
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 15 July 2021
Sec. Pediatric Cardiology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.682390
This article is part of the Research Topic Cardiovascular Genetics – Focus on Paediatric Cardiomyopathy View all 15 articles
 Laura Muiño-Mosquera1,2
Laura Muiño-Mosquera1,2 Julie De Backer2,3*
Julie De Backer2,3*Genetic aortic diseases are a group of illnesses characterized by aortic aneurysms or dissection in the presence of an underlying genetic defect. They are part of the broader spectrum of heritable thoracic aortic disease, which also includes those cases of aortic aneurysm or dissection with a positive family history but in whom no genetic cause is identified. Aortic disease in these conditions is a major cause of mortality, justifying clinical and scientific emphasis on the aorta. Aortic valve disease and atrioventricular valve abnormalities are known as important additional manifestations that require careful follow-up and management. The archetype of genetic aortic disease is Marfan syndrome, caused by pathogenic variants in the Fibrillin-1 gene. Given the presence of fibrillin-1 microfibers in the myocardium, myocardial dysfunction and associated arrhythmia are conceivable and have been shown to contribute to morbidity and mortality in patients with Marfan syndrome. In this review, we will discuss data on myocardial disease from human studies as well as insights obtained from the study of mouse models of Marfan syndrome. We will elaborate on the various phenotypic presentations in childhood and in adults and on the topic of arrhythmia. We will also briefly discuss the limited data available on other genetic forms of aortic disease.
Heritable Thoracic Aortic Diseases (HTAD) encompasses a spectrum of genetic conditions in which aortic disease (aneurysms and dissections) has an underlying genetic trigger or familial occurrence. HTAD is classified as syndromic and non-syndromic. The genetic causes fall into several distinct groups of genes coding for (I.) components of the extracellular matrix (ECM) (FBN1, COL3A1, LOX); (II.) components involved in the TGFβ pathway (TGFBR1 and 2, SMAD2 and 3 and TGFB2 and 3); and (III.) components of the vascular smooth muscle cell apparatus (ACTA2, MYLK, MYH11, PRKGA1) (1). The main clinical entities with their respective genes and clinical features are listed in Table 1. Here, only those genes with a definitive or strong association with HTAD are listed. There are many more candidate genes on the horizon, and this list keeps growing.
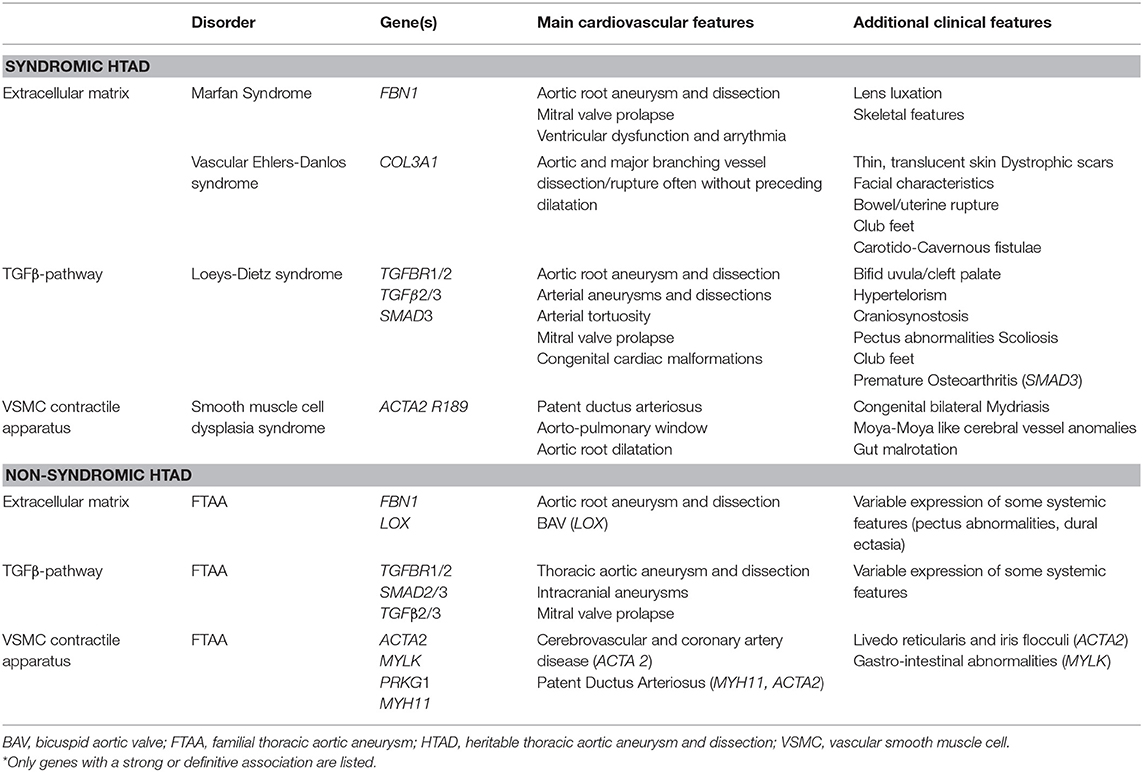
Table 1. Main clinical features and genes* associated with Heritable Thoracic Aortic Aneurysm and Dissection.
The paradigm syndromic HTAD entity is Marfan syndrome (MFS). MFS is an inherited connective tissue disease caused by pathogenic variants in the Fibrillin-1 gene (FBN1), which codes for the ECM protein fibrillin-1. The condition was first described in 1895 by the French pediatrician Antoine Bernard Marfan who described a constellation of skeletal abnormalities characterized by joint contractures and conspicuously long fingers (arachnodactyly) in a young girl (2). It took over 50 years for the clinical picture of Marfan syndrome to be more clearly defined in the seminal work of Victor McKusick. He described the condition as a connective tissue disease with cardiovascular involvement. Without knowing the underlying molecular defect, he very accurately reported that “Clinically, Marfan syndrome behaves as an abiotrophy of some connective tissue” (3). By this time, cardiovascular involvement had been consistently reported, along with the skeletal and ocular organ systems' involvement. The concept of an abnormality in elastic fibers in the aorta as a cause for the characteristic aortic aneurysms and dissections was put forward. In addition, involvement of the veins, the heart valves and also the endocardium and myocardium were suspected. The latter fact is of particular value in the context of this review.
