 Kaushal Sharma†
Kaushal Sharma† Pandiarajan Vignesh†
Pandiarajan Vignesh† Priyanka Srivastava
Priyanka Srivastava Jyoti Sharma
Jyoti Sharma Himanshi Chaudhary
Himanshi Chaudhary Sanjib MondalAnupriya KaurHarvinder Kaur
Sanjib MondalAnupriya KaurHarvinder Kaur Surjit Singh*
Surjit Singh*- Department of Pediatrics, Advanced Pediatrics Centre, Post Graduate Institute of Medical Education and Research, Chandigarh, India
Kawasaki disease (KD) is a common febrile multisystemic inflammatory illness in children that preferentially affects coronary arteries. Children with KD who develop coronary artery aneurysms have a life-long risk of premature coronary artery disease. Hypothesis of inherent predisposition to KD is supported by epidemiological evidence that suggests increased risk of development of disease in certain ethnicities and in children with a previous history of KD in siblings or parents. However, occurrence of cases in clusters, seasonal variation, and very low risk of recurrence suggests an acquired trigger (such as infections) for the development of illness. Epigenetic mechanisms that modulate gene expression can plausibly explain the link between genetic and acquired predisposing factors in KD. Analysis of epigenetic factors can also be used to derive biomarkers for diagnosis and prognostication in KD. Moreover, epigenetic mechanisms can also help in pharmacogenomics with the development of targeted therapies. In this review, we analysed the available literature on epigenetic factors such as methylation, micro-RNAs, and long non-coding RNAs in KD and discuss how these mechanisms can help us better understand the disease pathogenesis and advance the development of new biomarkers in KD.
Introduction
Kawasaki disease (KD) is one of the common vasculitides that usually affects children below the age of 5. This condition is eponymously named after Dr. Tomisaku Kawasaki who described the disorder in 1967. KD is characterised by oral mucosal redness, conjunctival injection, rash, and swelling in hands and feet (1). Diagnosis of KD is based on a set of clinical features as enumerated by the American Heart Association (AHA). Main complication of KD is development of coronary arteries abnormalities (CAAs) that are seen in 15–20% of children with KD who do not receive treatment with intravenous immunoglobulin (IVIg) (2). Risk of development of CAAs reduces to 3–5% when therapy is initiated on time. However, around 15–25% of patients with KD do not respond to IVIg treatment and are said to have IVIg–resistant KD. These patients are at an increased risk of development of CAAs (3).
Incidence of KD in North East Asian countries is higher compared to countries in Europe and North America. Japan records the highest incidence of KD in world (308.0 per 100,000) (4), followed by South Korea (199.7 per 100,000 <5) (5) and Taiwan (82.8 per 100,000) (6). Several epidemiological and clinical reports have suggested that KD can be triggered by infectious agents including Mycoplasma, Chlamydia, Streptococci or Staphylococci (7–10) and viruses like adenovirus, Epstein-Barr virus, and parvovirus (11–13). Increased incidence of KD in certain population groups and increased risk of development of KD phenotypes in siblings of children who are affected with KD suggests involvement of genetic factors in susceptibility to KD.
Epigenetics is widely explored in most of complex heterogenic diseases like KD where contribution of both genetic and environmental factors is equally shared (14–17). Genetic predisposition of complex disease phenotypes has been associated with substantial contribution of dysregulated epigenetic factors. Moreover, it has also been established that interplay of genetic, epigenetic, and environmental factors in several rheumatological illnesses play a crucial role in disease severity and treatment responses. Epigenetic factors can also potentially explain the phenotypic heterogeneity and differential treatment response.
Complex genetic architecture of KD suggests contribution of several innate and adaptive immune responses, extracellular matrix (ECM) components and proteins regulating its remodelling, and angiogenic proteins (14–17). Apart from widely explored genetic network in KD, a well-defined biomarker for diagnosis and prognostication has not yet been established. Exploration of epigenetic risk factors in KD, hitherto unexplored until date, could pave way for identification of potential biomarkers in future and help in advancement of precision medicine.
Advancement in molecular technologies has led to identification of several post-translational regulatory factors such as miRNA and long non-coding RNAs (lncRNA). Recently, involvement of gut microbiota and associated immune modulation has also been implicated in the pathogenesis of KD. In this review, we aim to focus on epigenetic studies that have been carried out in KD, discuss their potential contribution to our understanding of the disease pathogenesis, and their role in the development of biomarkers.
Search Strategy
We carried out literature search through PubMed/ Medline and Embase databases using the keywords in different combinations: Epigenetics, genetics, methylation, microRNAs, long non-coding RNA, Kawasaki disease. Original articles and reviews related to genetic and epigenetic studies in KD published in English language until 20 February 2021 were retrieved and analysed.
Pathophysiology and Genetic-Architecture of KD
Pathogenesis of KD involves several immunological and non-immunological genes. Activation of endothelial cells under the influence of S100 and HMGB1 in KD can be regulated through the action of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) suggesting involvement of innate immunity in the KD (18–23). Infiltration of CD14+ monocyte/macrophages in the coronary walls has also been noted in patients with KD (23). Identification of genetic variants of ITPKC, CASP3, CD40, and ORAI in KD signifies the involvement of T-cell activation and differentiation (24). Recent report from Onouchi et al. has demonstrated impaired development and function of B-cell (rs2254546 for FAM167A-BLK; src family B lymphoid tyrosine kinase) and augmented activation of T-cells (rs4813003 CD40) in KD. These results have also been supported by the studies carried out in Taiwanese and Han Chinese population groups (25–29). Moreover, six SNPs located in variable region of heavy chain of immunoglobulin gene were also shown to have a strong association with KD in Taiwanese population (22). Association between HLA genetic variants and formation of coronary artery lesions (CALs) in KD has been established in several ethnicities including Chinese (HLA-A, HLAB, HLADR), Japanese (e.g., Bw54, Bw22J, Bw22J2), and Caucasians (Bw51, Bw44, DRB3, DRB1*301, DRB1, DQA1, DQB1, DPB1) (30–36). All these studies suggest pathological significance of both adaptive and innate immunity in KD.
Wang et al. have studied the role of Th1/Th2 secreted cytokines in patients with KD. Levels of IL-6, IL-10, TNFα, and IFNγ were significantly elevated in acute stages of KD and all, except TNFα levels, decreased after IVIg treatment. On subgroup analysis, levels of TNFα were found to decrease in IVIg-responder group, and in patients with KD without CAAs. Higher TNFα levels were observed in IVIg resistant KD and in patients with CAAs. Results have also showed that patients with KD and CAAs exhibited significant elevation in levels of IL-4, IL-6, IL-10, and IFNγ. After IVIg therapy, IL-6 and IL-10 levels were significantly higher in IVIg-resistant group when compared with IVIg-responder group (37).
Higher proportion of Th17 (helper type 17) cells with concordant increased expression of cytokines IL-17, IL-6, IL-23, and transcription factors IL-17A/F and ROR-γt were observed in acute phase of KD. IVIg-resistant patients had higher proportion of Th17 cells compared to IVIg-responsive patients. These studies have also showed the pathological importance of regulatory T cells which were found to be decreased in children with acute KD (38). Moreover, decrease proportion of CD4 + CD25 + FOXP3 + Treg cells have been associated with IVIg resistance in KD (39, 40). In another study, significantly decreased levels of both IL-6 and IL-17A, and elevated levels of FOXP3, were reported in patients with KD after IVIg treatment. Increased levels of IL-17A and IL-10 levels were found to be associated with an IVIg resistant phenotype of KD (41). A decreased T-cell proliferative response (TCR/CD3 mediated) has also been observed in KD and this may affect the production of pro-inflammatory cytokines (24, 29, 38, 39, 42).
Extracellular matrix (ECM) proteins (including collagen and integrin proteins) have been widely studied in KD. Abnormalities of ECM remodelling have been linked with development of coronary artery lesions (CALs) in KD. MMPs play a vital role in ECM remodelling, neointimal proliferation, and initiation of angiogenesis (43–46). They also have an important role in maintaining the integrity of coronary vascular wall structure and function. MMPs also mediate macrophage invasion and tissue destruction. Differential expression of MMPs at systemic and micro-environmental levels has been documented in KD. Abolition of MMP-9 activity stimulates the inflammatory responses after exposure to Lactobacillus casei cell wall extract (LCWE) in a mice model of KD (47).
