 Niaz Muhammad Khan1†
Niaz Muhammad Khan1† Basharat Hussain2,3†
Basharat Hussain2,3† Chenqing Zheng4†
Chenqing Zheng4† Ayaz Khan5
Ayaz Khan5 Muhammad Shareef Masoud1Qingquan Gu4Linhui Qiu2Naveed Altaf Malik5
Muhammad Shareef Masoud1Qingquan Gu4Linhui Qiu2Naveed Altaf Malik5 Muhammad Qasim1*
Muhammad Qasim1* Muhammad Tariq5
Muhammad Tariq5 Junlei Chang2*
Junlei Chang2*- 1Department of Bioinformatics and Biotechnology, Government College University Faisalabad, Faisalabad, Pakistan
- 2Shenzhen Key Laboratory of Biomimetic Materials and Cellular Immunomodulation, Institute of Biomedicine and Biotechnology, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen, China
- 3University of Chinese Academy of Sciences, Beijing, China
- 4Shenzhen Real Omics Biotech Co., Ltd., Shenzhen, China
- 5National Institute for Biotechnology and Genetic Engineering (NIBGE-C), Faisalabad, Pakistan; Pakistan Institute of Engineering and Applied Sciences (PIEAS), Islamabad, Pakistan
Microcephaly (MCPH) is a genetically heterogeneous disorder characterized by non-progressive intellectual disability, small head circumference, and small brain size compared with the age- and sex-matched population. MCPH manifests as an isolated condition or part of another clinical syndrome; so far, 25 genes have been linked with MCPH. Many of these genes are reported in Pakistani population, but due to a high rate of consanguinity, a significant proportion of MCPH cohort is yet to be explored. MCPH5 is the most frequently reported type, accounting for up to 68.75% alone in a genetically constrained population like Pakistan. In the current study, whole exome sequencing (WES) was performed on probands from 10 families sampled from South Waziristan and two families from rural areas of the Pakistani Punjab. Candidate variants were validated through Sanger sequencing in all available family members. Variant filtering and in silico analysis identified three known mutations in ASPM, a MCPH5-associated gene. The founder mutation p.Trp1326* was segregating in 10 families, which further confirmed the evidence that it is the most prominent mutation in Pashtun ethnicity living in Pakistan and Afghanistan. Furthermore, the previously known mutations p.Arg3244* and p.Arg1019* were inherited in two families with Punjab ethnic profile. Collectively, this study added 12 more families to the mutational paradigm of ASPM and expanded the Pakistani MCPH cohort.
Introduction
Microcephaly (MCPH, OMIM#251200) is a neurodevelopmental disorder characterized by a small head circumference, non-progressive intellectual disability, and small brain size compared with the age- and sex-matched population. The prevalence of MCPH5, the most frequently reported type, is 1/10,000 in consanguineous populations, such as Pakistan's; sporadically, its probability is 1/1,000,000 in non-consanguineous European population (1, 2). Occipitofrontal circumference (OFC) ranges between −2 and −8 SD (standard deviation) at birth (32 and 26 cm, respectively) (3). Patients may exhibit mild to severe developmental delay, sloping forehead, epilepsy, and hereditary hearing loss. Predominantly, primary microcephaly shows an autosomal recessive mode of inheritance (4, 5). So far, mutations in 25 genes have been linked with MCPH. These genes mainly express during cell division and mutations lead to disruption in neurogenesis, cell cycle checkpoints, and centrosome and spindle positioning. The result is an architecturally normal brain with reduced volume, especially in the cerebral cortex region (6). Most of these genes were implicated in the last 10 years after the advent of cutting-edge sequencing technologies (7). Since ~85% of the known variants for rare genetic diseases occur in the coding subset of DNA, therefore, exome sequencing is a handy approach for quick genetic diagnosis, thereby reducing the number of variants for follow-up studies (8, 9). Exome sequencing has the advantage of sequencing the entire coding subset of the genome in a single experiment.
Biallelic mutations in ASPM (MCPH5 MIM #605481) occur more frequently than other genes in MCPH, underlying up to 40% of all the reported primary microcephaly cases. ASPM (MIM# 605481), located on chromosome 1q31.3, is 6.2 kb long with 28 exons, coding for 3,477 amino acids (Ensemble, GRCh38/hg38). This gene plays a vital role in the division of neural progenitor cells and in controlling the cell cycle by helping symmetric proliferative division, as well as asymmetric neurogenic divisions. ASPM knockdown in animal models leads to a decrease in cortical area and microcephaly as observed in humans (10, 11).
