
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 07 June 2021
Sec. Pediatric Rheumatology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.669116
 Emily Mirizio1
Emily Mirizio1 Christopher Liu1
Christopher Liu1 Qi Yan2Julia Waltermire1Roosha Mandel1
Qi Yan2Julia Waltermire1Roosha Mandel1 Kaila L. Schollaert1
Kaila L. Schollaert1 Liza Konnikova3Xinjun Wang2
Liza Konnikova3Xinjun Wang2 Wei Chen2
Wei Chen2 Kathryn S. Torok1,4*
Kathryn S. Torok1,4*The purpose of this study was to explore the skin transcriptional profile in pediatric localized scleroderma (LS) to provide a better understanding of the altered immune and fibrotic pathways promoting disease. LS is a progressive disease of the skin and underlying tissue that causes significant functional disability and disfigurement, especially in developing children. RNA sequencing (RNAseq) technology allows for improved understanding of relevant cellular expression through transcriptome analysis of phases during LS disease progression (more active/inflammatory vs. inactive/fibrotic) and also permits the use of RNA extracted from existing paraffin-embedded skin tissue, which is important in pediatrics. A strong correlation was observed between the comparison of genes expressed between fresh (RNAlater) and paraffinized skin in healthy and LS subjects, supporting the use of paraffinized tissue. LS gene signatures compared to healthy controls showed a distinct expression of an inflammatory response gene signature (IRGS) composed of IFNγ-, IFNα-, and TNFα-associated genes. GSEA© enrichment analysis showed that the IRGS, including interferon-inducible chemokines such as CXCL9, CXCL10, CXCL11, and IFNγ itself, was more highly expressed in LS patients with more inflammatory lesions. The use of paraffinized skin for sequencing was proven to be an effective substitute for fresh skin by comparing gene expression profiles. The prevalence of the IFNγ signature in the lesion biopsies of active LS patients indicates that these genes reflect clinical activity parameters and may be the promoters of early, inflammatory disease.
Localized scleroderma (LS), also known as morphea, is an autoimmune disease characterized by skin fibrosis and subsequent atrophy (typically in bands along the lines of Blaschko) in the absence of vascular and internal-organ involvement, with an annual incidence of 1–3 per 100,000 children (1). LS progression is biphasic, with an inflammatory active phase that is followed by a fibrotic damage phase distinguished by inflammatory infiltrate of the skin, collagen deposition, and subsequent thickening of the deep dermis and subcutis, respectively (2). Clinically, active disease is defined by cutaneous features, such as erythema, skin thickening, new lesions, and lesion expansion, all captured by validated cutaneous clinical outcome measures including the modified Localized Scleroderma Skin Severity Index (mLoSSI) and Physician Global Assessment of Activity (PGA-A) (3–5). Since juvenile LS presents during development (mean age of onset 8 years) and can persist for many years (mean disease duration of 13.5 years) (6), morbidity can be substantial. A recent review of 259 LS patients in the Childhood Arthritis and Rheumatology Research Alliance (CARRA) registry found that 38% had musculoskeletal damage and 25% had limited functional capacity (Figures 1A,C) (7). Damage is most prevalent in linear scleroderma, the pre-dominant juvenile LS subtype (60%) (8, 9).
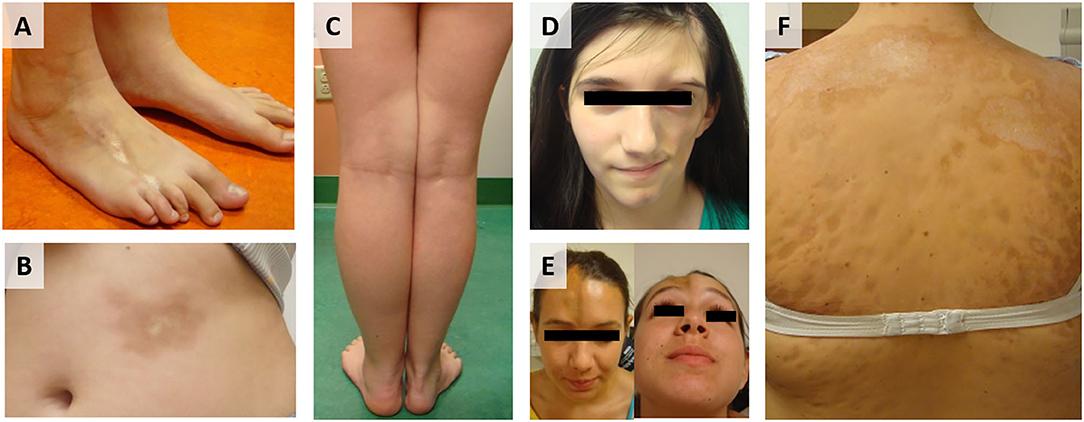
Figure 1. Juvenile Localized scleroderma (LS) subtypes: (A) Linear leg lesion with atrophy of the third toe, (B) Circumscribed plaque morphea lesion trunk, (C) Linear limb lesion with subsequent atrophy and limb length discrepancy, (D) LS of the head leading to hemi facial atrophy (Parry Romberg Syndrome), (E) LS of the head with en coupe de sabre (ECDS) linear depression forehead/skull, (F) Generalized morphea with hyperpigmentation and central sclerosis of lesions.
Biologically, active disease components are still being unraveled. The exact pathogenesis is unclear; however, translational peripheral blood and skin studies in LS support a predominance of CD4+ T cells, macrophages, fibroblasts, and TH1- and IFNγ-associated chemokines/cytokines (10). There is significant elevation of circulating CD4+ IFNγ + T cells (TH1) during active disease (2), along with IFNγ-related proteins CXCL9 [monokine induced by gamma interferon (MIG)] and CXCL10 [interferon gamma-induced protein 10 (IP-10)]. Both CXCL9 and CXCL10 were also present in active LS skin lesions within the perivascular lymphocytic infiltrate of the papillary and reticular dermis. CXCL9 also stained in close approximation to both CD4+TH cells and macrophages (11), suggesting potential interaction between lymphocytes and macrophages via IFNγ chemokine signaling. Overall, these interactions may synergistically promote fibroblasts to increase collagen expression in LS, leading to increased collagen deposition, and fibrosis.
Abrogating the inflammatory response during active LS is critical for limiting disease progression and damage; therefore, further identification of cellular components and molecules involved is paramount. A large-scale, unbiased approach to studying dysregulated IFNγ-mediated pathways and to identify additional pathways involved in LS is now available using large-scale next-generation sequencing (NGS), which is an unbiased method that provides a detailed exploration of dysregulated LS pathways (12). The purpose of this investigation is to further evaluate the LS skin transcriptome using RNA sequencing (RNAseq) to identify up- and downregulated pathways and serve as a platform for future mechanistic studies.