The sub-acute and chronic vasculitis in KD originates from smooth muscle cell-derived myofibroblasts under the differential expression of collagen type I, α1 (COL1A1), MMP7, and integrins α4 and αM (ITGA4, ITGAM). Study on LCWE induced murine model of KD has emphasised the augmented TGF-β signalling and proteolytic activity of elastin in wall of coronary arteries. Studies have also demonstrated the reduction of plasminogen activator inhibitor-1 (PAI-1, proteolytic inhibitor) as well as increased levels of MMP9 (matrix metalloproteinase-9) after administration of TGF-β inhibitor (48).
Interestingly, ECM components have also been shown to have immune-modulatory effects. Denatured collagen type 1 has showed the ability to adhere with scavenger receptors of innate immunity and found to modulate the secretion of MMP9 from macrophages (49, 50). Disintegrated ECM can also serve as catalyst for coronary aneurysm formation in KD with enhanced transmural infiltration of activated T-cells. Moreover, the infiltration of macrophages in the adventitious layer of coronary arteries has also been observed in patients with KD (51). Altered expression of adhesion molecules like E-selectin or VCAM-1 (vascular cell adhesion molecule-1) could also facilitate the infiltration of inflammatory cells at the site of inflammation in KD (52). Proteins involved in TGF-β pathway along with over-expression of IL-6 and IL-17 in infiltrating monocytes have been implicated in development of thickened intima of affected coronary arteries in patients with KD. Activated CD8+ (HLA-DR positive) and FOXP3+Treg cells were also observed in coronary arteries of patient group implicating the role of effector T cells and TGF-β signalling in KD (53).
Epigenetic Regulation in KD
Involvement of a wide spectrum of immunological and non-immunological factors indicates heterogenic genetic architecture that contributes to pathogenesis of KD. In addition to genetic factors, several environmental factors including geographical distribution of population and infections could also equally contribute to KD. Recently, epigenetic regulation of genes involved in the pathogenesis of KD has also been studied. Epigenetic modification can occur by several ways including covalent modification of cytosine (e.g., methylation by DNA methyltransferases), post-transcriptional modification of histone protein (e.g., acetylation, phosphorylation, methylation, citrullination, ubiquitylation, ribosylation, and sumoylation), and RNA based regulation of transcriptional machinery (54). Exploration of epigenetics especially in complex disease phenotypes like KD is warranted wherein both genetic (even genetic interactions) and environmental factors contribute to disease pathogenesis and can influence the treatment response.
Methylation and KD
Methylation is the most widely studied epigenetic factor in KD. Mechanism of methylation is regulated by a set of DNA methyltransferase (DNMTs) enzymes along with other assisting proteins. Covalent binding of a methyl group (-CH3) to CpG island of the genome (especially on promoter region) can modulate the expression of genes and subsequently influence protein expression without altering genetic sequence.
Huang et al. have shown that expression levels of DNMT1 and DNMT3A were significantly lower in patients with KD compared to normal controls. Authors concluded that transient DNA methylation occurs during acute stage of KD (55). Another study by Li et al. has analysed the changes in DNA methylation in patients with KD before and after IVIg administration. Treatment with IVIg has been shown to alter methylation mainly by hypomethylating CpG markers. Pathway enrichment analysis showed that genes regulated by altered methylation after IVIg administration are associated with inflammatory response (56). Chen et al. (57) studied difference in methylation between patients with KD and control population. Authors observed that 97% of the CpG regions that had more than 20% difference of methylation between KD patients and controls were hypomethylated (57). Most of the differentially methylated regions appear to be enriched in genes involved in inflammatory response, innate immunity, and chemokine signalling pathways. Epigenetic changes in patients of KD have been observed in genes such as FCGR2A, matrix metalloproteinases (MMPs), toll-like receptors (TLRs), NOD-like receptors, HAMP giving insight in pathophysiology of KD.
Toll-like receptors (TLRs) are involved in triggering a proinflamatory cytokine cascade in response to several antigens including bacterial lipopolysaccharide (LPS). TLRs widely recognise signature patterns presented on bacteria (lipopolysaccharide) and viruses (dsDNA/RNA, ssDNA/RNA), mediate inflammatory responses and biosynthesis of cytokines through the activation of protein kinases (e.g., IRAK1/4, TBK, and IKKi, etc.). Patients with KD were shown to have elevated mRNA expression of TLRs except for TLR3 and TLR7 in the acute stage. However, methylation patterns of CpG sites on TLR1, 2, 4, 6, 8, and 9 were found to be lower in acute stages of KD when compared to normal controls. These patterns increased significantly after IVIg treatment (58). Stimulation of TLR2 and TLR4 with LPS has been shown to modulate the expression of IL-2, IL-6, IL-10, MCP-1, TNF-α, and INF-β in mice models of KD (59, 60). This observation provides preliminary evidence that KD is triggered by a bacterial agent.
Polarisation of macrophages from M1 to M2 has been noted in acute stages of KD. Recently, transcriptomic profiling using GeneChip array in patients with acute phase of KD has showed elevated expression of TLR2 and IL2RA (M1 macrophage) in addition to markers associated with M2 macrophage (e.g., MS4A4A, MS4A6A, TLR1, TLR8, TLR5, CD36, CCR2, and ARG1). Authors have also noted hypomethylation of the promoter region of respective genes (17). IVIg treatment given to these patients has further enhanced the expression pattern of these genes by enriching the hypomethylation status. Trans-endothelial movement of leukocytes at site of inflammation is regulated by “S100A genes.” Increase in expression of these genes result in enhanced trans-endothelial migration of leukocytes. Hypomethylation and hypermethylation of S100A genes has been noted in acute and convalescent phases of KD, respectively (16). Abnormal neutrophil activation has also been associated with KD. Enhanced expression of neutrophil activation marker CD177 with abnormal methylation patterns has been observed in patients with KD. While the levels of CD177 were found to be decreased in patients with IVIg-responsive illness, the levels were increased in patients with IVIg-resistant KD (61).
NOD-like receptors (NLRs) are cytoplasmic sensors for endogenous or exogenous damaging molecules. They form part of the inflammasome structure, and downstream signalling through NLRs activates caspase (CASP) protein (especially CASP1). Activation of CASP1 has been noted in acute stages of KD, which further facilitates the synthesis of pro-inflammatory cytokines like IL-β1 and IL-18 (62–66). NLRC4 (Nucleotide-binding protein) is a key molecule to activate CASP1 protease to ultimately direct the synthesis of the IL-β1 from its precursor IL-β1 (pre-IL-β1). Experimental studies in mice have shown that caspase 1 and IL1-β are required for development of CAL in KD (67). Hypomethylation and hypermethylation of CpG (promoter region) have been observed in NLRC4 and NLRP12, respectively, in patients with KD. Subsequently, this resulted in increased expression of NLRC4 and IL-β1, and deceased expression of NLRP12, which were further found to be associated with formation of CAL in KD (15).
Fc receptors [Immuno-receptor tyrosine-based activation motif (ITAM)-associated receptor family] in the immunocytes bind to antibody or antigen-antibody complexes and help in regulation of opsonization, phagocytosis, degranulation, and cytokine biosynthesis (68, 69). Changes in expression levels or altered function of Fc receptors have been implicated in the immunopathogenesis of several rheumatological diseases including KD (70–72). Genome-wide association studies (GWAS) had identified FCGR2A gene as a susceptibility gene for KD. FCGR2A codes for the immunoglobulin IgG (gamma Fc region receptor II-a) receptor that is widely expressed in activated immune cells. Studies had shown that methylation level of this gene affects the binding of the IgG2 to the receptor (73). A recent GWAS conducted on the Han Chinese population has identified 10 SNPs that confer genetic susceptibility in KD patients. One of the SNPs identified in this study was FCGR2A (rs1801274) (74). Epigenetic regulation of FCGR2A with 15.54% hypomethylation pattern has also been demonstrated in KD patients in comparison to controls as reported by Kuo et al. (75). Significantly higher hypomethylation was also documented in patients with IVIg resistance compared to patients who responded to first dose of IVIg. Variations in gene copy numbers of FcγR2C and FcγR3B have also been shown to be associated with predisposition to KD and IVIg responsiveness (76). Chang et al. showed that methylation of FCER1A was lower in patients with KD compared to controls (77). This provides evidence for the role of IgG receptors in the pathogenesis of KD (Table 1).
