
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 11 June 2021
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.682846
 Jianlong Zhuang1†
Jianlong Zhuang1† Chunnuan Chen2†Jia Li3Yuying Jiang1Junyu Wang1
Chunnuan Chen2†Jia Li3Yuying Jiang1Junyu Wang1 Yuanbai Wang1Shuhong Zeng1Yiming Lin4*Yingjun Xie5,6*
Yuanbai Wang1Shuhong Zeng1Yiming Lin4*Yingjun Xie5,6*Background: Very few reports are available on human XX ovotesticular disorder of sex development involving SOX3 gene duplication. Here we aim to present a rare case of SOX3 gene duplication in a person from the Chinese population who exhibits XX ovotesticular disorder of sex development.
Case Presentation: A 7-year-old Chinese individual from Fujian province in Southeast China was recruited. The patient presented 46, XX karyotype, absence of sex-determining region Y, and was diagnosed with XX ovotesticular disorder of sex development. Furthermore, SNP array analysis demonstrated that the patient had a 2.2-Mb duplication in the Xq27.1q27.2 region (arr[hg19]Xq27.1q27.2:139,499,778-141,777,782) involving the SOX3 gene. Additionally, no SOX3 duplication was observed in the parents or the sibling, who displayed none of the clinical features.
Conclusion: We identified the first case of SOX3 duplication in a Chinese individual who exhibits ovotesticular disorder of sex development. Our study strengthens the link between the SOX3 duplication and XX ovotesticular disorder of sex development and indicates that SOX3 is the evolutionary antecedent of sex-determining region Y.
Sex-determining region Y (SRY) is the key gene in 46, XY normal males. SRY initiates a complex genetic cascade, promoting the differentiation of the testis. However, the coexistence of ovarian and testicular tissues is present in some 46, XX individuals, which refers to as ovotesticular disorder of sex development (OT-DSD) (1, 2). Studies have shown that the occurrence of 46, XX OT-DSD is related to the dislocation recombination on the X and Y chromosomes during the meiosis of the paternal chromosome, which transfers the SRY gene from the Y chromosome to X (3), but only few patients with 46, XX OT-DSD have a detectable SRY gene; most of the subjects show an absence of the SRY gene (4, 5). However, the SRY gene is present in most of (~80%) 46, XX testicular DSD cases (6). Currently, it is believed that sex determination and differentiation are processes of orderly and coordinated expression of autosomal and sex chromosomes, but with the SRY gene, abnormalities in any process can lead to sex abnormalities.
As we know, SRY up-regulates the expression of SRY-Box transcription factor 9 (SOX9) in bipotential gonads, leading to the differentiation of testicular cells and eventually testicular differentiation (7). Moreover, a study has shown that ectopic SOX9 expression induces the formation of mouse testis in XX gonads (8). Recently, several cases have been reported to carry SOX9 duplications (9–12), which have been proposed to be responsible for SOX9 expression during gonad development. SRY-Box transcription factor 3 (SOX3), located on the chromosome X (Xq27.1), is a member of the SRY-Box transcription factor family (13). Duplications involving the SOX3 gene have been reported to be associated with developmental delay, intellectual disability, growth hormone deficiency, infundibular hypoplasia and hypopituitarism, etc. (14, 15).
Recently, XX sex reversal has been reported in transgenic mice with ectopic SOX3 expression and observed in 46, XX DSD patients with duplications of SOX3 or genomic rearrangements within the SOX3 regulatory region (16). Few reports are available on 46, XX SRY-negative males with SOX3 duplications, though a recent study conducted by Tasic et al. revealed a 46, XX male who presented congenital anomalies of kidneys and the urinary tract and had a duplication on chromosome Xq27 involving the SOX3 gene, indicating links between SOX3 gene dosage and kidney malformations and sex determination (17). Moreover, a study has shown a 46, XX SRY-negative individual with duplication of the SOX3 gene exhibiting XX OT-DSD (18). In the present study, we describe a 7-year-old OT-DSD case with Xq27.1q27.2 duplication involving the SOX3 gene, which was first identified in Chinese individuals and additionally strengthened the pathogenic role of SOX3 duplication in XX OT-DSD.
The patient comes from Quanzhou City, Fujian province, in Southeast China. The child was delivered vaginally with a birth weight of 3.7 kg. There was no family history of DSD, and the parents denied any consanguinity. A physical examination showed the child presented ambiguous sex, coronal hypospadias, a penis or enlarged clitoris, and the presence of a scrotum but non-palpable gonads. Subsequent ultrasonography indicated that the patient might have the coexistence of testicular and ovarian tissues on the left side and testicular tissue on the right side. After clinical consultation, the family decided to raise the child as a male, and ovariectomy was performed at 10 months after birth, to remove the ovarian section from the left ovotesticular area. The subsequent histology analysis confirmed the presence of unilateral ovotestes tissues in the left side of the patient.