Traditionally, NGS techniques including RNAseq have employed fresh or fresh-frozen (FF) samples rather than formalin-fixed, paraffin-embedded (FFPE) samples due to the potential for degradation, with decreased exonic reads and increased intronic reads with FFPE samples (13). However, more recent studies of other human tissue, including brain, endometrial, lung, and breast cancers (14–22), demonstrated that FFPE samples stored for up to 32 years have been successfully analyzed with RNAseq, introducing the potential for future NGS analysis of widely available archival FFPE samples (23). FFPE samples are more readily available for children who have undergone a diagnostic biopsy, and eliminate the need for a separate research procedure. To our knowledge, skin FFPE vs. FF RNA and sequencing integrity have not been reported. Therefore, our initial goal was to examine the RNA integrity in FFPE skin, followed by RNAseq of LS and healthy FFPE skin specimens to study the differentially expressed genes of the LS transcriptome and their associated pathway(s) analyses.
After obtaining written informed consent, samples for LS subjects were collected through the National Registry for Childhood Onset Scleroderma (NRCOS, IRB #PRO11060222) and healthy controls through IRB #PRO12040127. Accompanying clinical measures and outcome data associated with the subjects' specimens were extracted from these registries. Demographic variables included sex, race, and age at sample visit. Healthy controls were age and sex matched with a 3:1 ratio. Additional clinical variables for LS subjects included LS disease subtype, number of affected body sites, and validated measures of disease activity and severity, which included the Localized Scleroderma Cutaneous Assessment Tool (LoSCAT) and physician global assessments (24, 25). The LoSCAT includes the modified Localized Scleroderma Skin Index (mLoSSI) which quantifies cutaneous disease activity (24). The mLoSSI and the physician global assessment of activity (PGA-A) are the core variables defining disease activity in LS (24) and have been found to be responsive to change (26). The PGA-A is graded on a 100-mm analog scale and includes consideration of the following cutaneous variables: new lesions within the previous month, erythema/violaceous color at the border of the lesion, and skin thickening/induration at the border of the lesion. Patients with mLoSSI > 3 and PGA-A > 10 were considered to have active disease; those with lower scores were considered clinically inactive with a PGA-A and mLoSSI score of 0 (2, 24). Physician documentation of overall judgment of disease state (active/inactive) was also obtained at the study visit.
RNA was extracted from paraffin-embedded skin and a subset of these subjects that had accompanying FF skin collected at the same time which was RNAlater preserved (n = 2 LS, n = 2 healthy) using the Qiagen AllPrep® DNA/RNA FFPE (Qiagen #80234) and the Qiagen RNeasy Mini (Qiagen #74014) extraction kits, respectively. RNAs were quantified using a Nanodrop ND-100 Spectrophotometer (Nanodrop Technologies, Wilmington, USA) and a 2100 Bioanalyzer (Agilent RNA 6000 Nano Kit, Waldbronn, Germany). Extracted RNA samples were only sequenced if they had a %DV200 (percentage of RNA fragments > 200 nucleotides) (27) >30% for FFPE samples and an RNA integrity number of ≥8 for RNAlater samples to ensure quality control.
Extracted RNA was prepared for sequencing using the Illumina HTS TrueSeq Access library preparation and sequenced on the Illumina NextSeq 500. FastQ files were generated via llumina bcl2fastq2 (Version 2.17.1.14—http://support.illumina.com/downloads/bcl2fastq-conversion-software-v217.html) starting from.bcl files produced by the Illumina NextSeq sequencer. The quality of individual sequences was evaluated using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
Paired-end RNA sequencing data was aligned using STAR (https://github.com/alexdobin/STAR/releases) and quantified using HTseq (https://htseq.readthedocs.io/en/release_0.11.1/). The human genome reference used for the alignment was hg38—Ensembl Transcripts release 93. Expressed transcripts per sample were evaluated imposing a minimum threshold of five counts per gene to consider it as expressed. Differential expression analysis for all transcripts was performed with the R package DESeq2 (http://bioconductor.org/packages/DESeq2). Genes were analyzed for differences between subject groups using differentially expressed gene (DEG) cutoffs of log2fold change > ±2.5, adjusted p <0.05, counts per gene > 20%, and a false discovery rate (FDR) cutoff of <0.1.
Gene enrichment analysis was performed on significant DEGs between subject groups. Enrichment software (GSEA©) with Broad Institute Hallmark gene lists was used to determine whether DEGs between subject groups show statistically significant overrepresentation in a set of genes and any association with disease phenotypes. Gene Ontology (GO) analysis for biological processes, cellular components, and molecular function was also used. PCA and hierarchical clustering of log2-transformed fragments-per-kilobase-per-million (FPKM) data were performed using Partek® software. Data was clustered linearly mean centered using Euclidian distance. Color scales were adjusted for presentation purposes. LS clinical subtype data (active/inflammatory vs. stable/disease damage) was applied to these clustering techniques.
The location of transcripts of interest from DEG and pathway analyses was analyzed in skin specimens. Formalin-fixed paraffin-embedded biopsies of two LS patients and two healthy controls were used for dual ISH (in situ hybridization) and immunofluorescence (IF) multicolor staining. Advanced Cell Diagnostics (ACD) (Newark, CA) RNAscope® LS Multiplex Fluorescent Assay Combined with IF was used. The assay was performed on 3-μm-thick sections using RNAscope® probes targeting CXCL9 (Cat No. 440161), IFNγ (Cat No. 310501-C2), CXCR3 (Cat No. 539251), and CD3 (Cat No. 599391-C2) which were developed by ACD and used according to the manufacturer's recommendations. For IF, primary antibodies against human CD163 as well as appropriate secondary antibodies were used. For multicolor fluorescence microscopy, sections were sequentially stained using the tyramide fluorescence assay. An example is incubation with primary anti-CD163 (clone 56C6; 1:50; Leica MS, Wetzlar, Germany), which was followed by horse anti-rabbit secondary with tyramide Cy5 amplification (PerkinElmer, Waltham, MA). Endogenous peroxidase was blocked with normal horse serum (Sigma-Aldrich, Saint Louis, MO) in Tris-buffered saline (TBS) with bovine serum albumin (BSA). 4′,6′-Diamidino-2-phenylindole (DAPI) counterstaining was performed to visualize the tissue structure. Fluorescence images were recorded using an Echo Revolve fluorescence microscope with filter combinations, specifically DAPI, Cyanine3, and Cy5. The total numbers of RNA and protein-positive cells were counted using ImageJ software (NIH, Bethesda, MD) at ×40 magnification. One slide per sample was stained, and at least three representative images were analyzed per tissue section.
Sequencing analyses included R package DESeq2 for DEG analyses and GSEA, GO, and Partek® for pathway analyses of DEGs of interest, with cutoffs for significance as mentioned above. Spearman's correlation coefficient analyses were performed to investigate the relationship between gene expression and clinical measures using GraphPad Prism (Version 7.0e, La Jolla, CA, USA). ImageJ software (NIH, Bethesda, MD) was used to count the number of stained transcripts in RNAscope® analyses with % difference between sample type run in GraphPad Prism (Version 7.0e, La Jolla, CA, USA).
