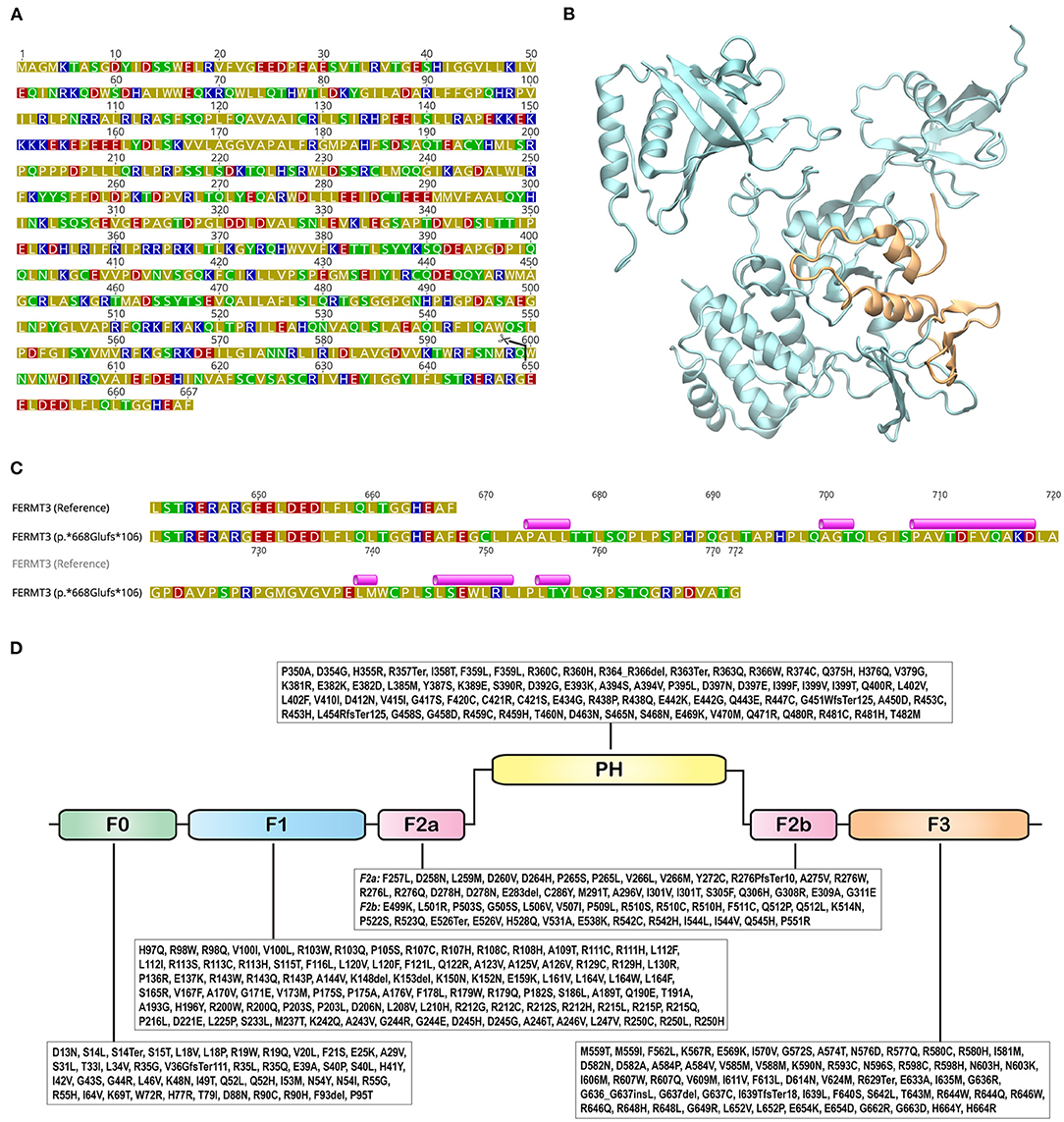
Figure 4. Structure and sequence of FERMT3. (A) Sequence of FERMT3 (NCBI Accession: NP_848537.1) with the site of truncation in p.Trp600* shown with a pair of scissors. Amino acids are colored based on their polarity: blue - basic; red - acidic; green - polar; olive – hydrophobic. (B) Three dimensional protein structure of FERMT3, generated using the Protein Data Bank structure 7C3M, depicted in cartoon representation. The region truncated in the p.Trp600* non-sense variant is shown in orange. (C) Protein sequence alignment of FERMT3 (NCBI Accession: NP_848537.1) and the p.*668Glufs*106 non-stop variant starting from position 640. Secondary structures, predicted using PSIPRED 4.0 (Predict Secondary Structure; http://bioinf.cs.ucl.ac.uk/psipred/), in the extended region of the variant are shown above the sequence. Violet cylinders represent α-helices. (D) Variations identified in the canonical Ensembl transcript (ENST00000279227.5) were from gnomAD v2.1.1 are shown with amino acids represented by their one letter codes. FERM domains (sub-domains F0, F1, F2a, F2b, and F3; InterPro classifies region 258-558 as FERM central domain) and pleckstrin homology (PH) are shown as rounded rectangles.