At age 7, the child's height (130 cm) and weight (23 kg) were within the normal ranges. Hormonal laboratory tests showed low luteinizing hormone (<0.20 mIU/ml), follicle-stimulating hormone (1.21 mIU/ml) and testosterone (<0.10 ng/ml). Serum progesterone and prolactin were normal. Currently, human menopausal gonadotropin (menotropins for injection, AnHui BBCA Pharmaceutical Co., Ltd.) is injected for treatment with 150 U a day. Regular follow-up showed normal penile and testicular development, with normal morning erection.
Chromosome G-banding analysis revealed a normal karyotype (46, XX) in the patient. The parental karyotypes were normal as well. Chromosomal microarray analysis demonstrated that the patient had a 2.2-Mb duplication in the Xq27.1q27.2 region (arr[hg19]Xq27.1q27.2:139,499,778-141,777,782) of the X chromosome (Figure 1). The duplication contains 12 Online Mendelian Inheritance in Man (OMIM) genes: CDR1, LDOC1, MAGEC1, MAGEC2, MAGEC3, SOX3, SPANXA1, SPANXA2, SPANXB1, SPANXB2, SPANXC, and SPANXD. SNP array analysis was also performed on parental and sibling blood samples. Chromosomal microarray results showed that none of the Y chromosome was observed, and further study indicated that no SRY gene was observed in the patient by polymerase chain reaction. Furthermore, no SOX3 duplication was observed in the parents or the sibling with normal phenotype.
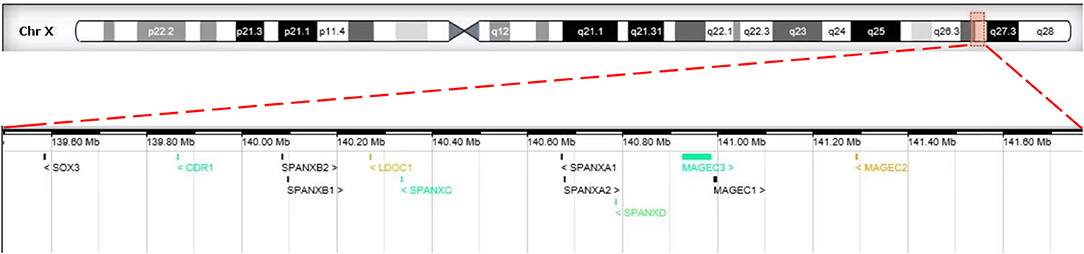
Figure 1. The breakpoint of Xq27.1q27.2 duplication in our study. The upper panel shows the schematic representation of chromosome X. The red rectangle displays the genomic location of Xq27.1q27.2 on the chromosome X. The lower panel presents a magnified view of the Xq27.1q27.2 duplication (arr[hg19] chrX: 139,499,778- 141,777,782) involving the SOX3 gene.
OT-DSD is the disease defined as presence of both male and female gonads. The SRY gene is present in few cases of 46, XX OT-DSD patients, which can explain testicular development (18). However, the SRY gene is absent in the most of 46, XX OT-DSD patients, and the mechanism underlying the testis development is not fully understood.
SOX genes are considered key players in the regulation of nervous system development and embryogenesis; they encode transcription factors that act as key regulators in a variety of developmental processes, including specification, gastrulation, cellular differentiation, and neural induction (19). SOX9 is critical to the human testis differentiation, while it is still poorly understood whether SOX3 expression affects sex differentiation. A study showed knockout of SOX3 did not cause any defects of sex determination; however, affected testis differentiation and oocyte development were observed in SOX3-null mice (20). Moreover, another study showed that SOX3 mutations were absent in the subjects diagnosed with 46, XY gonadal dysgenesis and 46, XX sex reversal, indicating that SOX3 might not be involved in testis differentiation (21). Recent studies have shown that several human XX male sex reversal cases present rearrangements of the SOX3 locus, suggesting that a defect in the SOX3 gene might result in XX male sex reversal in mice and humans. Therefore, researchers believe that SRY may arise from SOX3 and the two genes have interchangeable functions in sex determination (16, 22).