The integrity of RNA extracted from FFPE compared to FF skin was compared for paired samples. FastQC data results for RNA extracted from fresh frozen (RNAlater preserved) and paraffinized FFPE skin in healthy (n = 2) and LS (n = 2) subjects were comparable for measurements of RNA quality, such as total reads and read coverage (Table 1). Once sequenced, FF and FFPE samples were almost indistinguishable and downstream alignment revealed that 92% of mapped genes were conserved between sample types of paired samples (Figure 2A). Gene count data for FF and FFPE paired samples correlated significantly with a correlation coefficient of 0.91 (p ≤ 0.0001; Figure 2B). FPKM-normalized data correlated even better with a correlation coefficient of 0.94 (not shown). The high integrity and correlation of expressed genes using FFPE compared to FF-stored skin supported the utilization of FFPE specimens for the RNA Seq analyses of pediatric LS skin compared to healthy controls.

Table 1. FastQC analysis results for FFPE and RNAlater sample types support comparable coverage and quality.
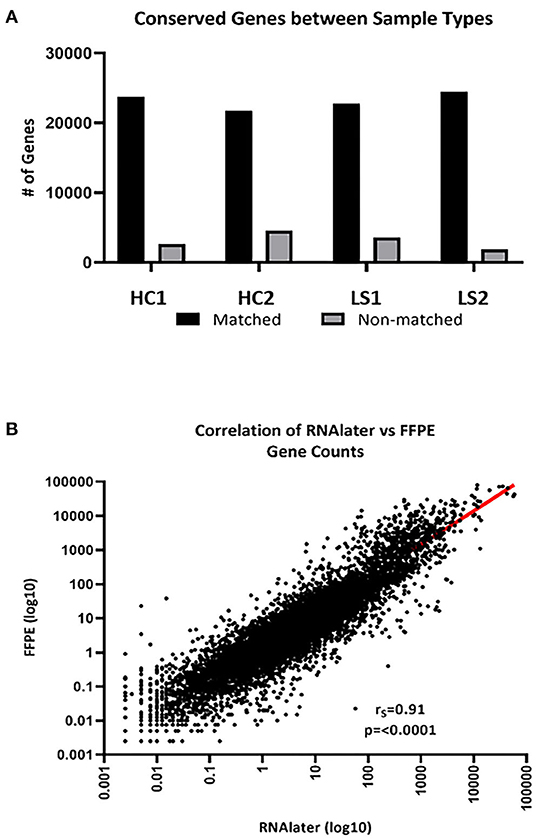
Figure 2. FFPE and FF (RNAlater) generated data sequencing and mapped well per sample type. (A) An average of 92% of total genes was conserved between sample types. HC, healthy control and LS, localized scleroderma. (B) Of the conserved genes, gene expression correlated significantly (rs = 0.91, p < 0.0001).
RNA extracted from FFPE samples was analyzed for differentially expressed genes (DEGs) between pediatric LS (n = 14 and age-matched healthy (n = 4) samples using the R program DESeq. Demographics and clinical variables are provided in Table 2. There were 3,753 DEGs (up and downregulated) between LS and healthy controls, and after applying expression cutoffs 1,302 genes remained significant as demonstrated in the MA plot, a scatterplot of M (log ratio) as the log2 fold changes (on the y-axis) vs. the A (mean average) as the mean of normalized counts (on the x-axis) (Figure 3A) (Supplementary Table 1 for full gene list). Visualization of genetic expression per subject using (1) t-Distributed Stochastic Neighbor Embedding (tSNE) clustering techniques (Figure 3B) and (2) heat map expression (Figure 3C) demonstrates that RNA transcript data showed a clear separation between the two groups, with relative homogeneity among LS patients compared to healthy.
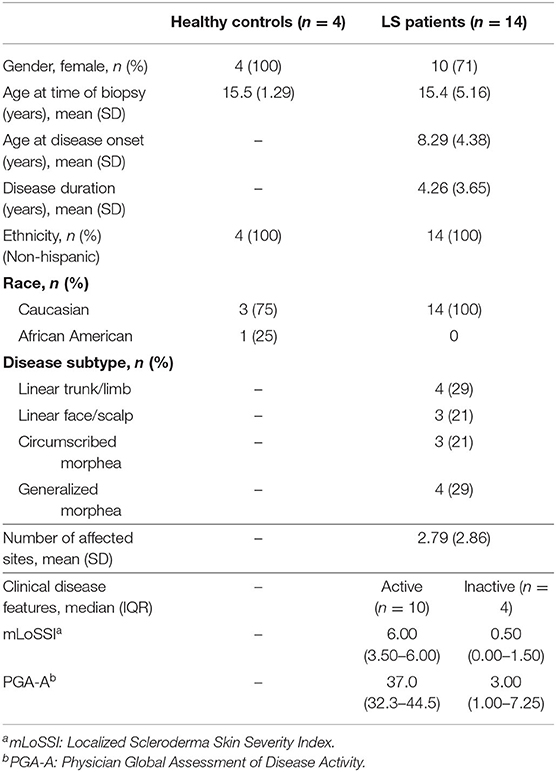
Table 2. Pediatric healthy control and localized scleroderma (LS) patient demographics and clinical measures.
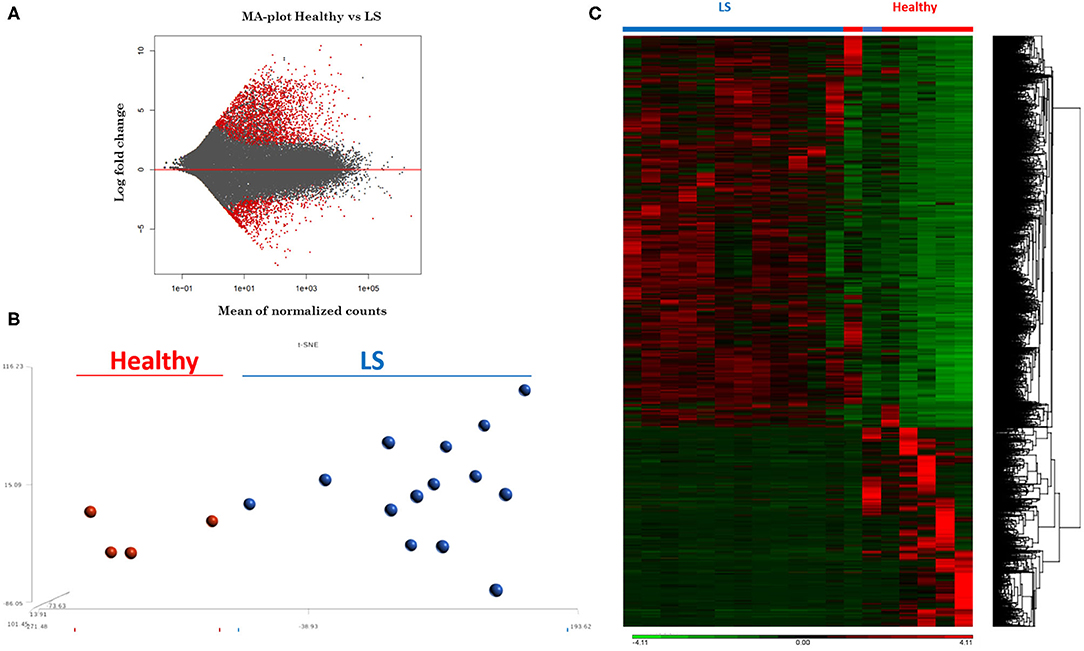
Figure 3. RNAseq expression differences between healthy and localized scleroderma (LS) subject samples. (A) Intensity-dependent ratios between two groups of data. Red points denote genes with significant differences. (B) tSNE clustering of genetic expression confirms significant differences between the two data groups. (C) Heat map showing the overall expression difference between all DEGs comparing LS to healthy samples.