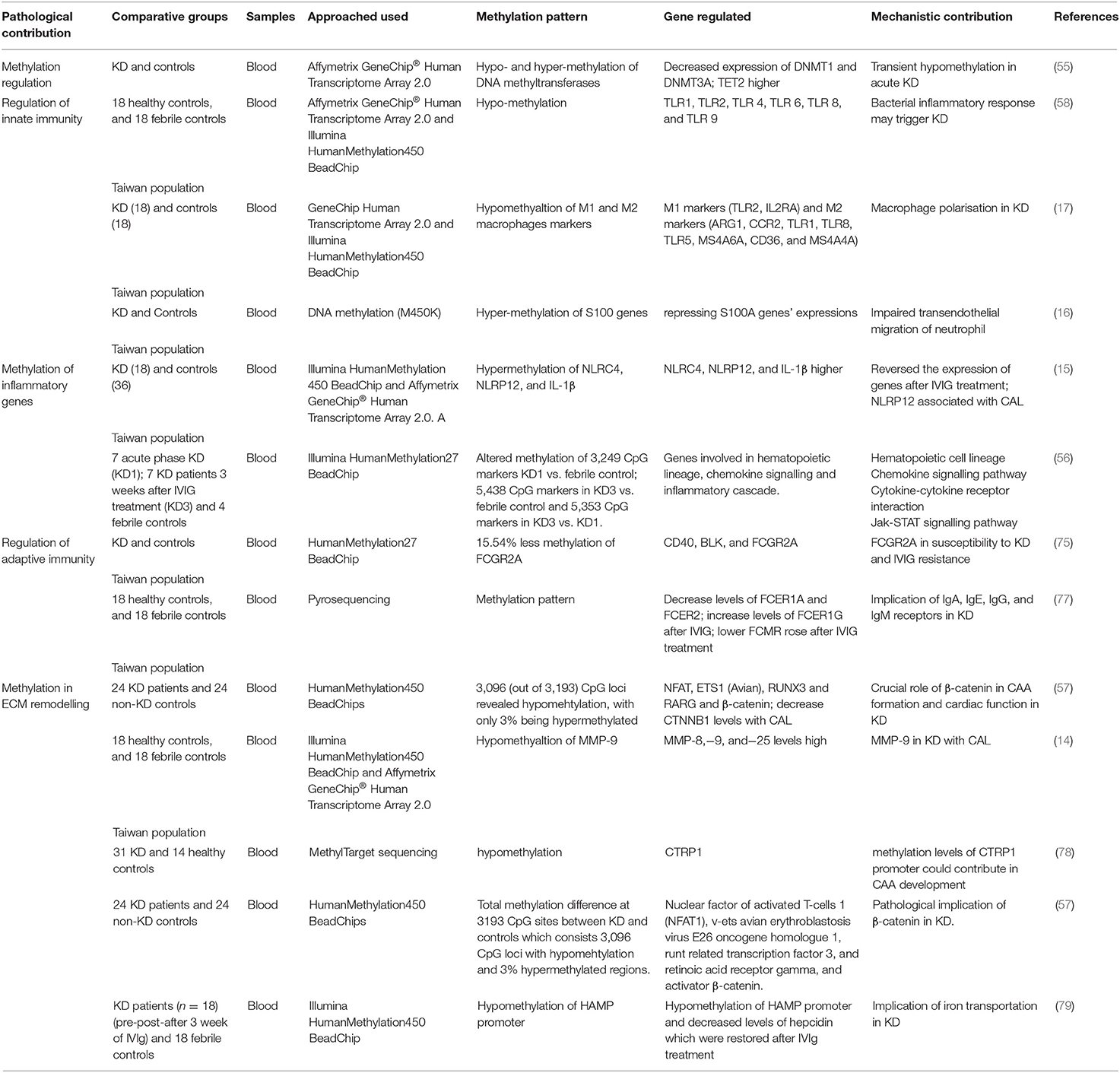
Table 1. Tabular representation of studies related to regulation of genes by methylation, downstream cellular mechanism and implication in KD.
Association of epigenetic regulation of ECM remodelling with CAA formation in KD has not been much explored. MMPs have an important role in maintaining coronary vascular wall structure and function. They also mediate macrophage invasion and tissue destruction. Kuo et al. found significant differential expression of MMPs 8, 9, and 25 in patients with KD in comparison to healthy and febrile controls (14). Methylation levels of these MMPs were significantly lower in KD patients. MMP-9 showed significant hypermethylation after IVIg treatment in patients with KD. IVIg resistant patients with CAL associated KD have exhibited higher expression of MMP9. Studies have demonstrated the association of various cytokines such as TNF-alpha and IFN-γ with MMPs suggesting that the MMPs can serve as KD biomarkers (14) (Table 1).
Similarly, collagen binding protein CTRP1 (C1q/tumour necrosis factor-related protein-1) has also demonstrated reduction in CpG methylation at multiple sites near promoter region (nucleotide sites at 34, 51, 69, 79, 176, and 206) whereas hypermethylation pattern was observed at the promoter sites 69 and 154 in CAA group (coronary artery aneurysms). Results have suggested the CpG methylation of CTRP1 as prominent marker to distinguish KD with and without CAA (74, 78). Huang et al. (79) in a cohort of patients with KD have also assessed the methylation and functional aspects of promoter region of HAMP that codes for hepcidin (regulate iron transportation). Hypomethylation of HAMP (at CpG sites cg23677000 and cg04085447) with subsequent elevation in hepcidin levels has been noted in patients with KD. Levels of hepcidin decreased after IVIg treatment in patient group (79). This study suggests that iron transportation is probably involved in ECM remodelling and CAA formation in KD.
Clusters of few hypermethylated regions have also been shown to positively correlate with KD susceptibility and CALs in KD. The biological context of these differentially methylated regions (DMRs) has been obtained by network and pathway analysis studies. One such analysis identified a network of genes in the β-catenin pathway which plays an important role in cardiac repair (80). Identified gene network consists of transcription activator β-catenin (CTNNB1- a core regulator in the network) along with associated transcription factors (NFATC1, ETS1, RUNX3, and RARG), correlated regulators CDC25B and PDCD1, and the related effectors LTA, BTLA, and CD247. mRNA levels of five genes (CTNNB1, RUNX3, ETS1, NFATC1, CD247) were considerably lower in KD patients as compared to controls, while expression levels of β-catenin (CTNNB1) were significantly lower in KD patients with CAL when compared to non-CAL patients. Decreased CTNNB1 expression in monocyte cell line has been shown to increase expression of CD40 and CD40L in human coronary artery endothelial cells (57).
Role of microRNA in Kawasaki Disease
MicroRNAs (miRNAs) are endogenous, small (18–25 nucleotides long) non-coding, single stranded RNAs which play an important role in controlling mRNA translation. Non-coding RNAs are transcribed but not translated into proteins, and act as an epigenetic mechanism to regulate gene expression. They regulate the gene expression after transcription by binding the translation section. This results in either mRNA degradation or translational inhibition (81, 82). miRNAs target the 3′ untranslated region (3′UTR) of their target mRNA molecule and control their stability and protein interactions (83). These are highly conserved, and their expression is time specific. miRNA expression is controlled by other epigenetic mechanisms and itself controls these mechanisms as an “epigenetics-miRNA regulatory circuit” that, when perturbed, can contribute to disease pathogenesis (84).