The p.*668Glufs*106 non-stop variant results in an extended polypeptide with an additional 105 amino acids, a 16% extension to the wild-type sequence. Basic Local Alignment Search Tool (BLAST; https://blast.ncbi.nlm.nih.gov) (22) searches did not identify any homologous sequences or structures. Secondary structure prediction with PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/) (23) indicated that this region could potentially harbor six α-helices (Figure 4C). While secondary structures have been predicted in the extended sequence of p.*668Glufs*106 (Figure 4C), it is unclear if this extended part will cooperatively fold to produce a functional domain. Nonetheless, even if it does, its close proximity to the integrin-binding pocket of the F3 subdomain, located near the C-terminal of the protein, is likely to disrupt its ability to bind to integrins.
Pathogenic variants of FERMT3 (MIM#607901) cause LAD3 (MIM#612840). GnomAD v2.1.1 lists 1,112 variations in FERMT31. Of these, 372 are start-loss, stop-loss, inframe deletion, inframe insertion, frameshift, or non-sense mutations (6, 24). Variations that map on to the FERM (F: 4.1 protein, E: ezrin, R: radixin, and M: moesin) and PH (pleckstrin homology) domains of FERMT3 are shown in Figure 4D.
As indicated above, LAD3 is joint defects of LAD1 (MIM#116920)-like immune deficiency and Glanzmann thrombasthenia (MIM#273800)-like bleeding. The defect involves the intracellular protein that interacts with β-integrins in hematopoietic cells, including granulocytes, lymphocytes, macrophages, and platelets. The predominant phenotypes result from failures to activate β1 and β2 integrins on the surface of granulocytes and lymphocytes and β3 integrin (αIIbβ3) on the surface of platelets (13, 14). As previously shown, integrins α2β1 and αIIbβ3 support the irreversible adhesive interactions of platelets with collagen and VWF; these receptors also support the platelet recruitment and spreading (25). Integrin β1 (CD29) also pairs with other α subunits, such as α4β1 (a vascular cell adhesion protein 1, VCAM-1, receptor), α5β1 (a fibronectin receptor), and α6β1 (a laminin receptor) (26). Integrin β2 (CD18), on the other hand, is exclusively expressed on the surface of leukocytes. Its pairing with one of a number of distinct α subunits (e.g., αM, or CD11b) enables cell-cell interactions, such as the binding to intercellular adhesion molecule 1 (ICAM-1) on an activated endothelial cell thus mediating neutrophil adhesion and spreading (27). Similarly, integrin β3 (CD61) was shown to pair with the αv subunit; the resulting receptor (αvβ3) binds the secreted protein vitronectin and mediates cell adhesion, spreading and signaling (28). Defective “inside-to-outside” signalings explain the hallmarks of the entity of granulocytosis (innate immune defects), lymphocytosis (defective adaptive immunity) and life-threatening mucocutaneous bleeding (impaired primary hemostasis) (14, 15, 29, 30).
Noticeable features of his phenotype (see reference 8, and Table 3) include: (1) Delayed umbilical cord separation for about 3 weeks (reference mean ± SD time = 6.61 ± 2.33 days; confidence of interval 95% = 6.16–7.05) (31); (2) Spontaneous petechiae in the neonatal period; (3) Severe epistaxis in early infancy (his first episode was at 8 mo of age, requiring transfusion); (4) Normal secondary hemostasis; (5) Normal platelet count; (6) Abnormal platelet function screen (PFA-100® test, >300 s for both channels); (7) Normal von Willebrand factor antigen and activity; (8) Impaired platelet aggregation with all agonists (ristocetin, thrombin, ADP, epinephrine, arachidonic acid, and collagen); (9) Normal expression of platelet receptors (e.g., CD41/CD61, or integrin αIIbβ3); (10) Normal expression of leukocyte adhesion molecules (e.g., CD18, or integrin β2); (11) Absent hepatosplenomegaly and lymphadenopathy; (12) Prominent thymus on the chest radiograph; (13) Absence of fever; (14) Normal inflammatory biomarkers (CRP and ferritin); (15) Persistent marked leukocytosis/ granulocytosis (most notably, high monocyte and eosinophil counts); and (16) Persistent lymphocytosis (high T cell and B cell counts; with C19 B cell count higher than CD3 T cell count).