GO enrichment and gene set enrichment (GSEA) analysis was performed on DEGs, and GO and GSEA terms with significant enrichment were selected. Significant enrichment groups (p ≤ 0.05 and FDR ≤ 0.05) were found to positively relate to TH1- and IFNγ-related immune responses, including the mediation and migration of leukocytes and KRAS signaling (Table 3). Significant groups were also found to negatively relate to DEGs, including the response to the epithelial mesenchymal transition processes (Table 3). Genes encoding for cytokine and inflammatory responses such as CXCL11-, CXCL10-, IL12B-, IFNG-, and IGKV1-related genes were upregulated in disease groups compared to control. Downregulated genes encoding for the epithelial transition stimuli were also found with similar magnitude including PRSS2, WNT5A, and IGFBP2.
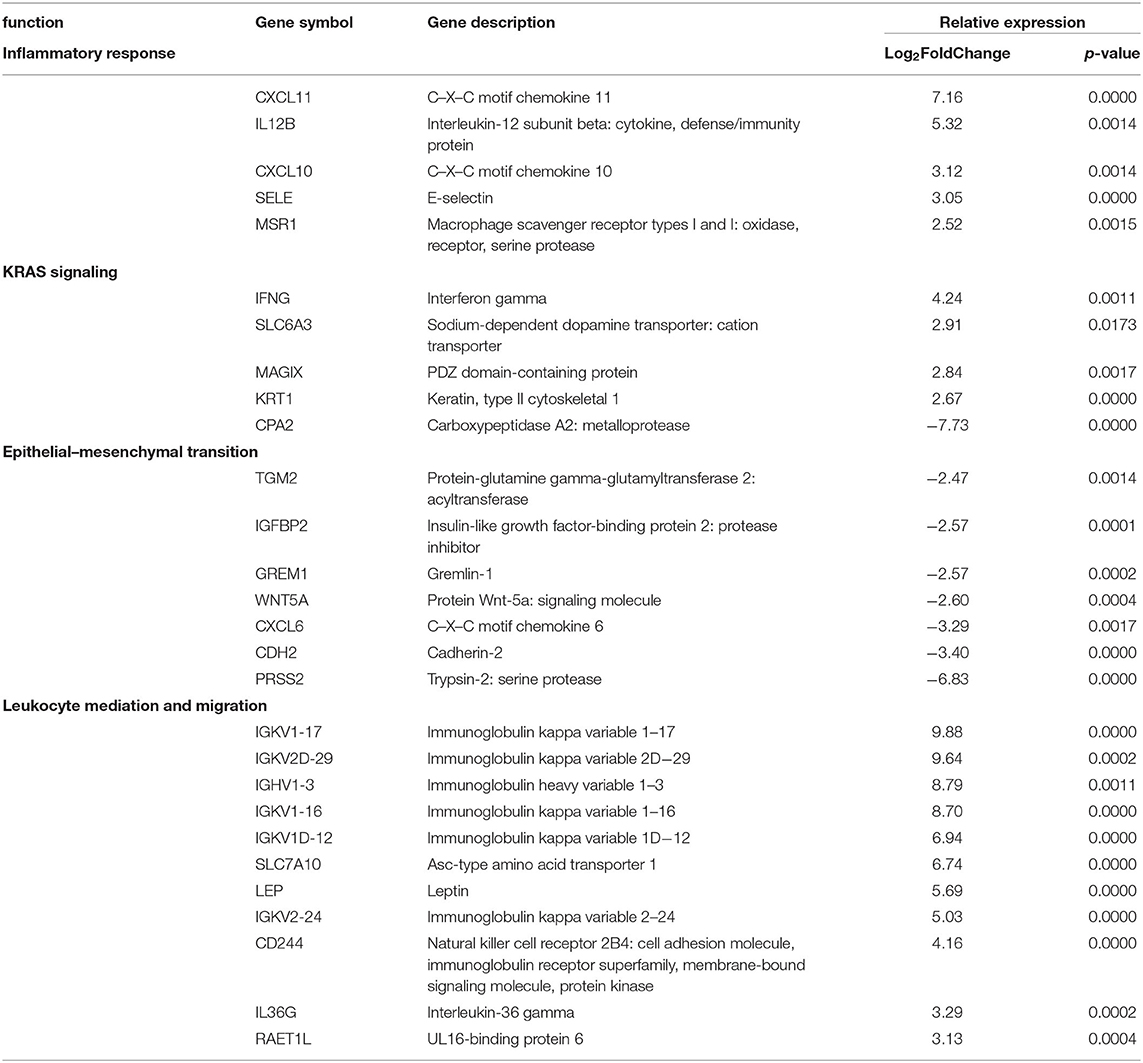
Table 3. Pediatric localized scleroderma vs. healthy control comparison identifies gene enrichment groups of interest relating to inflammatory and regulatory signatures.
Of note was the absence of certain cell type and pathway signatures. Genes relating to certain subsets of T cells that are often connected with autoimmune diseases such as Tregs (FOXP3, CD25) and TH17 (IL-17, RORC) were decreased in gene lists. Additionally, an immune response antagonist, transforming growth factor beta (TGFβ) which has been implicated in promoting activity in other forms of scleroderma, was relatively unaffected in our LS disease samples. While not significantly up or downregulated, the relatively low expression of these genes provides insight in the cells involved in pathogenesis.
Clinical activity, determined by mLoSSI and PGA-A scores, was then used to separate the LS samples into inactive and active groups. Ten active samples and four inactive samples were independently compared to the healthy controls and differential expression calculated. After applying expression cutoffs, 2,366 genes were differentially expressed in the active group compared to controls (see Supplementary Table 2 for a full gene list). Within the active LS subjects, DEG analyses demonstrated a distinct expression of genes encoding for inflammatory responses native to IFNγ/α and TNFα pathways and leukocyte activation and/or regulation, which include CXCL9, CXCL10, CXCL11, IFI27, STAT1, CXCL3, TNF, CSF2, GZMA, and IRF1. Also upregulated in active LS samples were MHC Class II genes, HLA-DQA1 and HLA-DRB1, reflecting activated immune response (Table 4). These pathways were further supported using GSEA© and GO enrichment software (Figures 4A,B). Predominant genes in these GSEA hallmarks were then complied into a master grouping we designated as the “inflammatory response gene signature (IRGS),” composed of 175 genes (see Supplementary Table 3 for full gene list). This gene signature had a high number of genes related to IFNγ that had a high log2 fold change including CXCL11, CXCL10, CXCL9, IRF1, CCL5, CMKLR1, BATF2, OASL, CMPK2, TRIM21, IFI27, GBP4, LAP3, ISG15, XCL1, CD274, GZMA, KLRK1, HLA-DQA1, IDO1, ZBP1, HLA-DRB1, SLAMF7, OAS3, and STAT1, indicating that this signature is highly related to IFNγ reflecting LS literature.