In humans, miRNAs are thought to play an important role in regulation of coding genes. miRNAs are considered to be a useful biomarker for many pathogenic states, aiding in both diagnosis and prognosis These are also involved in several cellular processes such as cell proliferation, apoptosis, migration, invasion, and stress response (85–87). Unique expression patterns of miRNAs may be used as a novel, non-invasive biomarker for disease diagnosis (88). miRNAs have been reported in tissues and also known to be released in peripheral blood. These circulating miRNAs have been found to be associated with a specific pathophysiological state (89). Therefore, peripheral serum microRNAs are widely used as diagnostic and therapeutic biomarkers for a variety of diseases. Aberrant miRNAs expression has been reported to be associated with several diseases including cancers, cardiovascular diseases, inflammatory conditions and immune regulation (90–93).
Increasing evidence suggests that miRNAs have a role in the pathogenesis of several human diseases, including KD (94). Previous reports on circulating miRNAs expression in KD are mentioned in Table 2.

Table 2. Regulation of various genetic and cellular networks involved in KD by miRNAs and lncRNAs.
Studies have shown that there were differential expressions of microRNAs in children with KD and during the acute stage of KD, miR-143, miR-199b-5p, miR-618, miR-223, and miR-145 were significantly higher (95, 97, 109, 110). Another study showed that serum miR-200c and miR-371-5p were significantly higher in KD patients as compared to healthy controls. These miRNAs may have an important role in disease pathogenesis and can be potential biomarkers for KD (97). Both exosomal and serum miRNAs e.g., miR-1246, miR-4436b-5p and miR-197-3p, and miR-671-5p have been studied for its role as biomarkers in KD (98, 107, 108). Zhang et al. studied 102 KD patients and showed that serum miR-200c and miR-371-5p were significantly elevated in sera in acute stages of KD. The levels were higher in the group with IVIG resistance who required further plasma therapy (100). They also showed that levels decreased significantly following therapy. In another study by Zhang et al., serum exosomal miR-328, miR-575, miR-134 and miR-671-5p have been shown to act as potential biomarkers for the diagnosis of KD and the prediction of outcomes of the IVIg therapy by influencing the expression of inflammatory genes (101). Wang et al. showed that hsa-let-7b-5p and hsa-miR-223-3p were slightly downregulated, while miR-200c, miR-197-3p and miR-671-5p was upregulated in KD (104).
Encapsulated miRNA-145-5p/miRNA-320a in endothelial particles has been shown to modulate inflammatory response and progression of vasculitis in patients with KD (99). Similarly, enhanced levels of miR-27a have been shown to induce inflammatory responses mediated by monocyte derived TNF-α synthesis in patients with KD. Results have also revealed negative regulation of IL-10 secretion from B cells under action of TNF-α (102). Many other miRNAs including miR-200c, miRNA-145-5p, miRNA-320a, and MiR-197-3p have also been shown to regulate the proliferation, migration and apoptosis/senescence of endothelial cells by moderating the action of various downstream molecules e.g. ZEB1 (Zinc finger E-box-binding homeobox 1, inhibit T cell specific IL2 expression), IGF1R (insulin-like growth factor 1) and BCL2 (96, 111).
As exogenous miR-223 has been shown to exert biological effects on endothelial cell (EC) functions via its target genes such as IGF1R (115). A study conducted by Chu et al assessed miRNAs profiles in patients with KD and found that miR-223 was significantly elevated in these patients (115). They also postulated that KD-induced EC injuries were related to increased miR-223 because they were inhibited by miR-223 knockdown. Increased miR-223 in ECs could work as a novel endocrine genetic signal and participate in vascular injury of KD. Li et al. have shown that 18 miRNAs were differentially expressed in plasma of patients with KD when compared with healthy controls (106). miR-125a-5p was significantly increased in plasma of KD patients and has been considered to play a role in pathogenesis of KD by regulating target gene MKK7 to induce apoptosis in vascular endothelial cells. Jia et al. have suggested a set of 4 miRNAs which could distinguish KD patients from other febrile patients as well as from healthy individuals in a single pass (106). They identified 4-miRNA set [namely, CT(miR-1246)-CT(miR-4436b-5p) and CT(miR-197-3p)-CT(miR-671-5p)] in 79 samples from two hospitals and proposed them as candidate diagnostic biomarkers for KD. Another study uncovered 7 miRNAs that were significantly upregulated (hsa-let-7b-5p, hsa-miR-223-3p, hsa-miR-4485, hsa-miR-4644, hsa-miR-4800-5p, hsamiR-6510-5p, and hsa-miR-765) and 3 that were significantly downregulated (hsa-miR-33b-3p, hsa-miR-4443, and hsa-miR-4515) in plasma of acute KD compared with healthy controls (105). MiR-93 may regulate vascular endothelial growth factor A (VEGFA) of circulating peripheral blood mononuclear cells in children with acute KD (85).
Previous studies have shown that in absence of specific miRNAs, forkhead box protein 3 (FOXP3)+ regulatory T cells (Treg) develop but fail to maintain immune homeostasis. Ni et al. (103) demonstrated that FoxP3 mRNA levels were primarily affected by the miR-155/SOCS1 (suppressors of cytokine signalling) and miR-31 signalling pathways. These results suggest that decrease in FoxP3+ Treg might be associated with decreased expression of miR-155, leading to aberrant SOCS1/STAT-5 signalling and overexpression of miR-31 in patients with acute KD. IVIg treatment may rescue Treg number and function by regulating miR-155 and miR-31 expression.
Role of Long Non-coding RNA in Kawasaki Disease
Long non-coding RNAs (lncRNAs) are a large class of ncRNAs (longer than 200 nucleotides in length) involved in many diverse biological processes through the regulation of gene transcription (116, 117). Over 27,000 lncRNAs have been predicted/annotated in the human genome and there is emerging evidence indicating their functional diversity and relevance in regulating human disease and development (117–119). Studies on lncRNA in KD are only handful.
Ko et al. (112) from China investigated changes in expression of lncRNAs in 37 patients with KD. Long non-coding RNA profiling revealed that the transcript XLOC_006277 was overrepresented in patients with acute KD and decreased after IVIg in IVIg-responsive patients. Notably, higher levels of XLOC_006277 transcript were found in patients with KD who later developed CAAs (112) (Table 2).
Many lncRNAs can enhance the inflammatory response by increasing the transcription of pro-inflammatory cytokines or other inflammatory target genes or by enhancing inflammatory signals. THRIL (TNF- and hnRNPL-related immunoregulatory lncRNA) is one of the many lncRNAs induced after TLR2 activation (113). CXCL10, one of the genes found to be regulated by THRIL, is up-regulated in KD patients in the acute phase and has been identified as a possible biomarker of the disease (120). In 17 patients with KD, linc1992/THRIL expression was lower during acute phase of disease when TNFα levels are elevated. Authors suggested linc1992/THRIL could be a new biomarker for immune activation in KD (113). In a study by Jiang et al., pregnancy-induced non-coding RNA (PINC) was shown to have an elevated expression in human umbilical vascular endothelial cells that are treated with TNF-α. The same study showed that PINC is involved in regulation of TNF-α induced vascular endothelial cell apoptosis (114).
In summary, lncRNAs are induced in response to inflammation and regulates gene transcription during the inflammatory response. Though there is limited literature available, yet there is compelling evidence that KD is associated with changes in expression of lncRNAs. However, further research is required to get an insight into the biological function and role of lncRNAs in KD for the development of new strategies for its treatment and as biomarker assessment.
Conclusion
Epigenetic studies have unravelled several new biomarkers for diagnosis and disease prognostication in KD. Majority of these are single-centre studies based on limited number of patients. These biomarkers, however, would need validation on larger cohorts and in different population groups before the results can be translated to clinical practise. Techniques and expertise required for carrying out epigenetic tests are demanding and the results may not be available quickly easily for bedside management of patients. However, with rapid decline in costs of genetic sequencing technology in recent years, it is possible such tests would be more utilised in future.