The study conducted by Sutton et al. (16) showed three patients with XX male sex reversal exhibiting rearrangements encompassing or in proximity of SOX3. Patient A had two microduplications, one of which covered the entire SOX3 gene; patient B carried a microdeletion located upstream of SOX3 in Xq27.1; patient C had a large duplication that encompassed the SOX3 gene and at least 18 additional genes, which might be responsible for the clinical phenotype (Table 1). Additionally, another study by Moalem et al. (22) showed de novo SOX3 gene duplication in XX male sex reversal with genital abnormalities. The patient exhibited a partial sex reversal with abnormal genitalia and had three copy number variants, the first of which was a 494-kb duplication in region Xq27.1, which encompassed the SOX3 gene. The phenotype might be associated with weak or slightly late ectopic expression of SOX3 in the early gonads. Subsequently, a study (18) identified the first 46, XX OT-DSD case who showed a SOX3 duplication, with absence of SRY, and presented hypospadias and bilateral cryptorchidism (Table 1).
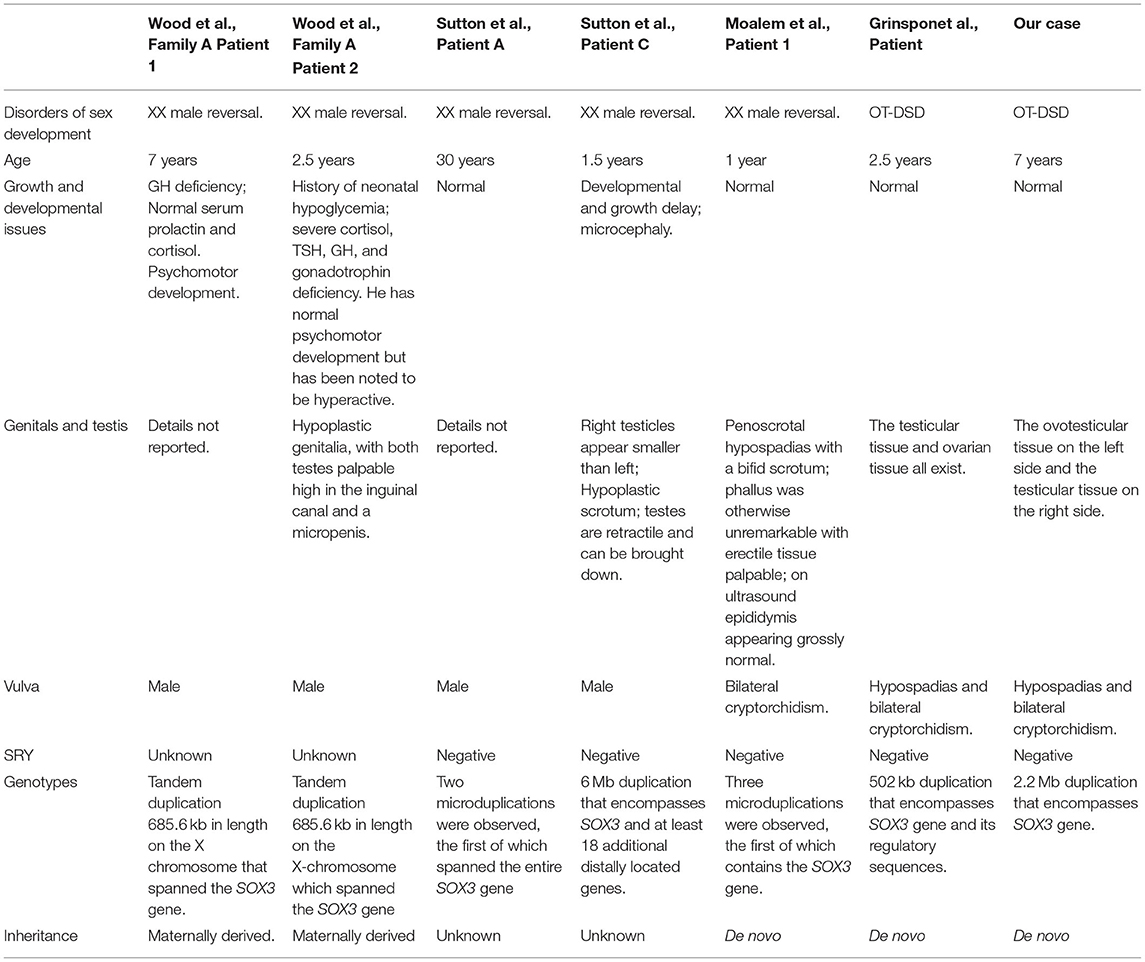
Table 1. Literature review of the involvement of SOX3 duplications in disorders of sex development.