It is worth emphasizing that the recurrent severe episodes of epistaxis were the foremost clinical manifestation. This problem triggered the extensive hemostatic evaluation and genetic testing. In our community, diagnostic exome sequencing provides about 50% yield in detecting inborn errors metabolism (36). Thus, this test was requested for his investigation. It is also worth noting that his hemostatic findings suggested variants in FERMT3 (see below).
The differential diagnosis of leukocytosis includes infection, leukemoid reaction [leukocytosis with circulating immature white and red blood cells (“left shift”), mimicking chronic myelomonocytic leukemia (CMML) and chronic myelogenous leukemia (CML)], and lymphoproliferation (including leukemia) (37). The eosinophilia may mimic atopy. The large thymus may mimic T cell lymphoma. Nevertheless, persistent marked elevations of granulocytes and lymphocytes suggest a leukocyte adhesion defect (8).
The differential diagnosis of impaired primary hemostasis (summarized in Table 2) includes von Willebrand disease, acquired platelet dysfunction (e.g., use of non-steroidal anti-inflammatory drugs), inherited platelet dysfunction [e.g., Glanzmann thrombasthenia (GT), Bernard-Soulier syndrome (BSS), ADP receptor defect, and gray platelet syndrome], and vascular anomaly (e.g., Ehlers-Danlos syndrome) (37). It is important to note, Glanzmann thrombasthenia is characterized by a diminished platelet aggregation with ADP, epinephrine, collagen, thrombin, and arachidonic acid, with normal agglutination with ristocetin. Bernard-Soulier syndrome, on the other hand, is associated with normal platelet aggregation with all agonists, except ristocetin. Diminished platelet aggregation in response to ADP alone is characteristic of ADP receptor defect. In gray platelet syndrome, platelet aggregation is variable and can be normal; however, thrombocytopenia is typically present (38).
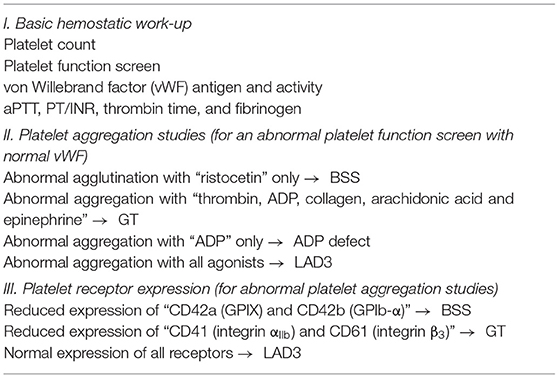
Table 2. Schema diagram summarizing the hemostatic assessment.
Defects in natural killer cell migration, adhesion and activation have been also reported in LAD3 (39). Thus, this entity represents a multifaceted inborn error of immunity. This consideration is especially important for countries which implement universal BCG vaccination at birth, such as India and United Arab Emirates. Furthermore, defects in bone-resorbing osteoclast adhesion have been reported in LAD3 (32, 40), justifying its inclusion in the differential diagnosis of impaired bone homeostasis (8).
The expediency of his abnormal “platelet function screen” is evident. This test assesses primary hemostasis when the platelet count is normal, and has replaced the old “bleeding time.” Thus, the platelet function screen test is essential in the current hemostatic evaluation (41).
Treatment of LAD3 is supportive and includes topical thrombin in conjunction with an anti-fibrinolytic agent, tranexamic acid or aminocaproic acid. The response to anti-fibrinolytic agents should be observed, and the dose should be adjusted as necessary. Platelet transfusion is indicated in severe bleeding, or it can be administered weekly as prophylaxis when necessary (8). Precautions with regard to circumcision and skin and mucosal (including dental) hygiene are necessary. Prompt antimicrobial therapy in case of infection is also vital, particularly as fever and inflammatory biomarkers may not be present or prominent (42). Prophylaxis for pneumocystis pneumonia (typically with the use of trimethoprim/sulfamethoxazole) is needed (see Table 3).