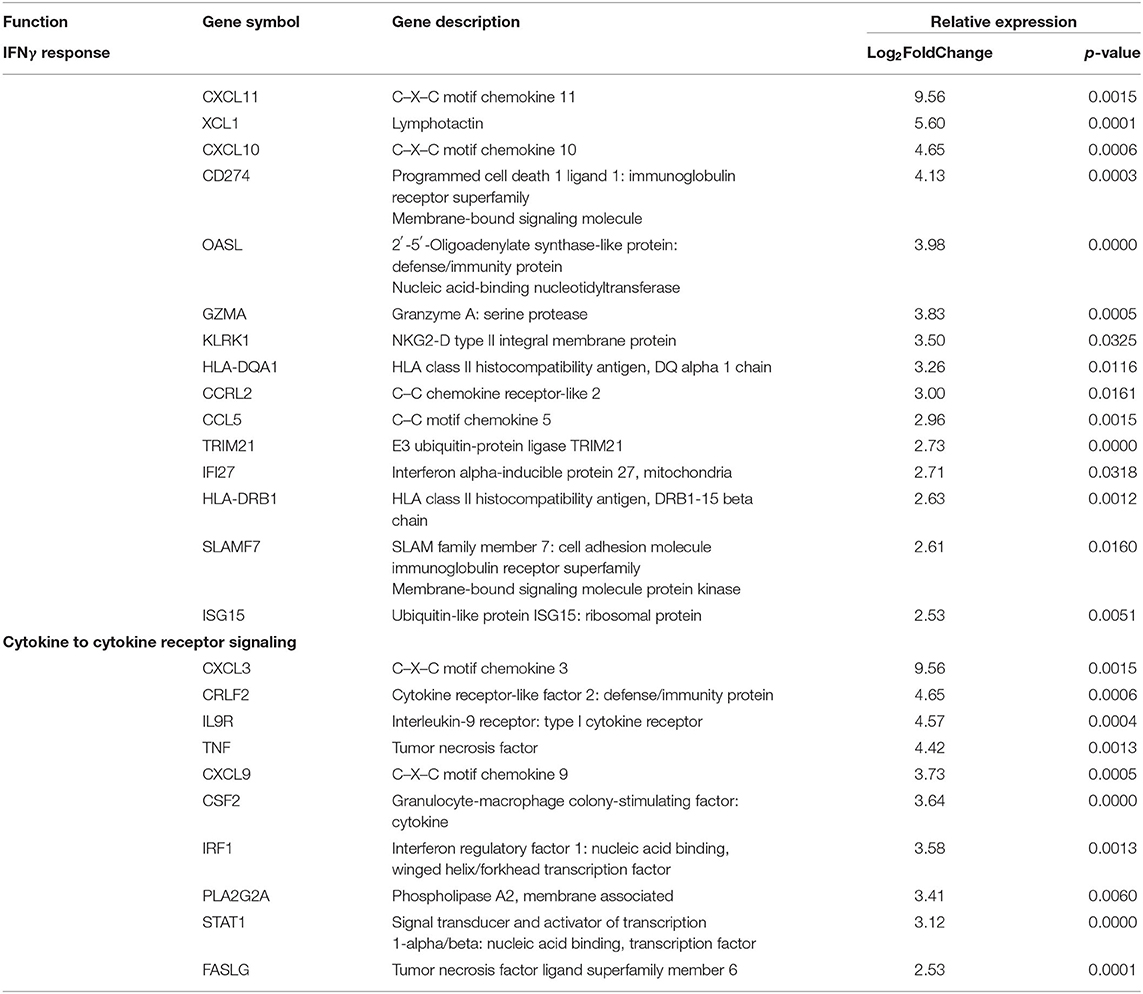
Table 4. Pediatric active localized scleroderma (LS) vs. healthy control sample comparison identifies gene enrichment groups of interest relating to inflammatory signatures that was used to develop the Inflammatory Response Gene Signature in LS.

Figure 4. Immune pathways of interest expressed in samples of active localized scleroderma subjects. Significant DEGs were related to known biological, molecular, and cellular processes including T cell migration, activation, and aggregation, and interferon gamma based on GSEA© (A) and GO (B) enrichment profiles.
Applying this evaluation of the IRGS gene expression to all LS samples compared to healthy with clinical features, using a cutoff of Spearman's ρ > 0.5 and p < 0.05, 57 of a total of 175 genes within the inflammatory signature group (ISRG) correlated strongly and positively with validated clinical disease activity measures, the mLoSSI and the PGA-A. Several interferon pathway-associated genes (inducible, regulatory, and receptor) and cytokine signaling genes were highly correlated, most with both measures. Notable were IFI44, IFT3, IRF5, IL-15RA, CXCL2, CD86, and STAT4 (Table 5). Applying hierarchal clustering of the genes included in the IRGS (Cluster 3.0 and Java TreeView) demonstrated that five extremely clinically active individuals out of the 14 total LS patient samples clustered together with an upregulated expression of this inflammatory signature (Figure 5). These subjects were composed of different clinically defined LS subtypes (two linear extremity, two circumscribed, one generalized morphea).