Unraveling the structural components of connective tissue again took several decades. Evidence for the link between connective tissue and the clinical entity of Marfan syndrome was first provided by immunohistochemic studies using antibodies for fibrillins, showing deficiencies in the amount and distribution of microfibrillar fibers in skin samples from patients with MFS (4). The identification of pathogenic missense variants in the FBN1 gene in two patients with Marfan syndrome in 1991 provided final confirmation (5). Fibrillins are large structural macromolecules that contribute to the integrity and function of all connective tissues. According to the initial concept, fibrillin microfibrils mainly served as a scaffold for elastic fiber formation. Biochemical investigations and genetic evidence from both humans and mice have now uncovered many more functions of fibrillin microfibrils. Today, we know that fibrillin microfibrils have essential tissue-specific architectural functions beyond serving as scaffolds for elastin deposition. More recently, an important functional role of fibrillin microfibrils has emerged: fibrillin microfibrils target and sequester members of the TGFβ superfamily of growth factors. In this manner, the structures of fibrillin microfibrils collaborate with biological functions to shape and maintain connective tissues (6). The combined structural and functional role of fibrillins nicely illustrates the current concept of mechanobiology underlying the pathophysiology of cardiovascular disease in MFS. Through interactions between vascular smooth muscle cells (in the aorta) or cardiomyocytes (in the myocardium) and the ECM, the cells can sense changes in mechanical forces of the ECM. These mechanical signals are converted into biochemical or electrical signals, thereby enabling a responsive cellular adaption and remodeling. This process, which is bidirectional, is called mechanobiology. The composition of the ECM and the proportion and the expression of each protein can have a profound influence on cardiac structure and compliance that will determine its hemodynamic functions. One of the major myocardial ECM components is collagen, which will, when present in excessive concentrations lead to myocardial fibrosis and distortion of the myocardial architecture. Fibrosis is prevalent in many acquired cardiac diseases and underlies several adverse cardiac events, such as heart failure, arrhythmia, and death. Increased fibrillin-1 expression has been reported in the context of myocardial fibrosis (7) and gene expression studies have targeted genes involved in the ECM as highly enriched in patients with cardiomyopathy (CMP) with fibrosis and cardiac remodeling (8). Monogenic forms of CMP caused by pathogenic variants in genes encoding for ECM structural components are scarce. Cases of non-compaction CMP caused by FBN1 pathogenic variants have been reported (9). Further research in this field is highly relevant, not only to identify potential other genes involved but also since several proteins represent candidate therapeutic targets to prevent or reverse fibrosis (10).
A detailed description of the role of the ECM in the myocardium can be found in a recent review by Frangogiannis (11).
The diagnosis of MFS is based on the identification of clinical manifestations, as defined in the revised Ghent nosology (12). The extent, severity and age of onset of clinical manifestations are highly variable, ranging from severe cardiovascular involvement at birth in the neonatal form to patients developing manifestations only in mid-life. The estimated prevalence of Marfan syndrome is 1 in 3.000–5.000 individuals, with no ethnic or sex predilection (13). Prognosis is mainly determined by progressive dilation of the aorta, leading to aortic dissection and death at a young age. Mean survival of untreated patients is about 40 years. Fortunately, improved management and ongoing research have led to a significant increase in life expectancy of at least 30 years (14, 15) which does not imply that life expectancy in MFS is normal. A recent population study demonstrated a median age at death in MFS patients of 50 years, which is 8–13 years lower than in the general population (16). A critical factor in improving prognosis is the early identification of patients with Marfan syndrome. Precipitating factors reported to accelerate progressive dilatation or dissection include elevated blood pressure, intense physical exercise and pregnancy (17, 18).
When referring to Marfan syndrome CMP, two different clinical entities should be distinguished: (1) Heart failure in very young children with MFS (neonates and infants) (2) CMP in classical MFS. We will discuss both entities separately in the next sections.
Neonatal MFS (nMFS) is a term usually reserved for very early clinical presentations of MFS even though some patients may present after the 1st month of life (19). The exact prevalence of nMFS is unknown but is much lower than the prevalence of classical Marfan syndrome. The majority of these patients (90–95%) carry a de novo variant in the so-called “neonatal region” (exons 24–32) with a cluster of variants in exons 25–26 (20). Children with nMFS have a typical appearance with dolichocephaly, progeroid appearance, arachnodactyly, crumpled ears, joint contractures and pectus abnormalities (Figure 1A). Some children will exhibit congenital lung emphysema and ocular abnormalities. Unlike classical MFS, the most prominent cardiac problem in children with nMFS is tricuspid and mitral valve prolapse, usually with severe progressive regurgitation leading to congestive heart failure (Figures 1B–E) (20–22). Aortic root dilatation is also commonly present in children with nMFS but does not account for the most significant morbidity and mortality in this age group. Most children with nMFS die within the 1st year of life of cardiac failure (20), although the number of survivors into teenage years is increasing thanks to improvement in care (23, 24).
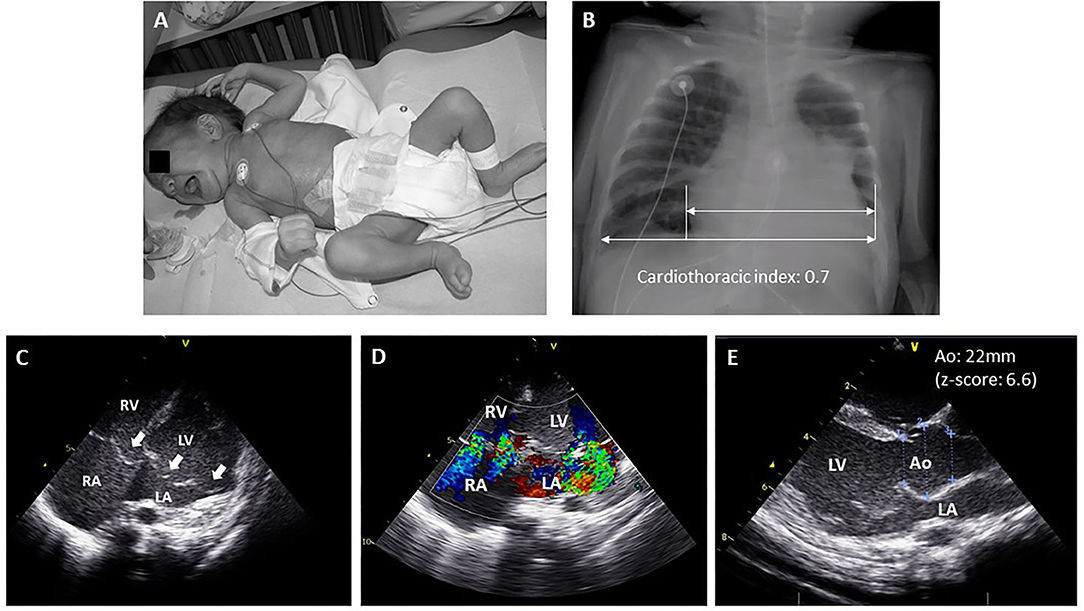
Figure 1. Typical manifestations of neonatal Marfan syndrome. (A) Child with neonatal MFS showing arachnodactyly, long feet, crumpled ears, lipodystrophy and mild pectus excavatum. (B) Chest X-ray showing cardiomegaly and mild scoliosis. (C) Echocardiographic four-chamber view showing mitral and tricuspid valve prolapse (white arrows). (D) Echocardiographic apical four-chamber color doppler view showing moderate-severe mitral and tricuspid valve regurgitation. (E) Echocardiographic parasternal long axis view showing enlargement of the sinus of Valsalva. Ao, aorta; LA, left atrium; LV, left ventricle; RV, right ventricle; RA, right atrium.
Children with the infantile form of MFS usually present a less severe course (25). These children might be diagnosed at a very young age (even during the first months of life), and the phenotype is similar to that of nMFS. However, from a cardiovascular perspective, the infantile form of MFS resembles a more severe subform of classical Marfan syndrome. They show severe aortic root dilation at a young age, but atrioventricular valve dysfunction is typically less prominent than in the neonatal form (21, 26). These children usually present left ventricular dilatation, even with mild valvular regurgitation, but preserved left ventricular function.
For obvious reasons, cardiovascular research and management recommendations for classic MFS have heavily focused on aortic disease. Interest in studying myocardial involvement was raised in the nineties with a study from Savolainen and colleagues, indicating abnormal diastolic function in children with MFS, assessed by cardiac magnetic resonance (CMR) (27).