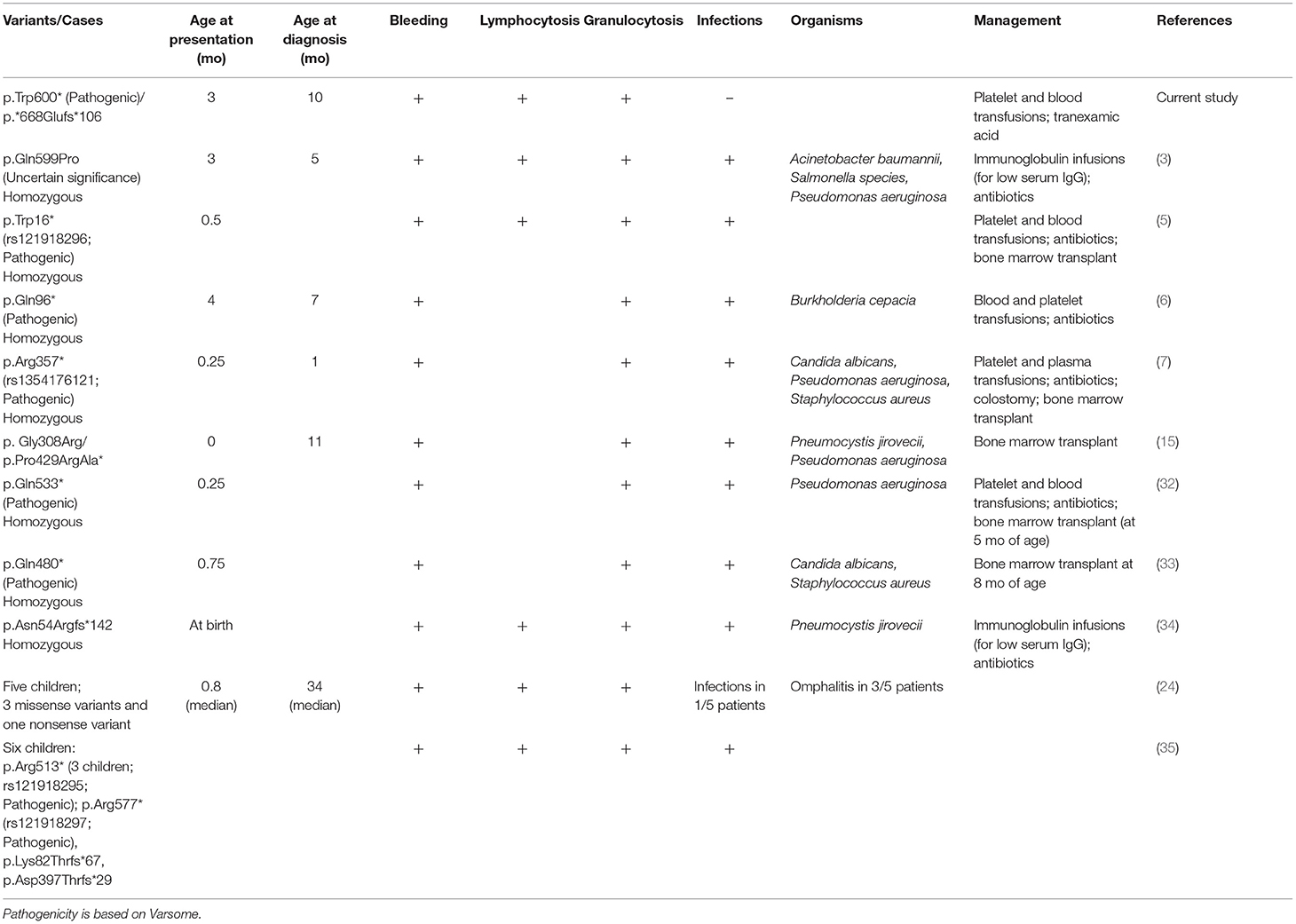
Table 3. Natural histories of the current and previously reported FERMT3 (Ensemble transcript: ENST00000279227.5) variants.