In the current study, 12 unrelated families were investigated. Of these, 10 families, with multiple patients, belong to the Wazir tribe of Pashtuns, located in the remote district of South Waziristan. The other two families are from rural areas of Pakistani Punjab and are mutually unrelated. Genetic analysis of these families revealed three known mutations in ASPM, a MCPH5-associated gene. In 10 Wazir families the variant c.3978G>A (p.Trp1326*) was segregating, whereas the two families from Punjab had two different ASPM variants c.9730C>T (p.Arg3244*) and c.3055C>T (p.Arg1019*). This study, thus, further endorses ASPM mutations as the most frequent cause of microcephaly. The p.Trp1326* variant has been previously reported as a founder mutation in more than 50 families among various Pashtun tribes including Wazir (4, 12). The segregation of this variant in 10 unrelated Wazir families in the current study suggests that this variant represents an old mutation and that mutations in this gene are a rare event.
Materials and Methods
Study Participants and Pedigree Construction
Patients were recruited according to the diagnostic criteria of reduced head circumference (HC) (≥3 SD), intellectual disability, and absence of brain malformation symptoms. These families were ascertained through field surveys and with the help of local healthcare workers. All the patients in participating families were characterized with primary microcephaly. Written informed consent was obtained from the guardians of patients and relevant family members. Pedigrees were drawn using HaploPainter v.2.0 (GPLv2) (13). Eighteen patients from the 12 families were male (53%), whereas 16 were female (47%). Ages of the patients range from 1 month to 40 years (Table 1). In seven of these families, patients were born to consanguineous parents. In the remaining five families, there was no immediate consanguinity; however, parents of the patients belong to same tribes (Supplementary Figure 1). HC was measured and compared with a graph of age- and sex-dependent HC values of the normal population (14). Peripheral blood samples were collected from 34 available affected individuals and their normal sibling in EDTA tubes. For molecular analysis, genomic DNA was extracted from blood leukocytes according to standard protocol (15). The Institutional Review Board of the Government College University Faisalabad (Pakistan) and Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences approved the study.
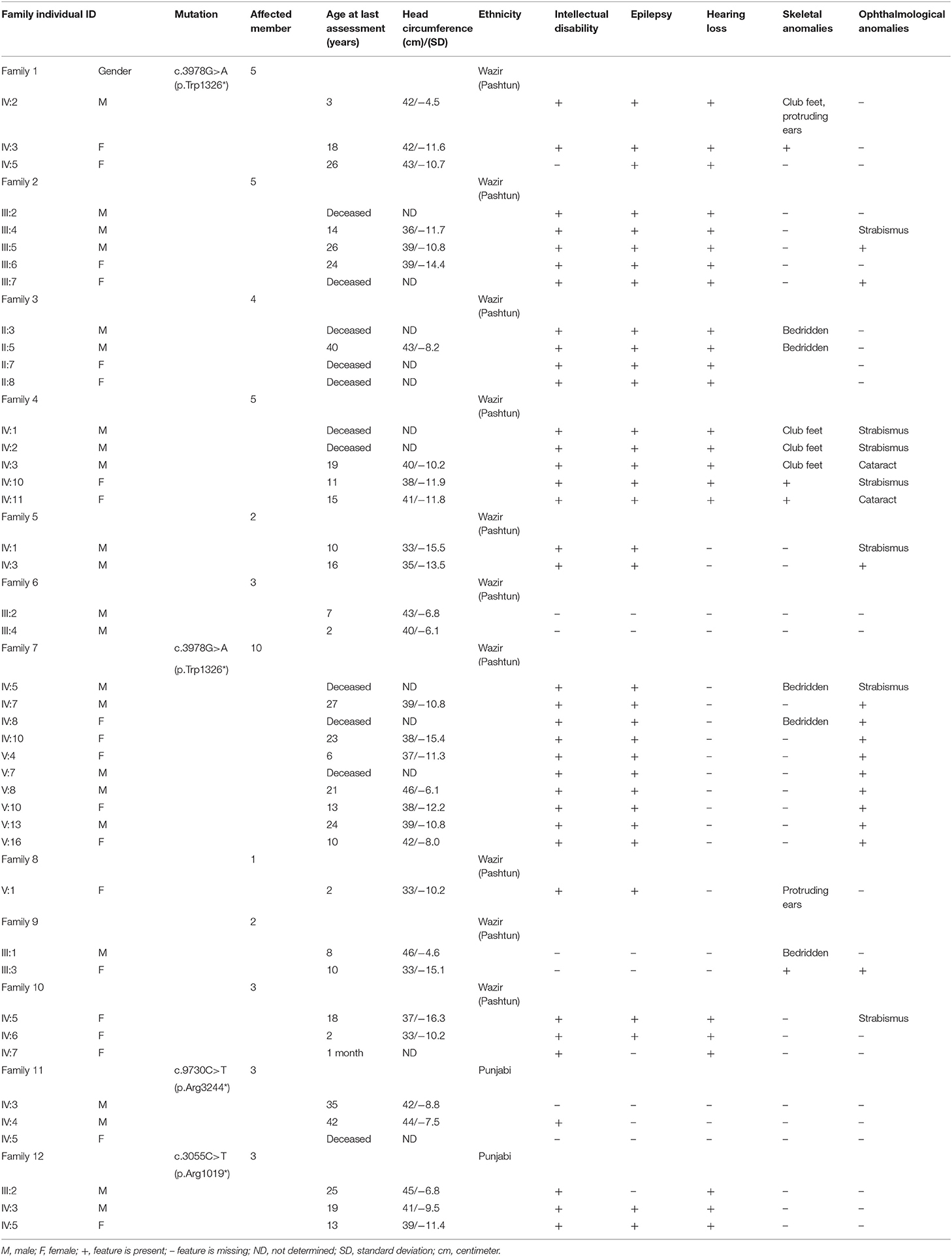
Table 1. Clinical and genetic manifestations in the MCPH cohort.