Moreover, several (historical) series on survival in MFS have listed heart failure as one of the leading causes of death. Estimates vary between 5 and 30% (16, 28) putting heart failure at least at an equal level as aortic dissection. End-stage heart failure necessitating heart transplantation in patients with MFS has been reported in several case reports and small series (29–31). In most of these patients, heart failure was triggered by underlying severe valvular disease (aortic and/or mitral valve regurgitation).
In addition to these extrinsic (secondary) causes of heart failure, several reports from independent researchers have shown intrinsic myocardial dysfunction in MFS. The reported prevalence of what is now known as Marfan CMP ranges from 3% (32) to 68% (33) across different series, depending on the definition and population characteristics. Involvement of both left and right ventricles with systolic and diastolic dysfunction has been reported (27, 34–39) (Table 2). Although myocardial dysfunction is mostly mild and subclinical and does not progress much over time, some patients will present in overt heart failure. A possible link between intrinsic CMP and an unfavorable course in the event of an additional hemodynamic trigger such as valvular dysfunction and/or aortic root replacement has not yet been demonstrated but seems plausible. Whether these hemodynamic changes trigger myocardial fibrosis, as seen in other types of CMP (50), is also not clear in MFS. A small study in children with MFS and Loeys-Dietz syndrome (LDS) showed increased left- and right ventricular volumes and diffuse myocardial fibrosis on CMR in comparison to healthy control subjects (51). Further evidence is necessary to elucidate whether diffuse fibrosis is present in MFS and influences clinical outcome. Whether the type of underlying pathogenic FBN1 variant plays a role in defining the risk for developing CMP is still unclear. Two independent studies observed a higher incidence of left ventricular dilation and decreased left ventricular function in patients carrying non-missense variants (46, 49), which is in line with recent data on genotype-phenotype correlations in patients with Marfan syndrome, indicating a worse cardiovascular phenotype in patients harboring non-missense variants predicted to result in a haploinsufficient effect (52).
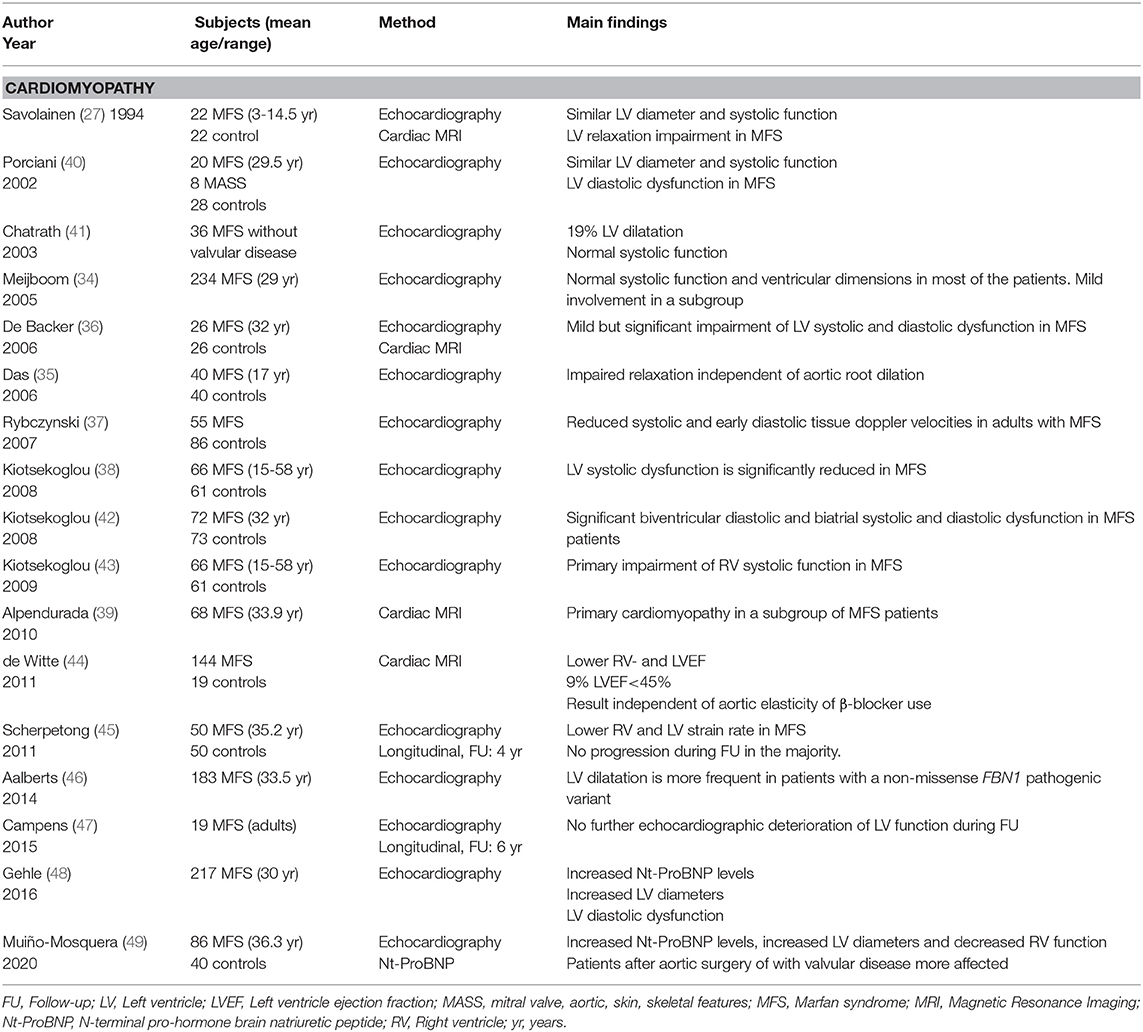
Table 2. Overview of the studies assessing cardiac function in Marfan syndrome.
Next to impaired function, arrhythmia may be considered as another manifestation of myocardial disease in MFS (Figure 2). Most of the evidence for arrhythmia in MFS comes from the study of adult cohorts, in which children were only occasionally included (Table 3). In these studies, significant ventricular ectopy (defined as >10 premature ventricular contractions per hour) was found in 20–35% of the patients (33, 49, 56). A slightly lower percentage of patients with MFS (10–25%) also present non-sustained ventricular tachycardia (NSVT) on 24h ambulatory monitoring (33, 49, 54, 56). Ventricular tachycardia (VT) and sudden cardiac death (SCD) have respectively been reported in 7–9 and 4% of the patients (33, 55, 56, 58).