In one report, platelet and granulocyte functions (including leukocytosis) and radiographic signs of osteosclerosis (osteopetrosis) were normalized after allogeneic hematopoietic stem cell transplantation (8). Thus, this therapeutic intervention is successful, especially if applied in infancy (43–46). Of note, our patient had no radiographic signs of osteopetrosis, as shown in Figure 1. Genetic prevention remains a foreseen goal.
As noted above, he had received the live vaccine BCG (Bacillus Calmette–Guérin). Leucocyte adhesion deficiencies are included in the list of inborn errors of immunity (IEI) that describes complications to the BCG vaccination in India (47). In a retrospective study, inborn errors of immunity were identified in 52 of 90 (58%) individuals who had a localized or disseminated BCG complication; these disorders included LAD1 (47). Although he had no evidence of BCG or TB (tuberculosis) disease, these complications (which are generally severe in infants) should be considered, especially in high TB burden countries such as India. Therefore, LAD3 should be added to the list of “defects in intrinsic and innate immunity” where BCG should be avoided as a precaution, and a prophylactic treatment for BCG disease should be considered (48, 49). Moreover, it is prudent to change the time of BCG administration from birth to 1 year of age, when most IEI become more recognizable (50). It is well to reemphasize that a cautious consideration of BCG complications in infants is necessary, especially in regions with a high prevalence of IEI such as UAE.
It is important to realize that leucocyte adhesion deficiencies are rarely considered in clinical practices, and are infrequently reported in the literature. They go underdiagnosed, mainly due to the infrequent genetic testing (51).
Table 3 compares the clinical findings of the studied toddler with a few of the previously reported children. Typically, these individuals show symptoms and are diagnosed in early infancy. Bleeding and granulocytosis are consistent findings. Infections are frequent and span a wide-spectrum of pathogens (bacterial and fungal), including Pneumocystis jirovecii, Acinetobacter baumannii, and Salmonella species (Table 3).
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by Tawam Human Research Ethics Committee. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
A-KS, RV, AY, SA-H, and LK: conceived, designed, and structured the report. RV: performed the variant analysis. A-KS, AY, AAA, HA, and AA: analyzed and interpreted the clinical data. AE: analyzed and interpreted the pathologic data. A-KS, AY, RV, and LK: wrote the original draft. A-KS, RV, and AY: reviewed and edited the paper. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.713921/full#supplementary-material
1. ^Available online at: https://gnomad.broadinstitute.org/gene/ENSG00000149781?dataset=gnomad_r2_1 (accessed June 19, 2021).
1. Sun J, Xiao D, Ni Y, Zhang T, Cao Z, Xu Z, et al. Structure basis of the FERM domain of kindlin-3 in supporting integrin αIIbβ3 activation in platelets. Blood Adv. (2020) 4:3128–35. doi: 10.1182/bloodadvances.2020001575
2. Li H, Deng Y, Sun K, Yang H, Liu J, Wang M, et al. Structural basis of kindlin-mediated integrin recognition and activation. Proc Natl Acad Sci USA. (2017) 114:9349–54. doi: 10.1073/pnas.1703064114
3. Suratannon N, Yeetong P, Srichomthong C, Amarinthnukrowh P, Chatchatee P, Sosothikul D, et al. Adaptive immune defects in a patient with leukocyte adhesion deficiency type III with a novel mutation in FERMT3. Pediatr Allergy Immunol. (2016) 27:214–7. doi: 10.1111/pai.12485
4. Bouti P, Zhao XW, Verkuijlen PJJH, Tool ATJ, van Houdt M, Köker N, et al. Kindlin3-dependent CD11b/CD18-integrin activation is required for potentiation of neutrophil cytotoxicity by CD47-SIRPα checkpoint disruption. Cancer Immunol Res. (2021) 9:147–55. doi: 10.1158/2326-6066.CIR-20-0491
5. Malinin NL, Zhang L, Choi J, Ciocea A, Razorenova O, Ma YQ, et al. A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat Med. (2009) 15:313–8. doi: 10.1038/nm.1917