Whole Exome Sequencing
DNA sample from a proband in each family was selected for whole exome sequencing (WES), using Illumina NovaSeq 6000 platform. Agilent V6 array was used to capture exons with ~100× depth of coverage, using paired-end 2 × 150 bp protocol (2021 Illumina, Inc., San Diego, CA, USA). Sequenced data were aligned and mapped to the human reference genome sequence (GRCh37) assembly. Variants were called using the Genome Analysis Tool Kit GATK, version 3.7 (16), and all variants were annotated and classified by utilizing SnpEff (version 4.2; http://snpeff.sourceforge.net/). After annotation, variants were filtered in public databases [1,000 Genomes Project and Genome Aggregation Database (gnomAD)] and those with MAF <0.005 were retained. Among these variants, homozygous and compound heterozygous variants were selected because the most likely inheritance pattern for these pedigrees was autosomal recessive. No further prediction tools were used as all the variants are known to result in premature termination of translation.
Sanger Sequencing
Candidate variants from exome sequencing were validated by bidirectional Sanger sequencing each variant in the proband. Validated variants were then bidirectionally Sanger sequenced in all the available family members for segregation analysis. Sanger sequencing was done using BigDye terminator sequencing chemistry in Genetic Analyzer 3730 (Applied Biosystems, Foster City, CA, USA). Sequence chromatograms were analyzed with sequence analysis software DNASTAR (Lasergene, Madison, WI, USA) and Sequencher 5.4.6 (Gene Codes Corporation, Ann Arbor, MI, USA).
Results
Clinical and Molecular Findings of MCPH Families
This study reports on three previously known ASPM variants in 12 Pakistani MCPH families with 34 affected individuals (18 males and 16 females). Clinical investigation of all affected individuals revealed reduced skull size (head circumference below −5 SD), mild to severe intellectual disability, slopping forehead, and protruding ears. In addition, speech impairment, ophthalmological problems, deafness, and skeletal defects were noted in the affected individuals (Supplementary Figures 1, 2 and Table 1).
WES was performed on the proband in each family. The resulting FASTQ files were transformed to BAM, and the BAM files were converted to variant call format (vcf) file. The resultant variants were utilized for the identification of the variant that may lead to the disease based on low frequency (MAF +0.01%). We applied various filters and bioinformatics tools to further narrow down the candidate variants and confirmed them in the large reference population cohort of the gnomAD (n > 120,000 exomes and >150,000 genomes). Variants that survived various bioinformatics filters/tools (as explained above) were analyzed, and the previously reported non-sense variants in ASPM gene were found in all the families under investigation. Mutations c.3978G>A (p.Trp1326*), c.9730C>T (p.Arg3244*), and c.3055C>T (p.Arg1019*) were found in homozygous form (Figure 1; Supplementary Figure 1). The sequences of oligos for Sanger confirmation are available in Supplementary Table 1.
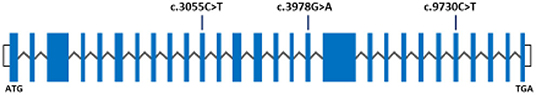
Figure 1. Schematic representation of the ASPM gene on 1q31, showing exons (blue rectangles) and introns (black zigzag lines). White blocks represent the location of the untranslated regions. ASPM is the main driver of primary microcephaly. The previously known mutations c.3055C>T, c.3978G>A, and c.9730C>T were found in this study.