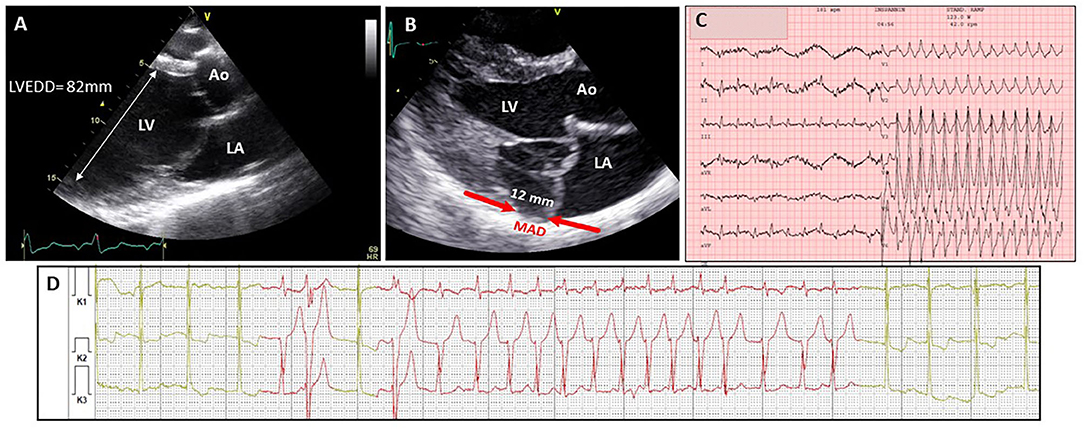
Figure 2. Different manifestations of myocardial disease in classical Marfan syndrome. (A) Echocardiographic image (2D TTE PSLAX view) in a 16 yr old male showing severe dilatation of the left ventricle. (B) Echocardiographic image (2D TTE PSLAX view) showing mitral annular disjunction. (C) ECG recordings in 28 yr old male showing ventricular tachycardia followed by presyncope during exercise test 6 m after mitral valve surgery and aortic root replacement (D) ambulatory electrocardiogram recording in a 57 yr old female with frequent episodes of non-sustained ventricular tachycardia. Ao, aorta; LA, left atrium; LV, left ventricle; LVEDD, left ventricular end-diastolic diameter; MAD, mitral annular disjunction.
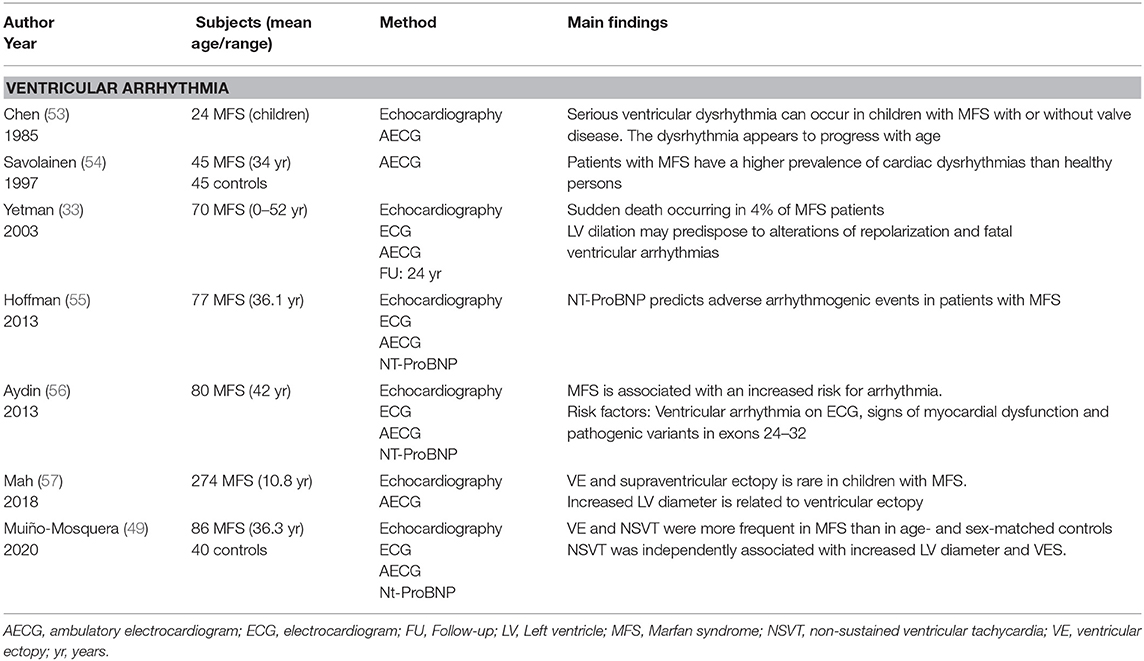
Table 3. Overview of the published papers evaluating ventricular arrhythmia in MFS.
One of the first studies of arrhythmia in children with MFS took place in the early 80s (53). In this study, eight of the 24 children with MFS (33.3%) presented with ventricular arrhythmia, three of whom showed ventricular tachycardia. Ventricular arrhythmia was associated with mitral valve prolapse and prolonged repolarization time. Another interesting early observation comes from the first (and only) randomized trial assessing the effect of propranolol on aortic root dilatation in patients with Marfan syndrome (59). Two deaths were observed in the control group of this trial, one 14 year old boy and one 18 year old women, both of which had mitral-valve prolapse and a history of paroxysmal tachyarrhythmia. Aortic dissection was excluded in both postmortem. While this study lacks power to draw meaningful conclusions from this observation, the history of arrhythmia is remarkable and one may even hypothesize that propranolol had a protective effect in the treatment group. A more recent study, an ancillary analysis of the Pediatric Heart Network (PHN) Marfan trial (57) also studied arrhythmia in children with MFS. The primary aim of the PHN Marfan trial was to compare aortic outcome in 608 children with MFS, randomly assigned to treatment with atenolol or losartan. As part of this study, a subgroup of patients (n = 274) underwent 24h ambulatory monitoring. Ventricular ectopy was present in 7% of these children, but (NS)VT was not observed. The prevalence of ventricular arrhythmia in these two studies is clearly different but probably reflects clinical variability among the cohorts. On the contrary, these studies show that ventricular arrhythmia, although uncommon, can present at an early age. Physicians taking care of children with MFS should also be vigilant for these complications.
The mechanisms underlying severe ventricular arrhythmia in MFS are not clear yet. Judging from the numbers above, it seems that age might play an important factor, although this has not been clearly shown (49). So far, an enlarged LV diameter appears to be the most consistent independent factor associated with an arrhythmic event (33, 49, 55). Other factors like mitral valve prolapse, mitral valve regurgitation and previous aortic surgery have also been associated with ventricular arrhythmia in a variable amount of studies (33, 49, 56, 60). Two studies from the Hamburg Marfan center indicated that NT-proBNP level is the strongest independent predictor of arrhythmogenic events (55, 56). In our own study, NT-pro-BNP levels were also elevated in patients presenting NSVT, although not significantly (49).
Lately, mitral annular disjunction (MAD), defined as the separation between the posterior mitral valve leaflet hinge point and the left ventricular myocardium, has gained interest in patients with MFS. A recent study shows that 46% of patients have MAD and that presence of MAD is associated with a worse aortic and mitral outcome (61). In non-MFS subjects, MAD has also received particular attention as a potential marker or substrate of ventricular arrhythmia and SCD (62). In our recent study in 142 patients with MFS, MAD was present in 36% of the cohort and was associated with the presence of VT and SCD (manuscript in press, JAMA Cardiology). Ventricular ectopy in patients with mitral valve prolapse and MAD is presumed to be partially due to regional stretch leading to fibrosis of the papillary muscles (63). Whether the same underlying mechanism is present in MFS is not clear yet and deserves further study.
Besides an abnormal substrate, triggering factors might also play a role in developing arrhythmia in MFS. Subtle ECG changes have been identified in patients with MFS independent of aortic root diameter, mitral and/or tricuspid valve prolapse or chamber dimension and function. Patients with MFS display slightly prolonged PQ- and QTc-intervals compared to healthy controls (49, 54).
Atrial arrhythmia in MFS has been given less attention. Atrial fibrillation seems to be more common in MFS than in the general population and seems to occur at a younger age (49, 64). Other types of atrial arrhythmias have been described in MFS, mainly re-entry tachycardia, but it does not seem more frequent in MFS than in the general population (57).