6. Shahid S, Zaidi S, Ahmed S, Siddiqui S, Abid A, Malik S, et al. A novel nonsense mutation in FERMT3 causes LAD-III in a Pakistani family. Front Genet. (2019) 10:360. doi: 10.3389/fgene.2019.00360
7. Wolach B, Gavrieli R, Wolach O, Stauber T, Abuzaitoun O, Kuperman A, et al. Leucocyte adhesion deficiency-A multicentre national experience. Eur J Clin Invest. (2019) 49:e13047. doi: 10.1111/eci.13047
8. Jurk K, Schulz AS, Kehrel BE, Räpple D, Schulze H, Möbest D, et al. Novel integrin-dependent platelet malfunction in siblings with leukocyte adhesion deficiency-III (LAD-III) caused by a point mutation in FERMT3. Thromb Haemost. (2010) 103:1053–64. doi: 10.1160/TH09-10-0689
9. Moser M, Nieswandt B, Ussar S, Pozgajova M, Fässler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. (2008) 14:325–30. doi: 10.1038/nm1722
10. Zimmerman GA. LAD syndromes: FERMT3 kindles the signal. Blood. (2009) 113:4485–6. doi: 10.1182/blood-2009-01-198853
11. Huang J, Li X, Shi X, Zhu M, Wang J, Huang S, et al. Platelet integrin αIIbβ3: signal transduction, regulation, and its therapeutic targeting. J Hematol Oncol. (2019) 12:26. doi: 10.1186/s13045-019-0709-6
12. Xu Z, Chen X, Zhi H, Gao J, Bialkowska K, Byzova TV, et al. Direct interaction of kindlin-3 with integrin αIIbβ3 in platelets is required for supporting arterial thrombosis in mice. Arterioscler Thromb Vasc Biol. (2014) 34:1961–1967. doi: 10.1161/ATVBAHA.114.303851
13. Rognoni E, Ruppert R, Fässler R. The kindlin family: functions, signaling properties and implications for human disease. J Cell Sci. (2016) 129:17–27. doi: 10.1242/jcs.161190
14. Svensson L, Howarth K, McDowall A, Patzak I, Evans R, Ussar S, et al. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat Med. (2009) 15:306–12. doi: 10.1038/nm.1931
15. McDowall A, Svensson L, Stanley P, Patzak I, Chakravarty P, Howarth K, et al. Two mutations in the KINDLIN3 gene of a new leukocyte adhesion deficiency III patient reveal distinct effects on leukocyte function in vitro. Blood. (2010) 115:4834–42. doi: 10.1182/blood-2009-08-238709
16. Bunting M, Harris ES, McIntyre TM, Prescott SM, Zimmerman GA. Leukocyte adhesion deficiency syndromes: adhesion and tethering defects involving beta 2 integrins and selectin ligands. Curr Opin Hematol. (2002) 9:30–5. doi: 10.1097/00062752-200201000-00006
17. Hidalgo A, Frenette PS. Leukocyte podosomes sense their way through the endothelium. Immunity. (2007) 26:753–5. doi: 10.1016/j.immuni.2007.06.002
18. Morikis VA, Masadeh E, Simon SI. Tensile force transmitted through LFA-1 bonds mechanoregulate neutrophil inflammatory response. J Leukoc Biol. (2020) 108:1815–28. doi: 10.1002/JLB.3A0520-100RR
19. Naeim FP, Nagesh R, Song SX, Phan RT. Atlas of Hematopathology: Morphology, Immunophenotype, Cytogenetics, and Molecular Approaches. 2nd ed. London: Academic Press (2018).
20. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. (2016) 17:122. doi: 10.1186/s13059-016-0974-4
21. Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Aguilera MA, Meyer R, et al. VarSome: the human genomic variant search engine. Oxford Bioinformatics. (2018) 35:1978–80. doi: 10.1093/bioinformatics/bty897
22. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. (1990) 215:403–10. doi: 10.1016/S0022-2836(05)80360-2
23. Buchan DWA, Jones DT. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. (2019) 47:W402–7. doi: 10.1093/nar/gkz297
24. Kambli PM, Bargir UA, Yadav RM, Gupta MR, Dalvi AD, Hule G, et al. Clinical and genetic spectrum of a large cohort of patients with leukocyte adhesion deficiency type 1 and 3: a multicentric study from India. Front Immunol. (2020) 11:612703. doi: 10.3389/fimmu.2020.612703
25. Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. (1998) 94:657–66. doi: 10.1016/S0092-8674(00)81607-4
26. Roberts K, Alberts B, Johnson A, Walter P, Hunt T. Molecular Biology of the Cell. 4th ed. New York, NY: Garland Science (2002). Integrins. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK26867/ (accessed July 05, 2021).