Discussion
We investigated 12 Pakistani families with primary microcephaly by WES. Sanger validation of the candidate variants and segregation analysis found a non-sense ASPM variant c.3978G>A (p.Trp1326*) in 10 of these families; the remaining two families had two different non-sense ASPM variants segregated, c.9730C>T (p.Arg3244*) and c.3055C>T (p.Arg1019*). The parents and some of the siblings of the homozygous patients are heterozygous carriers for these ASPM variants, whereas other siblings are homozygous for the wild-type allele, which corresponds to the recessive inheritance pattern of MCPH in these families. ASPM mutations result in abnormal spindle-like microcephaly-associated protein, which is responsible for 40 to 68% of MCPH incidence (17, 18).
The first variant c.3978G>A has been previously reported in only one Punjabi family and 49 Pashtun families, including three families from the Wazir tribes (4). A large cohort of non-Pashtun Pakistani families with MCPH did not carry this variant (6). The variant was, therefore, regarded as a founder mutation in Pashtun tribes of Pakistan and Afghanistan (4). With the 10 Wazir families included in this study, the total number of Pashtun families with this segregated variant rose to 59. Thus, our finding further endorses this variant as a founder mutation in various tribes of Pashtuns. Screening for this single variant alone can help in identifying a significant number of carriers in any prospective screening program.
The remaining two families were found to have two more ASPM variants, c.9730C>T, (p.Arg3244*) and c.3055C>T (p.Arg1019*). These variants have also been previously reported (19) and characterized. Our findings support a considerable clinical variability associated with ASPM mutations, and we do not see any clear-cut genotype–phenotype correlations in the patients. In conclusion, in 12 families from Pakistan, we found three known mutations in the well-characterized ASPM gene. The high frequency (10 out of 10 Pashtun families) of the ASPM mutation p.Trp1326* among Pashtun families hints on its role as a founder mutation among Pashtuns, especially the Wazir tribe, of Pakistan and Afghanistan. Therefore, testing for this mutation, among Pashtuns, as a first step will be a cost-effective and time-saving approach in the future while performing genetic analysis of MCPH families from this and other populations (20). Similarly, testing for this mutation can also help in population-wide screening for carrier screening and prenatal diagnosis to prevent further affected births.
Data Availability Statement
The sequencing data analyzed in this study is not readily accessible due to privacy restrictions with the patients. Requests to access these datasets should be directed to the corresponding author MQ ( cWFzaW1hd2FuJiN4MDAwNDA7Z2N1Zi5lZHU=.pk).
Ethics Statement
This study was approved from the ethical committee of Government College University, Faisalabad, Pakistan and Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
NK, BH, and CZ recruited the patients, gathered detailed clinical information for the study, and wrote the initial draft. MQ, JC, and MT designed the study. AK, MM, and NM critically reviewed and edited the manuscript. LQ and QG performed whole exome evaluation and mutational analyses. MQ and JC directed the project. All authors critically reviewed the paper. All authors contributed to the article and approved the submitted version.
Funding
The National Natural Science Foundation of China (81771293 to JC), Science Technology and Innovation Commission of Shenzhen Municipality (ZDSYS20190902093409851 and SGLH20180625142404672), International Collaboration project of Chinese Academy of Sciences (172644KYSB20200045), and Guangdong Innovation Platform of Translational Research for cerebrovascular diseases supported the work. The Chinese Government Scholarship (CSC) for International Students supports BH.
Conflict of Interest
CZ and QG were employed by the company Shenzhen Real Omics Biotech Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank all the participating patients, their families, and local healthcare workers for their cooperation in clinical characterization.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.695133/full#supplementary-material
References
1. Woods CG, Parker A. Investigating microcephaly. Arch Dis Child. (2013) 98:707–13. doi: 10.1136/archdischild-2012-302882
2. Moriwaki T, Yamazaki N, So T, Kosuga M, Miyazaki O, Narumi-Kishimoto Y, et al. Normal early development in siblings with novel compound heterozygous variants in ASPM. Hum Genome Var. (2019) 6:56. doi: 10.1038/s41439-019-0088-0
3. Verloes A, Drunat S, Passemard S. ASPM primary microcephaly. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al. editors. GeneReviews®. Seattle (WA): University of Washington, Seattle Copyright 1993-2021, University of Washington, Seattle (2020).