The presence of fibrillin-1 in the myocardium has clearly been evidenced in wild-type mice. Histology shows more abundant amounts in the atria compared to the ventricles and a distinct spatial arrangement in the ventricular myocardium with more fibrillin-1 in the inner trabeculated part when compared to the outer trabeculated part (65). The inner myocardium is more prone to shearing forces during ventricular contraction, and connective tissue aligning these lamellae play a role in providing mechanical coupling and preventing overextension (66). Based on the known elastic properties of fibrillin-1 and its observed spatial arrangement at the level of the inner myocardium, it is conceivable that fibrillin-1 provides the required elasticity to the myocardial tissue allowing shearing of the muscle lamellae. This hypothesis is supported by the limited presence of elastic fibers in the myocardium, making fibrillin-1 fibers the most important myocardial ECM component with elastic properties (67). In addition, the role of fibrillin-1 in providing elasticity to the myocardium is also observed in the fbn1mgR/mgR Marfan mouse model. A decrease in passive filling properties of the left ventricle in this model suggests an impaired elastic recoil of the left ventricle (68). It is assumed that the underlying abnormality in the FBN1 gene in MFS results in an impaired signaling function of fibrillin microfibrils in the ECM and that mechanical factors such as volume- or pressure overload are not correctly compensated which in turn leads to myocardial dysfunction. An example of pressure overload is provided by Rouf and colleagues who observed CMP after partial ligation of the aortic arch in fbn1C1039G/+ mice (69). Volume overload caused by valvular regurgitation (both mitral and aortic valve) results in dilated CMP in the same mouse model (70).
These observations support the concept of mechanobiology as a possible cause for CMP (71). As already mentioned, the model of abnormal mechanobiology has also been introduced in recent years to explain aortic pathology in MFS (72).
In addition to the evidence for functional impairment of the myocardium in mouse models for MFS, some interesting morphological alterations are also worth mentioning. In the fbn1mgR/mgR mice, an age-dependent decrease in myocardial compaction was noted on routine staining compared to WT mice sections (65). As mentioned above, fibrillin-1 fibers align the periphery of inner myocardial muscle lamellae in WT tissue. It appears that fibrillin-1 functions as a glue in the trabeculated myocardium, strengthening intercellular connections through cell-ECM-cell interactions by forming molecular bridges between the pericellular and interstitial ECM. Abnormal fibrillin-1 fibers may lead to loosened connections or non-compaction. Interestingly, left ventricular non-compaction has also been linked to pathogenic FBN1 variants in humans (9, 73). Next to the loss of myocardial compaction, macroscopic inspection of the right ventricle free wall in the fbn1mgR/mgR mouse model revealed the presence of (multiple) pseudoaneurysms. In WT mice, fibrillin-1 fibers cross the entire right ventricle free wall from lumen to pericardium. In the setting of MFS, reduced amounts of fibrillin-1 fibers may result in the formation of a gap crossing the entire right ventricle free wall. To our knowledge, there is no confirmation of such findings in human Marfan disease, but this definitely deserves further research.
Myocardial disease is an upcoming problem in MFS, especially in adults. As indicated above, a subgroup of children is also at risk. Careful monitoring of myocardial function and potential complications such as arrhythmia in patients with MFS is warranted. Yearly echocardiographic evaluation and follow-up should include assessment of biventricular systolic and diastolic function. Whether CMR will aid in risk stratification in patients with MFS still needs to be elucidated. Patients with enlarged left ventricular diameter, patients with MAD and patients with palpitations, (pre)syncope, or chest pain can benefit from an ambulatory electrocardiogram. As in general recommendations NT-pro-BNP levels are useful to monitor heart failure and may be useful for risk stratification for arrhythmia in Marfan syndrome (49, 55, 56).
There is currently no evidence that treatment of arrhythmia and heart failure in patients with MFS should differ from the guidelines for other non-MFS patients. If congestive heart failure is present as a result of valvular dysfunction, afterload-reducing agents can improve cardiovascular function. Whether losartan (or other angiotensin receptor blockers), drugs known for their beneficial effect in heart failure should be considered a first line choice in these patients is not known. In a small study with losartan in patients with MFS, we did not observe a significant effect on LV dimensions- and function (74). Indications for surgical intervention for valvular disease should follow the general guidelines (75).
End-stage heart failure is uncommon in patients with MFS but heart transplantation may be considered. Although early results with this procedure in MFS were not good (30), subsequent reports were more encouraging (29). A major concern relates to complications occurring in the distal aorta, which are mostly (but not exclusively) occurring in patients with pre-existing aortic complications (31, 76). Given the inherent fragility of the aortic tissue, the use of assist devices should be limited, although successful cases have been reported (77).
The role of β-blocker therapy for the treatment of arrhythmia or prevention of SCD in MFS is not clear yet. In one study, two of the three patients presenting SCD were under treatment with β-blocker (33). In our study, one patient presenting VT showed progressive ventricular ectopy under β-blocker treatment and episodes of sustained VT were only controlled after treatment with amiodarone (49). In the absence of specific risk factors to stratify patients at risk of VT or SCD, indications for implantable defibrillator should follow the general guidelines (78).
There is very scarce literature on myocardial disease and arrhythmia in HTADs and evidence of myocardial dysfunction in this group of diseases, mainly comes from case reports and casual description from registries.
LDS was first described in 2005 as a connective tissue disorder with important vascular involvement (79). Shortly thereafter, a case report describing a patient carrying a pathogenic variant in TGFBR1 with heart failure necessitating heart transplant was published (80). Several other case reports mentioned both heart failure and SCD in patients carrying variants in other genes: TGFB2, TGFBR2 and SMAD3 (81–84). Myocardial disease with left ventricular hypertrophy in 16% and VE in 19% of the patients carrying variants in SMAD3 was already reported in one of the first case series characterizing patients with pathogenic variants this gene (85). Only one systematic study evaluating repolarization abnormalities in patients carrying variants in TGFBR2 has been published (86). In this study, two patients presented SCD and 47% of patients presented abnormal repolarization characterized by slight prolongation of the QTc interval, abnormal ST-segment and abnormal T-U wave.
Findings based on human studies and from mouse models provide increasing evidence for the clinical relevance of CMP in genetic aortic disease, but more data are required for further confirmation and delineation.
Careful monitoring of myocardial function and potential consequences such as arrhythmia in patients with MFS and other HTAD is warranted. Further study to understand the underlying pathophysiology of myocardial disease is necessary to identify better treatment targets and improve patient's outcome.
JDB and LM-M wrote and reviewed this paper. Both authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SM declared a past co-authorship with one of the authors JDB.