27. Fagerholm SC, Guenther C, Llort Asens M, Savinko T, Uotila LM. Beta2-integrins and interacting proteins in leukocyte trafficking, immune suppression, and immunodeficiency disease. Front Immunol. (2019) 10:254. doi: 10.3389/fimmu.2019.00254
28. Suehiro K, Plow EF. Ligand recognition by beta 3 integrins. Keio J Med. (1997) 46:111–4. doi: 10.2302/kjm.46.111
29. Kinashi T, Aker M, Sokolovsky-Eisenberg M, Grabovsky V, Tanaka C, Shamri R, et al. LAD-III, a leukocyte adhesion deficiency syndrome associated with defective Rap1 activation and impaired stabilization of integrin bonds. Blood. (2004) 103:1033–6. doi: 10.1182/blood-2003-07-2499
30. Mory A, Feigelson SW, Yarali N, Kilic SS, Bayhan GI, Gershoni-Baruch R, et al. Kindlin-3: a new gene involved in the pathogenesis of LAD-III. Blood. (2008) 112:2591. doi: 10.1182/blood-2008-06-163162
31. López-Medina MD, López-Araque AB, Linares-Abad M, López-Medina IM. Umbilical cord separation time, predictors and healing complications in newborns with dry care. PLoS ONE. (2020) 15:e0227209. doi: 10.1371/journal.pone.0227209
32. Crazzolara R, Maurer K, Schulze H, Zieger B, Zustin J, Schulz AS. A new mutation in the KINDLIN-3 gene ablates integrin-dependent leukocyte, platelet, and osteoclast function in a patient with leukocyte adhesion deficiency-III. Pediatr Blood Cancer. (2015) 62:1677–9. doi: 10.1002/pbc.25537
33. Harris ES, Smith TL, Springett GM, Weyrich AS, Zimmerman GA. Leukocyte adhesion deficiency-I variant syndrome (LAD-Iv, LAD-III): molecular characterization of the defect in an index family. Am J Hematol. (2012) 87:311–3. doi: 10.1002/ajh.22253
34. Robert P, Canault M, Farnarier C, Nurden A, Grosdidier C, Barlogis V, et al. A novel leukocyte adhesion deficiency III variant: kindlin-3 deficiency results in integrin- and nonintegrin-related defects in different steps of leukocyte adhesion. J Immunol. (2011) 186:5273–83. doi: 10.4049/jimmunol.1003141
35. Van de Vijver E, Tool AT, Sanal Ö, Çetin M, Ünal S, Aytac S, Seeger K, Pagliara D, et al. Kindlin-3-independent adhesion of neutrophils from patients with leukocyte adhesion deficiency type III. J Allergy Clin Immunol. (2014) 133:1215–8. doi: 10.1016/j.jaci.2013.10.020
36. Al-Jasmi FA, Al-Shamsi A, Hertecant JL, Al-Hamad SM, Souid AK. Inborn errors of metabolism in the United Arab Emirates: disorders detected by newborn screening (2011-2014). JIMD Rep. (2016) 28:127–135. doi: 10.1007/8904_2015_512
37. Etzioni A. Leukocyte-adhesion deficiency. In: Notarangelo LD, Feldweg AM, editors. UptoDate. (2021) Available online at: https://www.uptodate.com/contents/leukocyte-adhesion-deficiency (accessed June 19, 2021).