In our study, we secondly identified a 46, XX OT-DSD case with SOX3 gene duplication, which was first found in Chinese individuals. The findings are in agreement with the previous studies, supporting the notion that duplication of SOX3 is responsible for partial testicular differentiation in the fetal XX gonads. In this study, apart from the SOX3 gene, 11 OMIM genes were also involved in this duplicated region. Previous studies indicated that the MAGE genes and SPANX genes are specifically expressed in tumors and testis (23–25), which needs further investigation as to the potential relationship between these genes and sex determination.
In conclusion, our study firstly identified a patient carrying the Xq27.1q27.2 duplication involving the SOX3 gene in a 46, XX OT-DSD Chinese individual, which provides additional evidence that the duplication of SOX3 is pathological in the XX OT-DSD and further indicates that SOX3 may be the evolutionary antecedent of SRY. However, more work can be done on the expression of SOX9 or other genes that affect the gonadal differentiation pathway, such as WNT4 or RSPO1.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Ethics Committee of Quanzhou women's and children's hospital. We confirmed that all subjects who participated in this study signed written informed consent for publishing their own and their children's genetic data and relevant information.
JZ and CC designed the study and wrote the article. JW, SZ, and YW performed the karyotype analysis and analyzed the data. YJ, JL, YL, and YX revised and polished the manuscript. All authors approved the final article.
This research was supported by the Fujian Provincial Health Commission Youth Science and Technology Project (2020QNB045) and Quanzhou City Science and Technology Project (2020C026R).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to express our appreciation to Fujian Provincial Health Commission and Quanzhou City Science and Technology Bureau for funding this work. We also express our appreciation to the patient and his family members who participated in this study.
1. Houk CP Hughes IA Ahmed SF Lee PA Writing Committee for the International Intersex Consensus Conference Participants. Summary of consensus statement on intersex disorders and their management. International Intersex Consensus Conference. Pediatrics. (2006) 118:753–7. doi: 10.1542/peds.2006-0737
2. Lee PA Houk CP Ahmed SF Hughes IA International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics. (2006) 118:e488–500. doi: 10.1542/peds.2006-0738
3. McElreavey K, Cortes LS. X-Y translocations and sex differentiation. Semin Reprod Med. (2001) 19:133–9. doi: 10.1055/s-2001-15393
4. Queipo G, Zenteno JC, Peña R, Nieto K, Radillo A, Dorantes LM, et al. Molecular analysis in true hermaphroditism: demonstration of low-level hidden mosaicism for Y-derived sequences in 46,XX cases. Hum Genet. (2002) 111:278–83. doi: 10.1007/s00439-002-0772-9
5. Krob G, Braun A, Kuhnle U. True hermaphroditism: geographical distribution, clinical findings, chromosomes and gonadal histology. Eur J Pediatr. (1994) 153:2–10. doi: 10.1007/BF02000779
6. Délot EC, Vilain EJ. Nonsyndromic46,XX testicular disorders of sex development. In: Adam MP, Ardinger HH, Pagon RA, Wallace, SE, Bean, LJH, Stephens, K, et al. GeneReviews®. Seattle, WA: University of Washington (2003).