1. Renard M, Francis C, Ghosh R, Scott AF, Witmer PD, Adès LC, et al. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J Am Coll Cardiol. (2018) 72:605–15. doi: 10.1016/j.jacc.2018.04.089
2. Marfan A. Un cas de déformation congénitale des quatre membres. Bull Mem Soc Med Hop Paris. (1896) 13:220–7.
3. McKusICK VA. The cardiovascular aspects of Marfan's syndrome: a heritable disorder of connective tissue. Circulation. (1955) 11:321–42. doi: 10.1161/01.CIR.11.3.321
4. Hollister DW, Godfrey M, Sakai LY, Pyeritz RE. Immunohistologic abnormalities of the microfibrillar-fiber system in the marfan syndrome. N Engl J Med. (1990) 323:152–159. doi: 10.1056/NEJM199007193230303
5. Dietz HC, Cutting CR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. (1991) 352:337–9. doi: 10.1038/352337a0
6. Sakai LY, Keene DR, Renard M, De Backer J. FBN1: the disease-causing gene for Marfan syndrome and other genetic disorders. Gene. (2016) 591:279–91. doi: 10.1016/j.gene.2016.07.033
7. Bouzeghrane F, Reinhardt DP, Reudelhuber TL, Thibault G. Enhanced expression of fibrillin-1, a constituent of the myocardial extracellular matrix in fibrosis. Am J Physiol-Heart Circ Physiol. (2005) 289:H982–91. doi: 10.1152/ajpheart.00151.2005
8. Zhao J, Lv T, Quan J, Zhao W, Song J, Li Z, et al. Identification of target genes in cardiomyopathy with fibrosis and cardiac remodeling. J Biomed Sci. (2018) 25:63. doi: 10.1186/s12929-018-0459-8
9. Parent JJ, Towbin JA, Jefferies JL. Fibrillin-1 gene mutations in left ventricular non-compaction cardiomyopathy. Pediatr Cardiol. (2016) 37:1123–6. doi: 10.1007/s00246-016-1404-9
10. Schelbert EB, Butler J, Diez J. Why clinicians should care about the cardiac interstitium. Spec Issue Imag Interstitium. (2019) 12:2305–18. doi: 10.1016/j.jcmg.2019.04.025
11. Frangogiannis NG. The extracellular matrix in ischemic and nonischemic heart failure. Circ Res. (2019) 125:117–46. doi: 10.1161/CIRCRESAHA.119.311148
12. Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. (2010) 47:476–85. doi: 10.1136/jmg.2009.072785
13. Judge DP, Dietz HC. Marfan's syndrome. Lancet. (2005) 366:1965–76. doi: 10.1016/S0140-6736(05)67789-6
14. Pyeritz RE. Marfan syndrome: 30 years of research equals 30 years of additional life expectancy. Heart. (2008) 95:173–175. doi: 10.1136/hrt.2008.160515
15. Silverman DI, Burton KJ, Gray J, Bosner MS, Kouchoukos NT, Roman MJ, et al. Life expectancy in the Marfan syndrome. Am J Cardiol. (1995) 75:157–60. doi: 10.1016/S0002-9149(00)80066-1
16. Groth KA, Stochholm K, Hove H, Andersen NH, Gravholt CH. Causes of mortality in the marfan syndrome(from a nationwide register study). Am J Cardiol. (2018) 122:1231–5. doi: 10.1016/j.amjcard.2018.06.034
17. Jondeau G, Detaint D, Tubach F, Arnoult F, Milleron O, Raoux F, et al. Aortic event rate in the marfan population. Circulation. (2012) 125:226–32. doi: 10.1161/CIRCULATIONAHA.111.054676
18. Roman MJ, Pugh NL, Hendershot TP, Devereux RB, Dietz H, Holmes K, et al. Aortic complications associated with pregnancy in marfan syndrome: the NHLBI national registry of genetically triggered thoracic aortic aneurysms and cardiovascular conditions (GenTAC). J Am Heart Assoc. (2016) 5:e004052. doi: 10.1161/JAHA.116.004052
19. Solé-Ribalta A, Rodríguez-Fanjul X, Carretero-Bellon JM, Pascual-Sala C, Martorell-Sampol L, Bobillo-Pérez S, et al. Neonatal marfan syndrome: a rare, severe, and life-threatening genetic disease. J Pediatr. (2019) 211:221–221.e2. doi: 10.1016/j.jpeds.2019.03.033
20. Stheneur C, Faivre L, Collod-Béroud G, Gautier E, Binquet C, Bonithon-Kopp C, et al. Prognosis Factors in Probands With an FBN1 Mutation Diagnosed Before the Age of 1 Year. Pediatr Res. (2011) 69:265–270. doi: 10.1203/PDR.0b013e3182097219
21. Faivre L, Masurel-Paulet A, Collod-Béroud G, Callewaert BL, Child AH, Stheneur C, et al. Clinical and molecular study of 320 children with marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics. (2009) 123:391. doi: 10.1542/peds.2008-0703
22. Peng Q, Deng Y, Yang Y, Liu H. A novel fibrillin-1 gene missense mutation associated with neonatal Marfan syndrome: a case report and review of the mutation spectrum. BMC Pediatr. (2016) 16:60. doi: 10.1186/s12887-016-0598-6
23. Ardhanari M, Barbouth D, Swaminathan S. Early-onset marfan syndrome: a case series. J Pediatr Genet. (2019) 8:86–90. doi: 10.1055/s-0038-1675338
24. Maeda J, Kosaki K, Shiono J, Kouno K, Aeba R, Yamagishi H. Variable severity of cardiovascular phenotypes in patients with an early-onset form of Marfan syndrome harboring FBN1 mutations in exons 24–32. Heart Vessels. (2016) 31:1717–23. doi: 10.1007/s00380-016-0793-2
25. Hennekam RCM. Severe infantile Marfan syndrome versus neonatal Marfan syndrome. Am J Med Genet A. (2005) 139A:1–8. doi: 10.1002/ajmg.a.30979
26. Morse RP, Rockenmacher S, Pyeritz RE, Sanders SP, Bieber FR, Lin A, et al. Diagnosis and management of infantile Marfan syndrome. Pediatrics. (1990) 86:888.
27. Savolainen A, Nisula L, Keto P, Hekali P, Viitasalo M, Kaitila L, et al. Left ventricular function in children with the Marfan syndrome. Eur Heart J. (1994) 15:625–30. doi: 10.1093/oxfordjournals.eurheartj.a060558
28. Diller GPl, Kempny A, Alonso-Gonzalez R, Swan L, Uebing A, Li W, et al. Survival prospects and circumstances of death in contemporary adult congenital heart disease patients under follow-up at a large tertiary centre. Circulation. (2015) 132:2118–25. doi: 10.1161/CIRCULATIONAHA.115.017202
29. Knosalla C, Weng Y, Hammerschmidt R, Pasic M, Schmitt-Knosalla I, Grauhan O, et al. Orthotopic heart transplantation in patients with marfan syndrome. Ann Thorac Surg. (2007) 83:1691–5. doi: 10.1016/j.athoracsur.2007.01.018
30. Kesler K, Hanosh J, O'Donnell J, Faust S, Turrentine M, Mahomed Y, et al. Heart transplantation in patients with Marfan's syndrome: a survey of attitudes and results. J Heart Lung Transplant. (1994) 13:899–904.