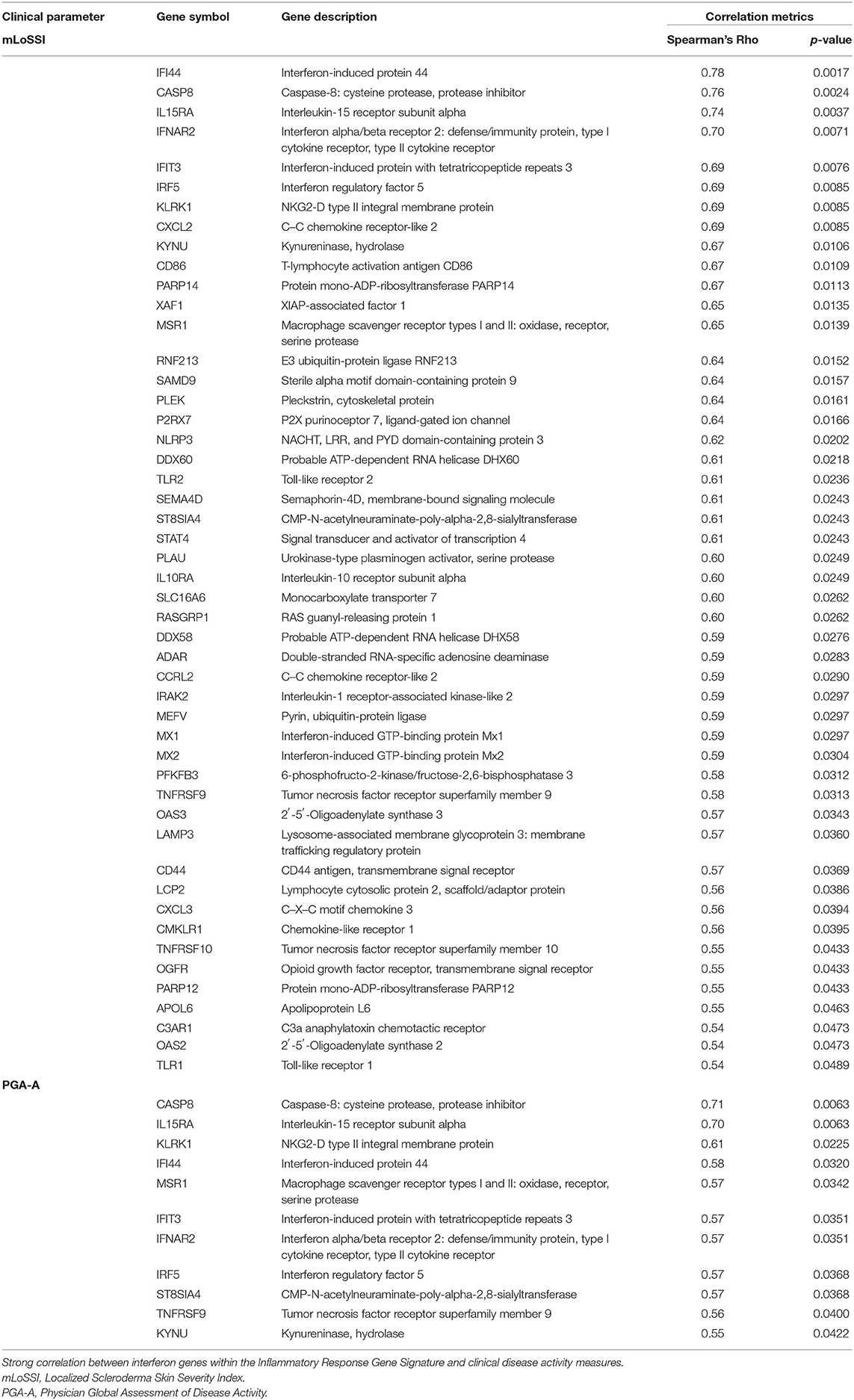
Table 5. Gene expression (FPKM) correlations with clinical parameters of disease activity.
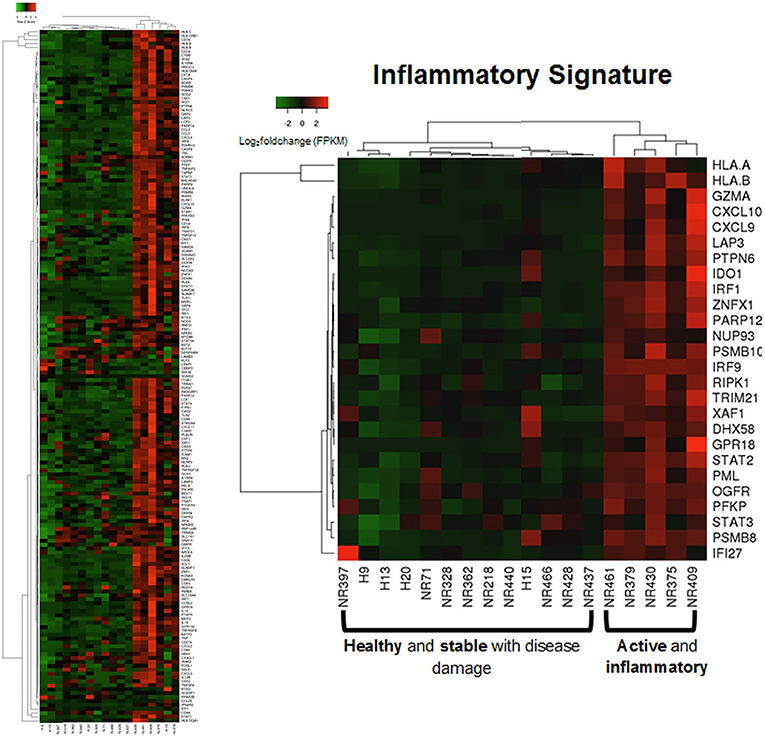
Figure 5. Supervised clustering using IRGS gene set identified a sub-clustering of 5 LS samples with high expression. These samples are considered to be in an active inflammatory disease state (via histological and clinical examination) suggesting the IRGS genes are indicators of active disease (see Supplementary Table 3 for IRGS gene set).
As described in the literature, IFNγ-related protein, CXCL9, had an upregulated transcriptional expression specifically in patients with active disease. Dual RNA and protein staining of LS and healthy negative controls showed localization of CXCL9 expression on CD163+ macrophages (Figure 6). Increased macrophage infiltration, with increased CXCL9 and IFNγ expression, was observed in LS tissue as compared to healthy tissue, especially in areas of inflammation when compared to H&E (Table 6). Macrophage-specific (CD163+) CXCL9 and IFNγ cells stained in close approximation to CXCR3+ T cells.