Author Contributions
KS, PV, PS, JS, AK, and HK preparation of manuscript. PV, PS, and SS editing and critical review of manuscript. HC and SM performed experiments and literature search. All authors approved the final version of manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rowley AH, Shulman ST. The epidemiology and pathogenesis of Kawasaki disease. Front Pediatr. (2018) 6:374. doi: 10.3389/fped.2018.00374
2. Newburger JW, Takahashi M, Burns JC. Kawasaki disease. J Am Coll Cardiol. (2016) 67:1738–49. doi: 10.1016/j.jacc.2015.12.073
3. Chantasiriwan N, Silvilairat S, Makonkawkeyoon K, Pongprot Y, Sittiwangkul R. Predictors of intravenous immunoglobulin resistance and coronary artery aneurysm in patients with Kawasaki disease. Paediatr Int Child Health. (2018) 38:209–12. doi: 10.1080/20469047.2018.1471381
4. Makino N, Nakamura Y, Yashiro M, Sano T, Ae R, Kosami K, et al. Epidemiological observations of Kawasaki disease in Japan, 2013-2014. Pediatr Int. (2018) 60:581–7. doi: 10.1111/ped.13544
5. Kim GB, Han JW, Park YW, Song MS, Hong YM, Cha SH, et al. Epidemiologic features of Kawasaki disease in South Korea: data from nationwide survey, 2009-2011. Pediatr Infect Dis J. (2014) 33:24–7. doi: 10.1097/INF.0000000000000010
6. Lin MC, Lai MS, Jan SL, Fu YC. Epidemiologic features of Kawasaki disease in acute stages in Taiwan, 1997-2010: effect of different case definitions in claims data analysis. J Chin Med Assoc. (2015) 78:121–6. doi: 10.1016/j.jcma.2014.03.009
7. Matsubara K, Fukaya T, Miwa K, Shibayama N, Nigami H, Harigaya H, et al. Development of serum IgM antibodies against superantigens of Staphylococcus aureus and Streptococcus pyogenes in Kawasaki disease. Clin Exp Immunol. (2006) 143:427–34. doi: 10.1111/j.1365-2249.2006.03015.x
8. Barton M, Melbourne R, Morais P, Christie C. Kawasaki syndrome associated with group A streptococcal and Epstein-Barr virus co-infections. Ann Trop Paediatr. (2002) 22:257–60. doi: 10.1179/027249302125001543
9. Wang JN, Wang SM, Liu CC, Wu JM. Mycoplasma pneumoniae infection associated with Kawasaki disease. Acta Paediatr. (2001) 90:594–5. doi: 10.1111/j.1651-2227.2001.tb00810.x
10. Normann E, Nääs J, Gnarpe J, Bäckman H, Gnarpe H. Demonstration of Chlamydia pneumoniae in cardiovascular tissues from children with Kawasaki disease. Pediatr Infect Dis J. (1999) 18:72–3. doi: 10.1097/00006454-199901000-00020
11. Embil JA, McFarlane ES, Murphy DM, Krause VW, Stewart HB. Adenovirus type 2 isolated from a patient with fatal Kawasaki disease. Can Med Assoc J. (1985) 132:1400.
12. Holm JM, Hansen LK, Oxhøj H. Kawasaki disease associated with parvovirus B19 infection. Eur J Pediatr. (1995) 154:633–4. doi: 10.1007/BF02079066
13. Kanegane H, Tsuji T, Seki H, Yachie A, Yokoi T, Miyawaki T, et al. Kawasaki disease with a concomitant primary Epstein-Barr virus infection. Acta Paediatr Jpn. (1994) 36:713–6. doi: 10.1111/j.1442-200X.1994.tb03277.x
14. Kuo HC, Li SC, Huang LH, Huang YH. Epigenetic hypomethylation and upregulation of matrix metalloproteinase 9 in Kawasaki disease. Oncotarget. (2017) 8:60875–91. doi: 10.18632/oncotarget.19650
15. Huang YH, Lo MH, Cai XY, Kuo HC. Epigenetic hypomethylation and upregulation of NLRC4 and NLRP12 in Kawasaki disease. Oncotarget. (2018) 9:18939–48. doi: 10.18632/oncotarget.24851
16. Huang LH, Kuo HC, Pan CT, Lin YS, Huang YH, Li SC. Multiomics analyses identified epigenetic modulation of the S100A gene family in Kawasaki disease and their significant involvement in neutrophil transendothelial migration. Clin Epigenetics. (2018) 10:135. doi: 10.1186/s13148-018-0557-1
17. Guo MM, Chang LS, Huang YH, Wang FS, Kuo HC. Epigenetic regulation of macrophage marker expression profiles in Kawasaki disease. Front Pediatr. (2020) 8:129. doi: 10.3389/fped.2020.00129
18. Nishio H, Kanno S, Onoyama S, Ikeda K, Tanaka T, Kusuhara K, et al. Nod1 ligands induce site-specific vascular inflammation. Arterioscler Thromb Vasc Biol. (2011) 31:1093–9. doi: 10.1161/ATVBAHA.110.216325
19. Motomura Y, Kanno S, Asano K, Tanaka M, Hasegawa Y, Katagiri H, et al. Identification of pathogenic cardiac CD11c+ macrophages in Nod1-mediated acute coronary arteritis. Arterioscler Thromb Vasc Biol. (2015) 35:1423–33. doi: 10.1161/ATVBAHA.114.304846
20. Ye F, Foell D, Hirono KI, Vogl T, Rui C, Yu X, et al. Neutrophil-derived S100A12 is profoundly upregulated in the early stage of acute Kawasaki disease. Am J Cardiol. (2004) 94:840–4. doi: 10.1016/j.amjcard.2004.05.076
21. Ebihara T, Endo R, Kikuta H, Ishiguro N, Ma X, Shimazu M, et al. Differential gene expression of S100 protein family in leukocytes from patients with Kawasaki disease. Eur J Pediatr. (2005) 164:427–31. doi: 10.1007/s00431-005-1664-5
22. Hoshina T, Kusuhara K, Ikeda K, Mizuno Y, Saito M, Hara T. High mobility group box 1 (HMGB1) and macrophage migration inhibitory factor (MIF) in Kawasaki disease. Scand J Rheumatol. (2008) 37:445–9. doi: 10.1080/03009740802144143
23. Foell D, Ichida F, Vogl T, Yu X, Chen R, Miyawaki T, et al. S100A12 (EN-RAGE) in monitoring Kawasaki disease. Lancet. (2003) 361:1270–2. doi: 10.1016/S0140-6736(03)12986-8
24. Kuijpers TW, Wiegman A, van Lier RA, Roos MT, Wertheim-van Dillen PM, Pinedo S, et al. Kawasaki disease: a maturational defect in immune responsiveness. J Infect Dis. (1999) 180:1869–77. doi: 10.1086/315111
25. Onouchi Y, Ozaki K, Burns JC, Shimizu C, Terai M, Hamada H, et al. A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat Genet. (2012) 44:517–21. doi: 10.1038/ng.2220
26. Lee YC, Kuo HC, Chang JS, Chang LY, Huang LM, Chen MR, et al. Two new susceptibility loci for Kawasaki disease identified through genome-wide association analysis. Nat Genet. (2012) 44:522–5. doi: 10.1038/ng.2227
27. Chang CJ, Kuo HC, Chang JS, Lee JK, Tsai FJ, Khor CC, et al. Replication and meta-analysis of GWAS identified susceptibility loci in Kawasaki disease confirm the importance of B lymphoid tyrosine kinase (BLK) in disease susceptibility. PLoS ONE. (2013) 8:e72037. doi: 10.1371/journal.pone.0072037
28. Tsai FJ, Lee YC, Chang JS, Huang LM, Huang FY, Chiu NC, et al. Identification of novel susceptibility Loci for kawasaki disease in a Han chinese population by a genome-wide association study. PLoS ONE. (2011) 6:e16853. doi: 10.1371/journal.pone.0016853
29. Furukawa S, Matsubara T, Yabuta K. Mononuclear cell subsets and coronary artery lesions in Kawasaki disease. Arch Dis Child. (1992) 67:706–8. doi: 10.1136/adc.67.6.706
30. Matsuda I, Hattori S, Nagata N, Fruse A, Nambu H. HLA antigens in mucocutaneous lymph node syndrome. Am J Dis Child. (1977) 131:1417–8. doi: 10.1001/archpedi.1977.02120250099033
31. Kato S, Kimura M, Tsuji K, Kusakawa S, Asai T, Juji T, et al. HLA antigens in Kawasaki disease. Pediatrics. (1978) 61:252–5.