31. Audenaert T, De Pauw M, François K, De Backer J. Type B aortic dissection triggered by heart transplantation in a patient with Marfan syndrome. BMJ Case Rep. (2015) 2015:bcr2015211138. doi: 10.1136/bcr-2015-211138
32. Hetzer R, Siegel G, Delmo Walter EM. Cardiomyopathy in Marfan syndrome†. Eur J Cardiothorac Surg. (2016) 49:561–8. doi: 10.1093/ejcts/ezv073
33. Yetman AT, Bornemeier RA, McCrindle BW. Long-term outcome in patients with Marfan syndrome: is aortic dissection the only cause of sudden death? J Am Coll Cardiol. (2003) 41:329–32. doi: 10.1016/S0735-1097(02)02699-2
34. Meijboom LJ, Timmermans J, van Tintelen JP, Nollen GJ, De Backer J, van den Berg MP, et al. Evaluation of left ventricular dimensions and function in Marfan's syndrome without significant valvular regurgitation. Am J Cardiol. (2005) 95:795–7. doi: 10.1016/j.amjcard.2004.11.042
35. Das BB, Taylor AL, Yetman AT. Left ventricular diastolic dysfunction in childre and young adults with marfan syndrome. Pediatr Cardiol. (2006) 27:26–8. doi: 10.1007/s00246-005-1139-5
36. De Backer JF, Devos D, Segers P, Matthys D, François K, Gillebert TC, et al. Primary impairment of left ventricular function in Marfan syndrome. Int J Cardiol. (2006) 112:353–8. doi: 10.1016/j.ijcard.2005.10.010
37. Rybczynski M, Koschyk DH, Aydin MA, Robinson PN, Brinken T, Franzen O, et al. Tissue Doppler imaging identifies myocardial dysfunction in adults with Marfan syndrome. Clin Cardiol. (2007) 30:19–24. doi: 10.1002/clc.3
38. Kiotsekoglou A, Bajpai A, Bijnens BH, Kapetanakis V, Athanassopoulos G, Moggridge JC, et al. Early impairment of left ventricular long-axis systolic function demonstrated by reduced atrioventricular plane displacement in patients with Marfan syndrome. Eur J Echocardiogr. (2008) 9:605–13. doi: 10.1093/ejechocard/jen003
39. Alpendurada F, Wong J, Kiotsekoglou A, Banya W, Child A, et al. Evidence for Marfan cardiomyopathy. Eur J Heart Fail. (2010) 12:1085–91. doi: 10.1093/eurjhf/hfq127
40. Porciani MC, Giurlani L, Chelucci A, Pepe G, Giusti BH, Brunelli T, et al. Diastolic subclinical primary alterations in Marfan syndrome and Marfan-related disorders. Clin Cardiol. (2002) 25:416–20. doi: 10.1002/clc.4960250905
41. Chatrath R, Beauchesne LM, Connolly HM, Michels VV, Driscoll DJ. Left ventricular function in the Marfan syndrome without significant valvular regurgitation. Am J Cardiol. (2003) 91:914–6. doi: 10.1016/S0002-9149(03)00039-0
42. Kiotsekoglou A, Sutherland GR, Moggridge JC, Kapetanakis V, Bajpai A, Bunce N, et al. Impaired right ventricular systolic function demonstrated by reduced atrioventricular plane displacement in adults with Marfan syndrome. Eur J Echocardiogr. (2008) 10:295–302. doi: 10.1093/ejechocard/jen239
43. Kiotsekoglou A, Moggridge JC, Bijnens BH, Kapetanakis V, Alpendurada F, Mullen MJ, et al. Biventricular and atrial diastolic function assessment using conventional echocardiography and tissue-Doppler imaging in adults with Marfan syndrome. Eur J Echocardiogr. (2009) 10:947–55. doi: 10.1093/ejechocard/jep110
44. de Witte P, Aalberts JJJ, Radonic T, Timmermans J, Scholte AJ, Zwinderman AH, et al. Intrinsic biventricular dysfunction in Marfan syndrome. Heart. (2011) 97:2063. doi: 10.1136/heartjnl-2011-300169
45. Scherptong RWC, Vliegen HW, van der Wall EE, Hilhorst-Hofstee Y, Bax JJ, Scholte AJ, et al. Biventricular performance in patients with marfan syndrome without significant valvular disease: comparison to normal subjects and longitudinal follow-up. J Am Soc Echocardiogr. (2011) 24:1392–9.e1. doi: 10.1016/j.echo.2011.09.004
46. Aalberts JJJ, van Tintelen JP, Meijboom LJ, Polko A, Jongbloed JDH, van der Wal H, et al. Relation between genotype and left-ventricular dilatation in patients with Marfan syndrome. Gene. (2014) 534:40–3. doi: 10.1016/j.gene.2013.10.033
47. Campens L, Renard M, Trachet B, Segers P, Muiño-Mosquera L, De Sutter J, et al. Intrinsic cardiomyopathy in Marfan syndrome: results from in-vivo and ex-vivo studies of the Fbn1C1039G/+ model and longitudinal findings in humans. Pediatr Res. (2015) 78:256–63. doi: 10.1038/pr.2015.110
48. Gehle P, Robinson PN, Heinzel F, Edelmann F, Yigitbasi M, Berger F, et al. NT-proBNP and diastolic left ventricular function in patients with Marfan syndrome. Int J Cardiol Heart Vasc. (2016) 12:15–20. doi: 10.1016/j.ijcha.2016.05.003
49. Muiño-Mosquera L, De Wilde H, Devos D, Babin D, Jordaens L, Demolder A, et al. Myocardial disease and ventricular arrhythmia in Marfan syndrome: a prospective study. Orphanet J Rare Dis. (2020) 15:300. doi: 10.1186/s13023-020-01581-8
50. Mewton N, Liu CY, Croisille P, Bluemke D, Lima JAC. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol. (2011) 57:891–903. doi: 10.1016/j.jacc.2010.11.013
51. Karur GR, Pagano JJ, Bradley T, Lam CZ, Seed M, Yoo S-J, et al. Diffuse myocardial fibrosis in children and adolescents with Marfan syndrome and loeys-dietz syndrome. J Am Coll Cardiol. (2018) 72:2279–81. doi: 10.1016/j.jacc.2018.07.095
52. Arnaud P, Milleron O, Hanna N, Ropers J, Ould Ouali N, Affoune A, et al. Clinical relevance of genotype–phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet Med. (2021) doi: 10.1038/s41436-021-01132-x. [Epub ahead of print].