38. Diz-Küçükkaya R. Inherited platelet disorders including Glanzmann thrombasthenia and Bernard-Soulier syndrome. Hematol Am Soc Hematol Educ Prog. (2013) 2013:268–75. doi: 10.1182/asheducation-2013.1.268
39. Gruda R, Brown AC, Grabovsky V, Mizrahi S, Gur C, Feigelson SW, et al. Loss of kindlin-3 alters the threshold for NK cell activation in human leukocyte adhesion deficiency-III. Blood. (2012) 120:3915–24. doi: 10.1182/blood-2012-02-410795
40. Schmidt S, Nakchbandi I, Ruppert R, Kawelke N, Hess MW, Pfaller K, et al. Kindlin-3-mediated signaling from multiple integrin classes is required for osteoclast-mediated bone resorption. J Cell Biol. (2011) 192:883–97. doi: 10.1083/jcb.201007141
41. Nieswandt B, Varga-Szabo D, Elvers M. Integrins in platelet activation. J Thromb Haemost. (2009) 7 (Suppl. 1):206–9. doi: 10.1111/j.1538-7836.2009.03370.x
42. Saultier P, Szepetowski S, Canault M, Falaise C, Poggi M, Suchon P, et al. Long-term management of leukocyte adhesion deficiency type III without hematopoietic stem cell transplantation. Haematologica. (2018) 103:e264–7. doi: 10.3324/haematol.2017.186304
43. Hamidieh AA, Pourpak Z, Hosseinzadeh M, Fazlollahi MR, Alimoghaddam K, Movahedi M, et al. Reduced-intensity conditioning hematopoietic SCT for pediatric patients with LAD-1: clinical efficacy and importance of chimerism. Bone Marrow Transplant. (2012) 47:646–50. doi: 10.1038/bmt.2011.140
44. Al-Dhekri H, Al-Mousa H, Ayas M, Al-Muhsen S, Al-Ghonaium A, Al-Ghanam G, et al. Allogeneic hematopoietic stem cell transplantation in leukocyte adhesion deficiency type 1: a single center experience. Biol Blood Marrow Transplant. (2011) 17:1245–9. doi: 10.1016/j.bbmt.2010.12.714
45. Stepensky PY, Wolach B, Gavrieli R, Rousso S, Ben Ami T, Goldman V, et al. Leukocyte adhesion deficiency type III: clinical features and treatment with stem cell transplantation. J Pediatr Hematol Oncol. (2015) 37:264–8. doi: 10.1097/MPH.0000000000000228
46. Aygun D, Nepesov S, Gershoni R, Camcioglu Y. Leukocyte adhesion deficiency III: report of two siblings. Pediatr Neonatol. (2017) 58:99–100. doi: 10.1016/j.pedneo.2016.07.006
47. Yadav RM, Dalvi A, Gupta M, Bargir UA, Shabrish S, Aluri J, et al. Spectrum of inborn errors of immunity in a cohort of 90 patients presenting with complications to BCG vaccination in India. Scand J Immunol. (2020) 93:e13010. doi: 10.1111/sji.13010
48. Boisson-Dupuis S. The monogenic basis of human tuberculosis. Hum Genet. (2020) 139:1001–9. doi: 10.1007/s00439-020-02126-6
49. Fekrvand S, Yazdani R, Olbrich P, Gennery A, Rosenzweig SD, Condino-Neto A, et al. Primary immunodeficiency diseases and bacillus Calmette-Guérin (BCG)-vaccine-derived complications: a systematic review. J Allergy Clin Immunol Pract. (2020) 8:1371–86. doi: 10.1016/j.jaip.2020.01.038
50. Al-Hammadi S, Yahya AM, Al-Amri A, Shibli A, Balhaj GB, Tawil MI, et al. Case report: BCG-triggered hemophagocytic lymphohistiocytosis in an infant with X-linked recessive Mendelian susceptibility to mycobacterial disease due to variant of chronic granulomatous disease. Front Pediatr. (2021) 9:687538. doi: 10.3389/fped.2021.687538
Keywords: platelet dysfunction, epistaxis, Glanzmann thrombasthenia, leukocytosis, inborn error of immunity, BCG vaccine, FERMT3, kindlin-3
Citation: Yahya AM, AlMulla AA, AlRufaye HJ, Al Dhaheri A, Elomami AS, Al-Hammadi S, Kailas L, Vijayan R and Souid A-K (2021) Case Report: A Case of Leukocyte Adhesion Deficiency, Type III Presenting With Impaired Platelet Function, Lymphocytosis and Granulocytosis. Front. Pediatr. 9:713921. doi: 10.3389/fped.2021.713921
Received: 24 May 2021; Accepted: 19 July 2021;
Published: 16 August 2021.
Edited by:
Claudio Pignata, University of Naples Federico II, ItalyReviewed by:
Ana Kasirer-Friede, University of California, San Diego, United StatesCopyright © 2021 Yahya, AlMulla, AlRufaye, Al Dhaheri, Elomami, Al-Hammadi, Kailas, Vijayan and Souid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ranjit Vijayan, cmFuaml0LnZAdWFldS5hYy5hZQ==; Abdul-Kader Souid, YXNvdWlkQHVhZXUuYWMuYWU=
†ORCID: Amal M. Yahya orcid.org/0000-0002-7654-0367
Suleiman Al-Hammadi orcid.org/0000-0003-2664-8246
Ranjit Vijayan orcid.org/0000-0002-3830-7409
Abdul-Kader Souid orcid.org/0000-0002-8562-4757
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.