32. Krensky AM, Berenberg W, Shanley K, Yunis EJ. HLA antigens in mucocutaneous lymph node syndrome in New England. Pediatrics. (1981) 67:741–3.
33. Krensky AM, Grady S, Shanley KM, Berenberg W, Yunis EJ. Epidemic and endemic HLA-B and DR associations in mucocutaneous lymph node syndrome. Hum Immunol. (1983) 6:75–7. doi: 10.1016/0198-8859(83)90063-0
34. Chang CC, Hawkins BR, Kao HK, Chow CB, Lau YL. Human leucocyte antigens in southern Chinese with Kawasaki disease. Eur J Pediatr. (1992) 151:866. doi: 10.1007/BF01957943
35. Kaslow RA, Bailowitz A, Lin FY, Koslowe P, Simonis T, Israel E. Association of epidemic Kawasaki syndrome with the HLA-A2, B44, Cw5 antigen combination. Arthritis Rheum. (1985) 28:938–40. doi: 10.1002/art.1780280814
36. Barron KS, Silverman ED, Gonzales JC, St Clair M, Anderson K, Reveille JD. Major histocompatibility complex class II alleles in Kawasaki syndrome–lack of consistent correlation with disease or cardiac involvement. J Rheumatol. (1992) 19:1790–3.
37. Wang Y, Wang W, Gong F, Fu S, Zhang Q, Hu J, et al. Evaluation of intravenous immunoglobulin resistance and coronary artery lesions in relation to Th1/Th2 cytokine profiles in patients with Kawasaki disease. Arthritis Rheum. (2013) 65:805–14. doi: 10.1002/art.37815
38. Jia S, Li C, Wang G, Yang J, Zu Y. The T helper type 17/regulatory T cell imbalance in patients with acute Kawasaki disease. Clin Exp Immunol. (2010) 162:131–7. doi: 10.1111/j.1365-2249.2010.04236.x
39. Furuno K, Yuge T, Kusuhara K, Takada H, Nishio H, Khajoee V, et al. CD25+CD4+ regulatory T cells in patients with Kawasaki disease. J Pediatr. (2004) 145:385–90. doi: 10.1016/j.jpeds.2004.05.048
40. Hirabayashi Y, Takahashi Y, Xu Y, Akane K, Villalobos IB, Okuno Y, et al. Lack of CD4*CD25*FOXP3* regulatory T cells is associated with resistance to intravenous immunoglobulin therapy in patients with Kawasaki disease. Eur J Pediatr. (2013) 172:833–7. doi: 10.1007/s00431-013-1937-3
41. Guo MM, Tseng WN, Ko CH, Pan HM, Hsieh KS, Kuo HC. Th17- and Treg-related cytokine and mRNA expression are associated with acute and resolving Kawasaki disease. Allergy. (2015) 70:310–8. doi: 10.1111/all.12558
42. Olivito B, Taddio A, Simonini G, Massai C, Ciullini S, Gambineri E, et al. Defective FOXP3 expression in patients with acute Kawasaki disease and restoration by intravenous immunoglobulin therapy. Clin Exp Rheumatol. (2010) 28(Suppl. 57):93–7.
43. Takeshita S, Tokutomi T, Kawase H, Nakatani K, Tsujimoto H, Kawamura Y, et al. Elevated serum levels of matrix metalloproteinase-9 (MMP-9) in Kawasaki disease. Clin Exp Immunol. (2001) 125:340–4. doi: 10.1046/j.1365-2249.2001.01608.x
44. Chua PK, Melish ME, Yu Q, Yanagihara R, Yamamoto KS, Nerurkar VR. Elevated levels of matrix metalloproteinase 9 and tissue inhibitor of metalloproteinase 1 during the acute phase of Kawasaki disease. Clin Diagn Lab Immunol. (2003) 10:308–14. doi: 10.1128/CDLI.10.2.308-314.2003
45. Gavin PJ, Crawford SE, Shulman ST, Garcia FL, Rowley AH. Systemic arterial expression of matrix metalloproteinases 2 and 9 in acute Kawasaki disease. Arterioscler Thromb Vasc Biol. (2003) 23:576–81. doi: 10.1161/01.ATV.0000065385.47152.FD
46. Wang L, Yang Y, Cui Q, Cui Y, Li Q, Che X, et al. Evaluating the added predictive ability of MMP-9 in serum for Kawasaki disease with coronary artery lesions. J Investig Med. (2021) 69:13–9. doi: 10.1136/jim-2020-001281
47. Lau AC, Duong TT, Ito S, Yeung RS. Matrix metalloproteinase 9 activity leads to elastin breakdown in an animal model of Kawasaki disease. Arthritis Rheum. (2008) 58:854–63. doi: 10.1002/art.23225
48. Reindel R, Baker SC, Kim KY, Rowley CA, Shulman ST, Orenstein JM, et al. Integrins α4 and αM, collagen1A1, and matrix metalloproteinase 7 are upregulated in acute Kawasaki disease vasculopathy. Pediatr Res. (2013) 73:332–6. doi: 10.1038/pr.2012.185
49. Gowen BB, Borg TK, Ghaffar A, Mayer EP. Selective adhesion of macrophages to denatured forms of type I collagen is mediated by scavenger receptors. Matrix Biol. (2000) 19:61–71. doi: 10.1016/S0945-053X(99)00052-9
50. Veres SP, Brennan-Pierce EP, Lee JM. Macrophage-like U937 cells recognize collagen fibrils with strain-induced discrete plasticity damage. J Biomed Mater Res A. (2015) 103:397–408. doi: 10.1002/jbm.a.35156
51. Brown TJ, Crawford SE, Cornwall ML, Garcia F, Shulman ST, Rowley AH. CD8 T lymphocytes and macrophages infiltrate coronary artery aneurysms in acute Kawasaki disease. J Infect Dis. (2001) 184:940–3. doi: 10.1086/323155
52. Miura M, Garcia FL, Crawford SE, Rowley AH. Cell adhesion molecule expression in coronary artery aneurysms in acute Kawasaki disease. Pediatr Infect Dis J. (2004) 23:931–6. doi: 10.1097/01.inf.0000142171.91235.fc
53. Shimizu C, Oharaseki T, Takahashi K, Kottek A, Franco A, Burns JC. The role of TGF-β and myofibroblasts in the arteritis of Kawasaki disease. Hum Pathol. (2013) 44:189–98. doi: 10.1016/j.humpath.2012.05.004
54. Gibney ER, Nolan CM. Epigenetics and gene expression. Heredity (Edinb). (2010) 105:4–13. doi: 10.1038/hdy.2010.54
55. Huang YH, Chen KD, Lo MH, Cai XY, Chang LS, Kuo YH, et al. Decreased DNA methyltransferases expression is associated with coronary artery lesion formation in Kawasaki disease. Int J Med Sci. (2019) 16:576–82. doi: 10.7150/ijms.32773
56. Li SC, Chan WC, Huang YH, Guo MM, Yu HR, Huang FC, et al. Major methylation alterations on the CpG markers of inflammatory immune associated genes after IVIG treatment in Kawasaki disease. BMC Med Genomics. (2016) 9(Suppl. 1):37. doi: 10.1186/s12920-016-0197-2
57. Chen KD, Huang YH, Ming-Huey Guo M, Lin TY, Weng WT, Yang HJ, et al. The human blood DNA methylome identifies crucial role of β-catenin in the pathogenesis of Kawasaki disease. Oncotarget. (2018) 9:28337–50. doi: 10.18632/oncotarget.25305
58. Huang YH, Li SC, Huang LH, Chen PC, Lin YY, Lin CC, et al. Identifying genetic hypomethylation and upregulation of Toll-like receptors in Kawasaki disease. Oncotarget. (2017) 8:11249–58. doi: 10.18632/oncotarget.14497
59. Lin IC, Kuo HC, Lin YJ, Wang FS, Wang L, Huang SC, et al. Augmented TLR2 expression on monocytes in both human Kawasaki disease and a mouse model of coronary arteritis. PLoS ONE. (2012) 7:e38635. doi: 10.1371/journal.pone.0038635