7. Piprek RP. Genetic mechanisms underlying male sex determination in mammals. J Appl Genet. (2009) 50:347–60. doi: 10.1007/BF03195693
8. Vidal VP, Chaboissier MC, de Rooij DG, Schedl A. Sox9 induces testis development in XX transgenic mice. Nat Genet. (2001) 28:216–7. doi: 10.1038/90046
9. Croft B, Ohnesorg T, Hewitt J, Bowles J, Quinn A, Tan J, et al. Human sex reversal is caused by duplication or deletion of core enhancers upstream of SOX9. Nat Commun. (2018) 9:5319. doi: 10.1038/s41467-018-07784-9
10. Cox JJ, Willatt L, Homfray T, Woods CG. A SOX9 duplication and familial 46,XX developmental testicular disorder. N Engl J Med. (2011) 364:91–3. doi: 10.1056/NEJMc1010311
11. Huang B, Wang S, Ning Y, Lamb AN, Bartley J. Autosomal XX sex reversal caused by duplication of SOX9. Am J Med Genet. (1999) 87:349–53. doi: 10.1002/(SICI)1096-8628(19991203)87:4 <349::AID-AJMG13>3.0.CO;2-N
12. Lee GM, Ko JM, Shin CH, Yang SW. A Korean boy with 46,XX testicular disorder of sex development caused by SOX9 duplication. Ann Pediatr Endocrinol Metab. (2014) 19:108–12. doi: 10.6065/apem.2014.19.2.108
13. Kamachi Y, Uchikawa M, Kondoh H. Pairing SOX off: with partners in the regulation of embryonic development. Trends Genet. (2000) 16:182–7. doi: 10.1016/S0168-9525(99)01955-1
14. Laumonnier F, Ronce N, Hamel BC, Thomas P, Lespinasse J, Raynaud M, et al. Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am J Hum Genet. (2002) 71:1450–5. doi: 10.1086/344661
15. Woods KS, Cundall M, Turton J, Rizotti K, Mehta A, Palmer R, et al. Over- and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am J Hum Genet. (2005) 76:833–49. doi: 10.1086/430134
16. Sutton E, Hughes J, White S, Sekido R, Tan J, Arboleda V, et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest. (2011) 121:328–41. doi: 10.1172/JCI42580
17. Tasic V, Mitrotti A, Riepe FG, Kulle AE, Laban N, Polenakovic M, et al. Duplication of the SOX3 gene in an Sry-negative 46,XX male with associated congenital anomalies of kidneys and the urinary tract: Case report and review of the literature. Balkan J Med Genet. (2019) 22:81–8. doi: 10.2478/bjmg-2019-0006
18. Grinspon RP, Nevado J, Mori Alvarez Mde L, Del Rey G, Castera R, Venara M, et al. 46,XXovotesticular DSD associated with a SOX3 gene duplication in a SRY-negative boy. Clin Endocrinol. (2016) 85:673–5. doi: 10.1111/cen.13126
19. Stagi S, Lapi E, Pantaleo M, Traficante G, Giglio S, Seminara S, et al. A SOX3 (Xq26.3-27.3) duplication in a boy with growth hormone deficiency, ocular dyspraxia, and intellectual disability: a long-term follow-up and literature review. Hormones. (2014) 13:552–60. doi: 10.14310/horm.2002.1523
20. Weiss J, Meeks JJ, Hurley L, Raverot G, Frassetto A, Jameson JL. Sox3 is required for gonadal function, but not sex determination, in males and females. Mol Cell Biol. (2003) 23:8084–91. doi: 10.1128/MCB.23.22.8084-8091.2003
21. Lim HN, Berkovitz GD, Hughes IA, Hawkins JR. Mutation analysis of subjects with 46, XX sex reversal and 46, XY gonadal dysgenesis does not support the involvement of SOX3 in testis determination. Hum Genet. (2000) 107:650–2. doi: 10.1007/s004390000428
22. Moalem S, Babul-Hirji R, Stavropolous DJ, Wherrett D, Bägli DJ, Thomas P, et al. XX male sex reversal with genital abnormalities associated with a de novo SOX3 gene duplication. Am J Med Genet A. (2012) 158A:1759–64. doi: 10.1002/ajmg.a.35390
23. Lucas S, De Plaen E, Boon T. MAGE-B5, MAGE-B6, MAGE-C2, and MAGE-C3: four new members of the MAGE family with tumor-specific expression. Int J Cancer. (2000) 87:55–60. doi: 10.1002/1097-0215(20000701)87:1<55::AID-IJC8>3.0.CO;2-J
24. Lucas S, De Smet C, Arden KC, Viars CS, Lethé B, Lurquin C, et al. Identification of a new MAGE gene with tumor-specific expression by representational difference analysis. Cancer Res. (1998) 58:743–52.
Keywords: disorder of sex development, ovotestis, Xq27.1q27.2 duplication, SOX3 gene, chromosomal microarray analysis
Citation: Zhuang J, Chen C, Li J, Jiang Y, Wang J, Wang Y, Zeng S, Lin Y and Xie Y (2021) The 46, XX Ovotesticular Disorder of Sex Development With Xq27.1q27.2 Duplication Involving the SOX3 Gene: A Rare Case Report and Literature Review. Front. Pediatr. 9:682846. doi: 10.3389/fped.2021.682846
Received: 19 March 2021; Accepted: 26 April 2021;
Published: 11 June 2021.
Edited by:
Lingqian Wu, Central South University, ChinaReviewed by:
Corrado Romano, Oasi Research Institute (IRCCS), ItalyCopyright © 2021 Zhuang, Chen, Li, Jiang, Wang, Wang, Zeng, Lin and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yiming Lin, bGlueWltaW5nMDgxOUBzaW5hLmNvbQ==; Yingjun Xie, eGlleWp1bkBtYWlsMi5zeXN1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.