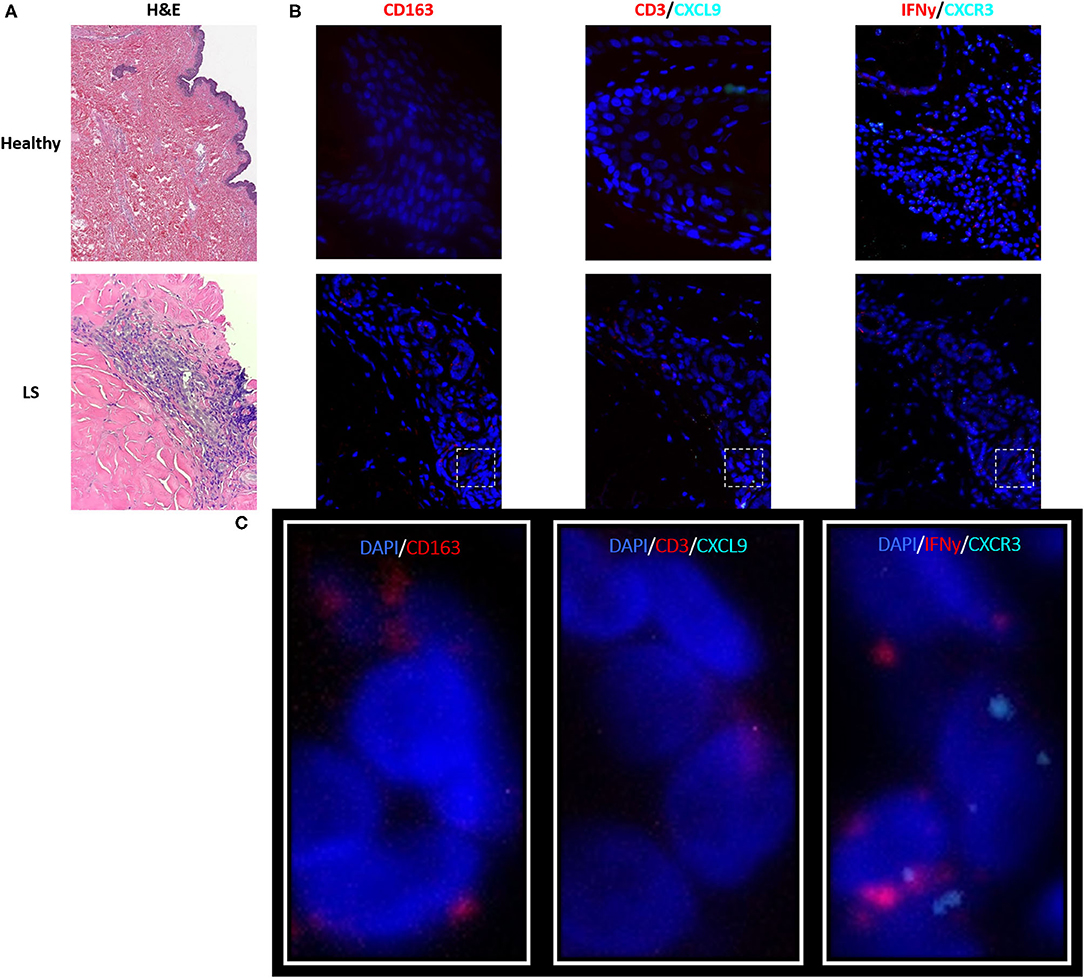
Figure 6. Visualization in the skin identifies T cell and macrophage co-localization, with CXCL9+ and IFNγ+ macrophages adjacent to CXCR3+ T cells in localized scleroderma (LS) skin. (A) H&E demonstrates inflammatory infiltrate extending into the lower reticular dermis in LS while the healthy sample demonstrates basal levels of minimal inflammatory cells that are typically localized to periadnexal structures. (B) RNAscope® fluorescent multiplex imaging showed increased macrophages (CD163+), CXCL9, and IFNy transcript in LS skin as compared to controls. (C) Macrophage-specific (CD163+) CXCL9 and IFNγ staining in close approximation to CXCR3+T cells.
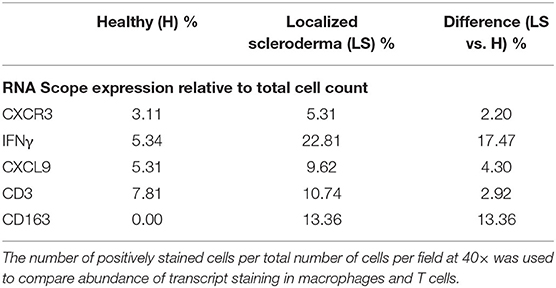
Table 6. Immunofluorescent and RNAscope® fluorescent multiplex imaging of localized scleroderma and control skin.
The remaining inactive samples were then investigated. After applying expression cutoffs, 2,247 genes were differentially expressed in the inactive group (see Supplementary Table 4 for full gene list). Genes encoding for ECM formation and dermal restructuring such as development and differentiation of the epidermis, ECM organization, and keratinization were the most significant after GO and GSEA enrichment analysis (Table 7). The genes included in these enrichment groups included COL17A1, KRT173, FLG, and COL17A. Additionally, transcription factors that are important in regulating the production of factors that control epithelial–mesenchymal interactions, cellular proliferation, and extracellular matrix production such as WNT, ERK, PI3K-TBX, FOX, RUNX, and SRF were demonstrated to be highly expressed. The inactive sample DEGs contain a much higher prevalence of collagen and keratin genes with significantly fewer genes relating to the IGRS inflammatory signature.
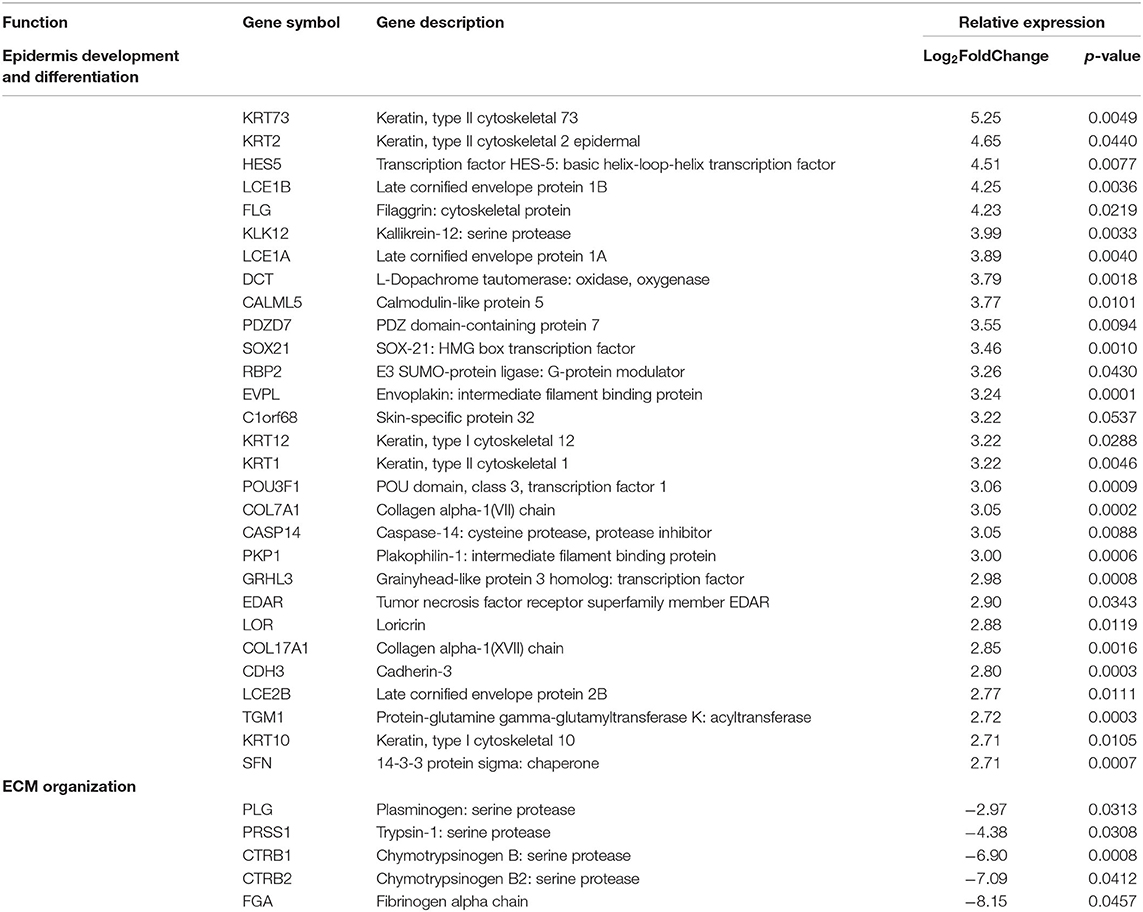
Table 7. Pediatric inactive localized scleroderma vs. healthy control sample comparison identifies gene enrichment groups of interest related to fibrotic signatures.