60. Habib A, Polavarapu R, Karmali V, Guo L, Van Dam R, Cheng Q, et al. Hepcidin-ferroportin axis controls toll-like receptor 4 dependent macrophage inflammatory responses in human atherosclerotic plaques. Atherosclerosis. (2015) 241:692–700. doi: 10.1016/j.atherosclerosis.2015.06.025
61. Huang YH, Lo MH, Cai XY, Liu SF, Kuo HC. Increase expression of CD177 in Kawasaki disease. Pediatr Rheumatol Online J. (2019) 17:13. doi: 10.1186/s12969-019-0315-8
62. Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, Alnemri ES. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J Biol Chem. (2001) 276:28309–13. doi: 10.1074/jbc.C100250200
63. Damiano JS, Stehlik C, Pio F, Godzik A, Reed JC. CLAN, a novel human CED-4-like gene. Genomics. (2001) 75:77–83. doi: 10.1006/geno.2001.6579
64. Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science. (1997) 275:206–9. doi: 10.1126/science.275.5297.206
65. Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. (1997) 386:619–23. doi: 10.1038/386619a0
66. Duncan JA, Canna SW. The NLRC4 Inflammasome. Immunol Rev. (2018) 281:115–23. doi: 10.1111/imr.12607
67. Lee Y, Schulte DJ, Shimada K, Chen S, Crother TR, Chiba N, et al. Interleukin-1β is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation. (2012) 125:1542–50. doi: 10.1161/CIRCULATIONAHA.111.072769
68. Ben Mkaddem S, Benhamou M, Monteiro RC. Understanding Fc receptor involvement in inflammatory diseases: from mechanisms to new therapeutic tools. Front Immunol. (2019) 10:811. doi: 10.3389/fimmu.2019.00811
69. Menikou S, Langford PR, Levin M. Kawasaki disease: the role of immune complexes revisited. Front Immunol. (2019) 10:1156. doi: 10.3389/fimmu.2019.01156
70. Shrestha S, Wiener H, Shendre A, Kaslow RA, Wu J, Olson A, et al. Role of activating FcγR gene polymorphisms in Kawasaki disease susceptibility and intravenous immunoglobulin response. Circ Cardiovasc Genet. (2012) 5:309–16. doi: 10.1161/CIRCGENETICS.111.962464
71. Chang LS, Lo MH, Li SC, Yang MY, Hsieh KS, Kuo HC. The effect of FcγRIIA and FcγRIIB on coronary artery lesion formation and intravenous immunoglobulin treatment responses in children with Kawasaki disease. Oncotarget. (2017) 8:2044–52. doi: 10.18632/oncotarget.13489
72. Biezeveld M, Geissler J, Merkus M, Kuipers IM, Ottenkamp J, Kuijpers T. The involvement of Fc gamma receptor gene polymorphisms in Kawasaki disease. Clin Exp Immunol. (2007) 147:106–11. doi: 10.1111/j.1365-2249.2006.03266.x
73. Warmerdam PA, van de Winkel JG, Vlug A, Westerdaal NA, Capel PJ. A single amino acid in the second Ig-like domain of the human Fc gamma receptor II is critical for human IgG2 binding. J Immunol. (1991) 147:1338–43.
74. Lou J, Zhong R, Shen N, Lu XZ, Ke JT, Duan JY, et al. Systematic confirmation study of GWAS-identified genetic variants for Kawasaki disease in a Chinese population. Sci Rep. (2015) 5:8194. doi: 10.1038/srep08194
75. Kuo HC, Chang JC, Kuo HC, Yu HR, Wang CL, Lee CP, et al. Identification of an association between genomic hypomethylation of FCGR2A and susceptibility to Kawasaki disease and intravenous immunoglobulin resistance by DNA methylation array. Arthritis Rheumatol. (2015) 67:828–36. doi: 10.1002/art.38976
76. Makowsky R, Wiener HW, Ptacek TS, Silva M, Shendre A, Edberg JC, et al. FcγR gene copy number in Kawasaki disease and intravenous immunoglobulin treatment response. Pharmacogenet Genomics. (2013) 23:455–62. doi: 10.1097/FPC.0b013e328363686e
77. Chang LS, Ming-Huey Guo M, Lo MH, Kuo HC. Identification of increased expression of activating Fc receptors and novel findings regarding distinct IgE and IgM receptors in Kawasaki disease. Pediatr Res. 2019. doi: 10.1038/s41390-019-0707-y
78. Weng H, Pei Q, Yang M, Zhang J, Cheng Z, Yi Q. Hypomethylation of C1q/tumor necrosis factor-related protein-1 promoter region in whole blood and risks for coronary artery aneurysms in Kawasaki disease. Int J Cardiol. (2020) 307:159–63. doi: 10.1016/j.ijcard.2020.02.002
79. Huang YH, Kuo HC, Li SC, Cai XY, Liu SF, Kuo HC. HAMP promoter hypomethylation and increased hepcidin levels as biomarkers for Kawasaki disease. J Mol Cell Cardiol. (2018) 117:82–7. doi: 10.1016/j.yjmcc.2018.02.017
80. Duan J, Gherghe C, Liu D, Hamlett E, Srikantha L, Rodgers L, et al. Wnt1/βcatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. (2012) 31:429–42. doi: 10.1038/emboj.2011.418
81. Flynt AS, Lai EC. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet. (2008) 9:831–42. doi: 10.1038/nrg2455
82. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. (2010) 11:597–610. doi: 10.1038/nrg2843
83. Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. (2011) 12:861–74. doi: 10.1038/nrg3074
84. Sato F, Tsuchiya S, Meltzer SJ, Shimizu K. MicroRNAs and epigenetics. FEBS J. (2011) 278:1598–609. doi: 10.1111/j.1742-4658.2011.08089.x
85. Saito K, Nakaoka H, Takasaki I, Hirono K, Yamamoto S, Kinoshita K, et al. MicroRNA-93 may control vascular endothelial growth factor A in circulating peripheral blood mononuclear cells in acute Kawasaki disease. Pediatr Res. (2016) 80:425–32. doi: 10.1038/pr.2016.93
86. Kaneda R, Fukuda K. MicroRNA is a new diagnostic and therapeutic target for cardiovascular disease and regenerative medicine. Circ J. (2009) 73:1397–8. doi: 10.1253/circj.CJ-09-0435
87. Kawashima T, Shioi T. MicroRNA, emerging role as a biomarker of heart failure. Circ J. (2011) 75:268–9. doi: 10.1253/circj.CJ-10-1254
88. Reid G, Kirschner MB, van Zandwijk N. Circulating microRNAs: association with disease and potential use as biomarkers. Crit Rev Oncol Hematol. (2011) 80:193–208. doi: 10.1016/j.critrevonc.2010.11.004
89. Cortez MA, Calin GA. MicroRNA identification in plasma and serum: a new tool to diagnose and monitor diseases. Expert Opin Biol Ther. (2009) 9:703–11. doi: 10.1517/14712590902932889
90. Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. (2006) 6:259–69. doi: 10.1038/nrc1840
91. van Rooij E, Olson EN. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest. (2007) 117:2369–76. doi: 10.1172/JCI33099
92. O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA. (2007) 104:1604–9. doi: 10.1073/pnas.0610731104
93. Creemers EE, Tijsen AJ, Pinto YM. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res. (2012) 110:483–95. doi: 10.1161/CIRCRESAHA.111.247452
94. Rowley AH, Pink AJ, Reindel R, Innocentini N, Baker SC, Shulman ST, et al. A study of cardiovascular miRNA biomarkers for Kawasaki disease. Pediatr Infect Dis J. (2014) 33:1296–9. doi: 10.1097/INF.0000000000000449