53. Chen S, Fagan LF, Nouri S, Donahoe JL. Ventricular dysrhythmias in children with Marfan's syndrome. JAMA Pediatr. (1985) 139:273–6. doi: 10.1001/archpedi.1985.02140050067024
54. Savolainen A, Kupari M, Toivonen L, Kaitila I, Viitasalo M. Abnormal ambulatory electrocardiographic findings in patients with the Marfan syndrome. J Intern Med. (1997) 241:225–30. doi: 10.1046/j.1365-2796.1997.115125000.x
55. Hoffmann BA, Rybczynski M, Rostock T, Servatius H, Drewitz I, Steven D, et al. Prospective risk stratification of sudden cardiac death in Marfan's syndrome. Int J Cardiol. (2013) 167:2539–45. doi: 10.1016/j.ijcard.2012.06.036
56. Aydin A, Adsay BA, Sheikhzadeh S, Keyser B, Rybczynski M, Sondermann C, et al. Observational cohort study of ventricular arrhythmia in adults with Marfan syndrome caused byFBN1 mutations. PLoS ONE. (2013) 8:e81281. doi: 10.1371/journal.pone.0081281
57. Mah DY, Sleeper LA, Crosson JE, Czosek RJ, Love BA, McCrindle BW, et al. Frequency of ventricular arrhythmias and other rhythm abnormalities in children and young adults with the Marfan syndrome. Am J Cardiol. (2018) 122:1429–36. doi: 10.1016/j.amjcard.2018.07.006
58. Schaeffer BN, Rybczynski M, Sheikhzadeh S, Akbulak RÖ, Moser J, Jularic M, et al. Heart rate turbulence and deceleration capacity for risk prediction of serious arrhythmic events in Marfan syndrome. Clin Res Cardiol. (2015) 104:1054–63. doi: 10.1007/s00392-015-0873-9
59. Shores J, Berger K, Murphy EA, Pyeritz RE. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan's syndrome. NEJM. (1994) 330:1335–41. doi: 10.1056/NEJM199405123301902
60. Gott VL, Greene PS, Alejo DE, Cameron DE, Naftel DC, Miller DC, et al. Replacement of the aortic root in patients with Marfan's syndrome. N Engl J Med. (1999) 340:1307–13. doi: 10.1056/NEJM199904293401702
61. Motwani M, Venetucci L. Mitral annular disjunction arrhythmia syndrome in Marfan syndrome. Eur Heart J Case Rep. (2020) 4:1–2. doi: 10.1093/ehjcr/ytaa306
62. Bennett S, Thamman R, Griffiths T, Oxley C, Khan JN, Phan T, et al. Mitral annular disjunction: a systematic review of the literature. Echocardiography. (2019) 36:1549–58. doi: 10.1111/echo.14437
63. Basso C, Perazzolo Marra M, Rizzo S, De Lazzari M, Giorgi B, Cipriani A, et al. Arrhythmic mitral valve prolapse and sudden cardiac death. Circulation. (2015) 132:556–66. doi: 10.1161/CIRCULATIONAHA.115.016291
64. Bunch TJ, Connolly HM, Asirvatham SJ, Brady PA, Gersh BJ, Munger TM, et al. Catheter ablation for atrial fibrillation in patients with the Marfan and Marfan-like syndromes. J Interv Card Electrophysiol. (2007) 20:15–20. doi: 10.1007/s10840-007-9162-5
65. Steijns F, van Hengel J, Sips P, De Backer J, Renard M. A heart for fibrillin: spatial arrangement in adult wild-type murine myocardial tissue. Histochem Cell Biol. (2018) 150:271–80. doi: 10.1007/s00418-018-1686-5
66. Pope AJ, Sands GB, Smaill BH, LeGrice IJ. Three-dimensional transmural organization of perimysial collagen in the heart. Am J Physiol Heart Circ Physiol. (2008) 295:H1243–52. doi: 10.1152/ajpheart.00484.2008
67. Takayama Y, Costa KD, Covell JW. Contribution of laminar myofiber architecture to load-dependent changes in mechanics of LV myocardium. Am J Physiol-Heart Circ Physiol. (2002) 282:H1510–20. doi: 10.1152/ajpheart.00261.2001
68. Steijns F, Renard M, Vanhomwegen M, Vermassen P, Desloovere J, Raedt R, et al. Spontaneous right ventricular pseudoaneurysms and increased arrhythmogenicity in a mouse model of Marfan syndrome. Int J Mol Sci. (2020) 21:7024. doi: 10.3390/ijms21197024
69. Rouf R, MacFarlane EG, Takimoto E, Chaudhary R, Nagpal V, Rainer PP, et al. Nonmyocyte ERK1/2 signaling contributes to load-induced cardiomyopathy in Marfan mice. JCI Insight. (2017) 2:e91588. doi: 10.1172/jci.insight.91588
70. Tae H-J, Petrashevskaya N, Marshall S, Krawczyk M, Talan M. Cardiac remodeling in the mouse model of Marfan syndrome develops into two distinctive phenotypes. Am J Physiol-Heart Circ Physiol. (2016) 310:H290–9. doi: 10.1152/ajpheart.00354.2015
71. Cook JR, Carta L, Bénard L, Chemaly ER, Chiu E, Rao SK, et al. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J Clin Invest. (2014) 124:1329–39. doi: 10.1172/JCI71059
72. Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res. (2015) 116:1448–61. doi: 10.1161/CIRCRESAHA.114.304936
73. Kwiatkowski D, Hagenbuch S, Meyer R. A teenager with Marfan syndrome and left ventricular noncompaction. Pediatr Cardiol. (2010) 31:132–5. doi: 10.1007/s00246-009-9552-9
74. Muiño-Mosquera L, De Nobele S, Devos D, Campens L, De Paepe A, De Backer J. Efficacy of losartan as add-on therapy to prevent aortic growth and ventricular dysfunction in patients with Marfan syndrome: a randomized, double-blind clinical trial. Acta Cardiol. (2017) 72:616–24. doi: 10.1080/00015385.2017.1314134
75. Baumgartner H, Falk V, Bax JJ, De Bonis M, Hamm C, Holm PJ, et al. 2017 ESC/EACTS Guidelines for the management of valvular heart disease. Eur Heart J. (2017) 38:2739–91. doi: 10.1093/eurheartj/ehx391
76. Rajagopal K, Rogers JG, Lodge AJ, Gaca JG, McCann RL, Milano CA, et al. Two-stage total cardioaortic replacement for end-stage heart and aortic disease in Marfan syndrome: case report and review of the literature. J Heart Lung Transplant. (2009) 28:958–63. doi: 10.1016/j.healun.2009.05.012
77. Pyeritz RE. The marfan syndrome. Annu Rev Med. (2000) 51:481–510. doi: 10.1146/annurev.med.51.1.481
78. Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. (2015) 36:2793–867. doi: 10.1093/eurheartj/ehv316
79. Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. (2005) 37:275–81. doi: 10.1038/ng1511
80. Eckman PM, Hsich E, Rodriguez ER, Gonzalez-Stawinski GV, Moran R, Taylor DO. Impaired systolic function in loeys-dietz syndrome. Circ Heart Fail. (2009) 2:707–8. doi: 10.1161/CIRCHEARTFAILURE.109.888636
81. Neubauer J, Haas C, Bartsch C, Medeiros-Domingo A, Berger W. Post-mortem whole-exome sequencing (WES) with a focus on cardiac disease-associated genes in five young sudden unexplained death (SUD) cases. Int J Legal Med. (2016) 130:1011–21. doi: 10.1007/s00414-016-1317-4
82. De Backer J, Braverman AC. Heart failure and sudden cardiac death in heritable thoracic aortic disease caused by pathogenic variants in the SMAD3 gene. Mol. Genet Genomic Med. (2018) 6:648–52. doi: 10.1002/mgg3.396
83. Feroe AG, Fiedler AG. Successful heart transplantation in a patient with loeys-dietz syndrome. Ann Thorac Surg. (2019) 107:e379–80. doi: 10.1016/j.athoracsur.2018.10.058
84. Krohg-Sørensen K, Lingaas PS, Lundblad R, Seem E, Paus B, Geiran OR. Cardiovascular surgery in loeys–dietz syndrome types 1–4. Eur J Cardiothorac Surg. (2017) 52:1125–31. doi: 10.1093/ejcts/ezx147
85. van der Linde D, Bekkers JA, Mattace-Raso FUS, van de Laar IMBH, Moelker A, van den Bosch AE, et al. Progression rate and early surgical experience in the new aggressive aneurysms-osteoarthritis syndrome. Ann Thorac Surg. (2013) 95:563–9. doi: 10.1016/j.athoracsur.2012.07.009
Keywords: Marfan syndrome, HTAD, cardiomyopathy, arrhythmia, myocardial disease, FBN1 gene
Citation: Muiño-Mosquera L and De Backer J (2021) Cardiomyopathy in Genetic Aortic Diseases. Front. Pediatr. 9:682390. doi: 10.3389/fped.2021.682390
Received: 18 March 2021; Accepted: 22 June 2021;
Published: 15 July 2021.
Edited by:
Elena Cervi, Great Ormond Street Hospital for Children NHS Foundation Trust, United KingdomReviewed by:
Shaine Morris, Baylor College of Medicine, United StatesCopyright © 2021 Muiño-Mosquera and De Backer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie De Backer, SnVsaWUuZGViYWNrZXJAdWdlbnQuYmU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.