4. Ahmad I, Baig SM, Abdulkareem AR, Hussain MS, Sur I, Toliat MR, et al. Genetic heterogeneity in Pakistani microcephaly families revisited. Clin Genet. (2017) 92:62–8. doi: 10.1111/cge.12955
5. Bazgir A, Agha Gholizadeh M, Sarvar F, Pakzad Z. A Novel Frameshift mutation in Abnormal Spindle-Like Microcephaly (ASPM) gene in an Iranian patient with primary microcephaly: a case report. Iran J Public Health. (2019) 48:2074–8. doi: 10.18502/ijph.v48i11.3528
6. Rasool S, Baig JM, Moawia A, Ahmad I, Iqbal M, Waseem SS, et al. An update of pathogenic variants in ASPM, WDR62, CDK5RAP2, STIL, CENPJ, and CEP135 underlying autosomal recessive primary microcephaly in 32 consanguineous families from Pakistan. Mol Genet Genomic Med. (2020) 8:e1408. doi: 10.1002/mgg3.1408
7. Jean F, Stuart A, Tarailo-Graovac M. Dissecting the genetic and etiological causes of primary microcephaly. Front Neurol. (2020) 11:570830. doi: 10.3389/fneur.2020.570830
8. Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet. (2003) 33:228–37. doi: 10.1038/ng1090
9. Gilissen C, Hoischen A, Brunner HG, Veltman JA. Disease gene identification strategies for exome sequencing. Eur J Hum Genet. (2012) 20:490–7. doi: 10.1038/ejhg.2011.258
10. Fish JL, Kosodo Y, Enard W, Pääbo S, Huttner WB. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci. (2006) 103:10438–43. doi: 10.1073/pnas.0604066103
11. Garrett L, Chang YJ, Niedermeier KM, Heermann T, Enard W, Fuchs H, et al. A truncating ASPM allele leads to a complex cognitive phenotype and region-specific reductions in parvalbuminergic neurons. Transl Psychiatry. (2020) 10:66. doi: 10.1038/s41398-020-0686-0
12. Ahmed J, Windpassinger C, Salim M, Wiener M, Petek E, Schaflinger E, et al. Genetic study of Khyber-Pukhtunkhwa resident Pakistani families presenting primary microcephaly with intellectual disability. J Pak Med Assoc. (2019) 69:1812–6. doi: 10.5455/JPMA.300681
13. Thiele H, Nürnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. (2005) 21:1730–2. doi: 10.1093/bioinformatics/bth488
14. Nellhaus G. Head circumference from birth to eighteen years. Practical composite international and interracial graphs. Pediatrics. (1968) 41:106–14.
15. Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. (1989) 17:8390. doi: 10.1093/nar/17.20.8390
16. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
17. Picher-Martel V, Labrie Y, Rivest S, Lace B, Chrestian N. Whole-exome sequencing identifies homozygous mutation in TTI2 in a child with primary microcephaly: a case report. BMC Neurol. (2020) 20:58. doi: 10.1186/s12883-020-01643-1
18. Zaqout S, Morris-Rosendahl D, Kaindl AM. Autosomal recessive primary microcephaly (MCPH): an update. Neuropediatrics. (2017) 48:135–42. doi: 10.1055/s-0037-1601448
19. Muhammad F, Mahmood Baig S, Hansen L, Sajid Hussain M, Anjum Inayat I, Aslam M, et al. Compound heterozygous ASPM mutations in Pakistani MCPH families. Am J Med Genet A. (2009) 149a:926–30. doi: 10.1002/ajmg.a.32749
Keywords: primary microcephaly, MCPH5, whole exome sequencing, Pakistani population, founder effect
Citation: Khan NM, Hussain B, Zheng C, Khan A, Masoud MS, Gu Q, Qiu L, Malik NA, Qasim M, Tariq M and Chang J (2021) Updates on Clinical and Genetic Heterogeneity of ASPM in 12 Autosomal Recessive Primary Microcephaly Families in Pakistani Population. Front. Pediatr. 9:695133. doi: 10.3389/fped.2021.695133
Received: 14 April 2021; Accepted: 27 May 2021;
Published: 06 July 2021.
Edited by:
Mahmood Rasool, King Abdulaziz University, Saudi ArabiaReviewed by:
Shahid Khan, Johns Hopkins Medicine, United StatesMohsin Shahzad, Shaheed Zulfiqar Ali Bhutto Medical University (SZABMU), Pakistan
Muhammad Saif Ur Rahman, Zhejiang University, China
Copyright © 2021 Khan, Hussain, Zheng, Khan, Masoud, Gu, Qiu, Malik, Qasim, Tariq and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Muhammad Qasim, cWFzaW1hd2FuJiN4MDAwNDA7Z2N1Zi5lZHUucGs=; Junlei Chang, amwuY2hhbmcmI3gwMDA0MDtzaWF0LmFjLmNu
†These authors have contributed equally to this work and share first authorship