95. Shimizu C, Kim J, Stepanowsky P, Trinh C, Lau HD, Akers JC, et al. Differential expression of miR-145 in children with Kawasaki disease. PLoS ONE. (2013) 8:e58159. doi: 10.1371/journal.pone.0058159
96. Magenta A, Cencioni C, Fasanaro P, Zaccagnini G, Greco S, Sarra-Ferraris G, et al. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. (2011) 18:1628–39. doi: 10.1038/cdd.2011.42
97. Yun KW, Lee JY, Yun SW, Lim IS, Choi ES. Elevated serum level of microRNA (miRNA)-200c and miRNA-371-5p in children with Kawasaki disease. Pediatr Cardiol. (2014) 35:745–52. doi: 10.1007/s00246-013-0846-6
98. Jone PN, Korst A, Karimpour-Fard A, Thomas T, Dominguez SR, Heizer H, et al. Circulating microRNAs differentiate Kawasaki Disease from infectious febrile illnesses in childhood. J Mol Cell Cardiol. (2020) 146:12–8. doi: 10.1016/j.yjmcc.2020.06.011
99. Nakaoka H, Hirono K, Yamamoto S, Takasaki I, Takahashi K, Kinoshita K, et al. MicroRNA-145-5p and microRNA-320a encapsulated in endothelial microparticles contribute to the progression of vasculitis in acute Kawasaki Disease. Sci Rep. (2018) 8:1016. doi: 10.1038/s41598-018-19310-4
100. Zhang W, Wang Y, Zeng Y, Hu L, Zou G. Serum miR-200c and miR-371-5p as the useful diagnostic biomarkers and therapeutic targets in Kawasaki disease. Biomed Res Int. (2017) 2017:8257862. doi: 10.1155/2017/8257862
101. Zhang X, Xin G, Sun D. Serum exosomal miR-328, miR-575, miR-134 and miR-671-5p as potential biomarkers for the diagnosis of Kawasaki disease and the prediction of therapeutic outcomes of intravenous immunoglobulin therapy. Exp Ther Med. (2018) 16:2420–32. doi: 10.3892/etm.2018.6458
102. Luo Y, Yang J, Zhang C, Jin Y, Pan H, Liu L, et al. Up-regulation of miR-27a promotes monocyte-mediated inflammatory responses in Kawasaki disease by inhibiting function of B10 cells. J Leukoc Biol. (2020) 107:133–44. doi: 10.1002/JLB.5A0919-075RR
103. Ni FF, Li CR, Li Q, Xia Y, Wang GB, Yang J. Regulatory T cell microRNA expression changes in children with acute Kawasaki disease. Clin Exp Immunol. (2014) 178:384–93. doi: 10.1111/cei.12418
104. Wang B, Wang LN, Cheng FF, Lv HT, Sun L, Wei DK, et al. MiR-222-3p in platelets serves as a distinguishing marker for early recognition of Kawasaki disease. Front Pediatr. (2019) 7:237. doi: 10.3389/fped.2019.00237
105. Chen Y, Ding YY, Ren Y, Cao L, Xu QQ, Sun L, et al. Identification of differentially expressed microRNAs in acute Kawasaki disease. Mol Med Rep. (2018) 17:932–8. doi: 10.3892/mmr.2017.8016
106. Li Z, Jiang J, Tian L, Li X, Chen J, Li S, et al. A plasma mir-125a-5p as a novel biomarker for Kawasaki disease and induces apoptosis in HUVECs. PLoS ONE. (2017) 12:e0175407. doi: 10.1371/journal.pone.0175407
107. Jia HL, Liu CW, Zhang L, Xu WJ, Gao XJ, Bai J, et al. Sets of serum exosomal microRNAs as candidate diagnostic biomarkers for Kawasaki disease. Sci Rep. (2017) 7:44706. doi: 10.1038/srep44706
108. Kuo HC, Hsieh KS, Ming-Huey Guo M, Weng KP, Ger LP, Chan WC, et al. Next-generation sequencing identifies micro-RNA-based biomarker panel for Kawasaki disease. J Allergy Clin Immunol. (2016) 138:1227–30. doi: 10.1016/j.jaci.2016.04.050
109. Wu R, Shen D, Sohun H, Ge D, Chen X, Wang X, et al. miR-186, a serum microRNA, induces endothelial cell apoptosis by targeting SMAD6 in Kawasaki disease. Int J Mol Med. (2018) 41:1899–908. doi: 10.3892/ijmm.2018.3397
110. Rong X, Ge D, Shen D, Chen X, Wang X, Zhang L, et al. miR-27b Suppresses endothelial cell proliferation and migration by targeting Smad7 in Kawasaki disease. Cell Physiol Biochem. (2018) 48:1804–14. doi: 10.1159/000492354
111. Li Y, Wu X, Gao F, Wang X. MiR-197-3p regulates endothelial cell proliferation and migration by targeting IGF1R and BCL2 in Kawasaki disease. Int J Clin Exp Pathol. (2019) 12:4181–92.
112. Ko TM, Chang JS, Chen SP, Liu YM, Chang CJ, Tsai FJ, et al. Genome-wide transcriptome analysis to further understand neutrophil activation and lncRNA transcript profiles in Kawasaki disease. Sci Rep. (2019) 9:328. doi: 10.1038/s41598-018-36520-y
113. Li Z, Chao TC, Chang KY, Lin N, Patil VS, Shimizu C, et al. The long noncoding RNA THRIL regulates TNFα expression through its interaction with hnRNPL. Proc Natl Acad Sci USA. (2014) 111:1002–7. doi: 10.1073/pnas.1313768111
114. Jiang C, Fang X, Jiang Y, Shen F, Hu Z, Li X, et al. TNF-α induces vascular endothelial cells apoptosis through overexpressing pregnancy induced noncoding RNA in Kawasaki disease model. Int J Biochem Cell Biol. (2016) 72:118–24. doi: 10.1016/j.biocel.2016.01.011
115. Chu M, Wu R, Qin S, Hua W, Shan Z, Rong X, et al. Bone marrow-derived microRNA-223 works as an endocrine genetic signal in vascular endothelial cells and participates in vascular injury from Kawasaki disease. J Am Heart Assoc. (2017) 6:e004878. doi: 10.1161/JAHA.116.004878
116. Ernst C, Morton CC. Identification and function of long non-coding RNA. Front Cell Neurosci. (2013) 7:168. doi: 10.3389/fncel.2013.00168
117. Miao Y, Ajami NE, Huang TS, Lin FM, Lou CH, Wang YT, et al. Enhancer-associated long non-coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat Commun. (2018) 9:292. doi: 10.1038/s41467-017-02113-y
118. Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJ, Gough J, et al. An atlas of human long non-coding RNAs with accurate 5' ends. Nature. (2017) 543:199–204. doi: 10.1038/nature21374
119. Barangi S, Hayes AW, Reiter R, Karimi G. The therapeutic role of long non-coding RNAs in human diseases: a focus on the recent insights into autophagy. Pharmacol Res. (2019) 142:22–9. doi: 10.1016/j.phrs.2019.02.010
Keywords: Kawasaki disease, methylation, microRNA, long non-coding RNA, biomarker
Citation: Sharma K, Vignesh P, Srivastava P, Sharma J, Chaudhary H, Mondal S, Kaur A, Kaur H and Singh S (2021) Epigenetics in Kawasaki Disease. Front. Pediatr. 9:673294. doi: 10.3389/fped.2021.673294
Received: 27 February 2021; Accepted: 01 June 2021;
Published: 25 June 2021.
Edited by:
Michael Portman, University of Washington, United StatesReviewed by:
Lovro Lamot, University of Zagreb, CroatiaEdoardo Marrani, Meyer Children's Hospital, Italy
Copyright © 2021 Sharma, Vignesh, Srivastava, Sharma, Chaudhary, Mondal, Kaur, Kaur and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Surjit Singh, c3Vyaml0c2luZ2hhcGNAZ21haWwuY29t
†These authors have contributed equally to this work and share first authorship