The 10 active and four inactive samples were then compared to each other using differential gene analysis. Due to sample similarity, the magnitude of expression was decreased in this comparison so an adjusted cutoff to log2fold change > ±0.5 was used. After applying adjusted expression cutoffs, 1,213 genes were differentially expressed in the active group compared to the inactive group (see Supplementary Table 5 for full gene list). The enrichment groups discovered after GO and GSEA enrichment analysis were much broader in category, including epithelial–mesenchymal transition. Enrichment groups specifically related to immune or functional pathways including genes encoding for IL2 STAT5, IL6 JAK/STAT3, TNFα via NFKB, and KRAS signaling were the most significant (Table 8). The genes included in these enrichment groups included IL1B, NFKBIA, DUSP1, and JUNB. A positive correlation of some of the genes included in the enrichment groups, such as IFI44, CASP8, IFNAR2, IL15RA, and IFNAR2, among others, with disease activity scores of mLoSSI and PGA-A was seen (Table 5).
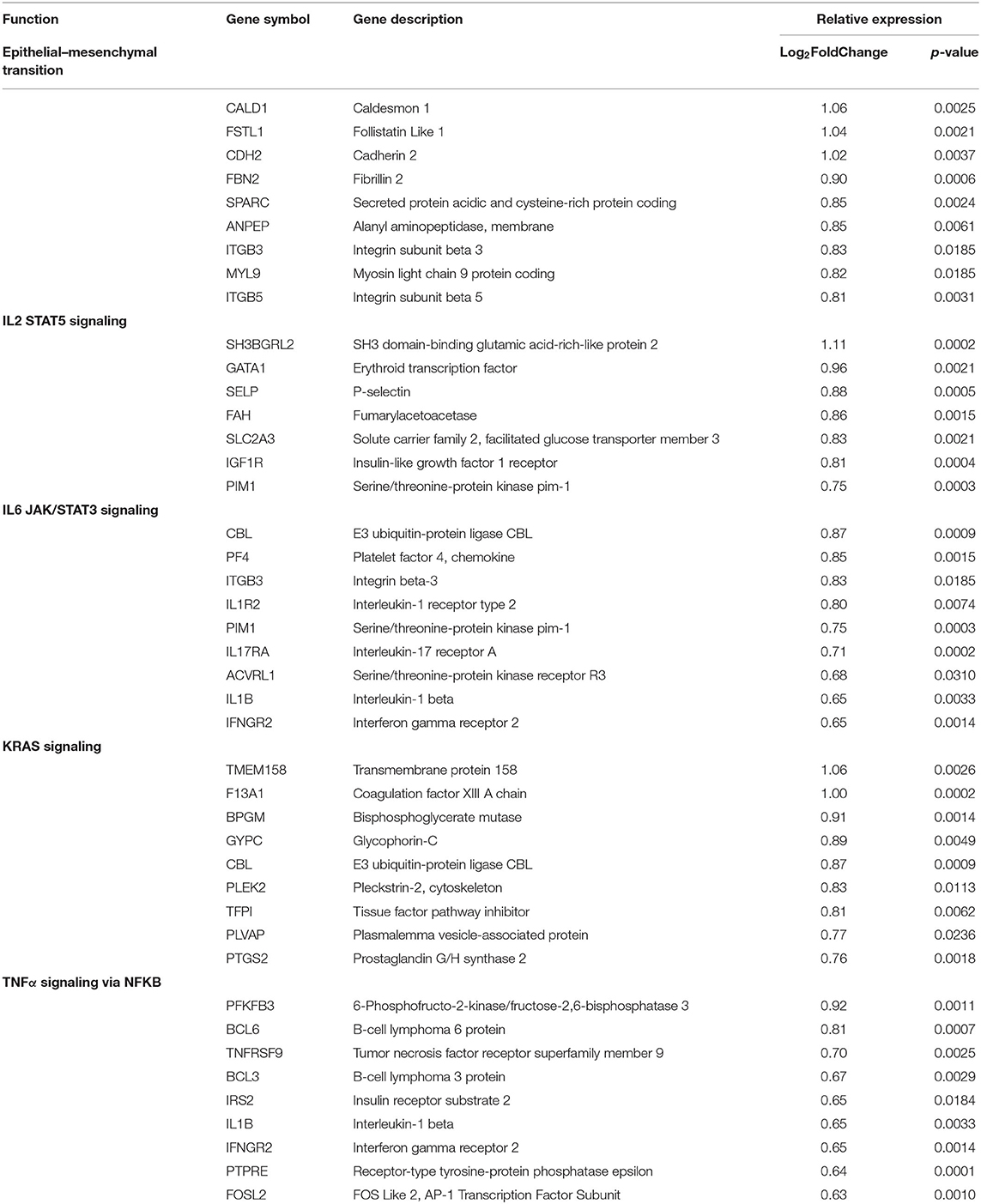
Table 8. Pediatric active vs. inactive localized scleroderma sample comparison identifies gene enrichment groups of interest.
Samples were then separated based on clinical subtype of disease (Figure 1), which included four linear scleroderma of the trunk and limb, three linear scleroderma of the face and scalp, three circumscribed morphea, and four generalized morphea samples, and then compared to healthy controls independently. For each comparison, around 50 genes were differentially expressed. Of these DEGs, many overlapped between subtypes with some specific expression for each group (Figure 7A). Clustering of all samples showed that even within LS, distinct subgroupings occurred. While many genes were common compared to healthy controls, each subtype has unique genetic traits correlating to cutaneous disease manifestations and different immune profiles (Figure 7A). Enrichment of subtype-specific DEGs showed that generalized morphea and circumscribed morphea subtypes (more common in adult-onset) had increased activity to the inflammasome pathway in cellular response categories (GSEA), but only generalized morphea had elevated TNF-related molecular adhesion and migration activity (GSEA) as well as T-cell activator differentiation (GO) (Figure 7B). Circumscribed morphea had elevated interferon and ubiquitin-related cell activation pathways in addition to seven inflammatory activation genetic profiles. Linear scleroderma (more common in pediatric onset) had increased activity of the immune activation-related genetic profile and high expression of cell killing, immune response, and chemical synapse pathways (GO) (Figure 7B). Linear scleroderma of the face and scalp had increased activity of the neural inflammatory signaling pathways (GSEA), which may correspond to the brain lesions associated with this subtype (28). Linear scleroderma of the trunk and limb subtype shared the most genes with the other subtypes, especially circumscribed, and individually had increased collagen deposition in the intrinsic prothrombin activation pathway (GSEA). An important limitation of this subtype subanalysis is the low sample number and an unequal distribution of active and inactive samples within each subtype group.
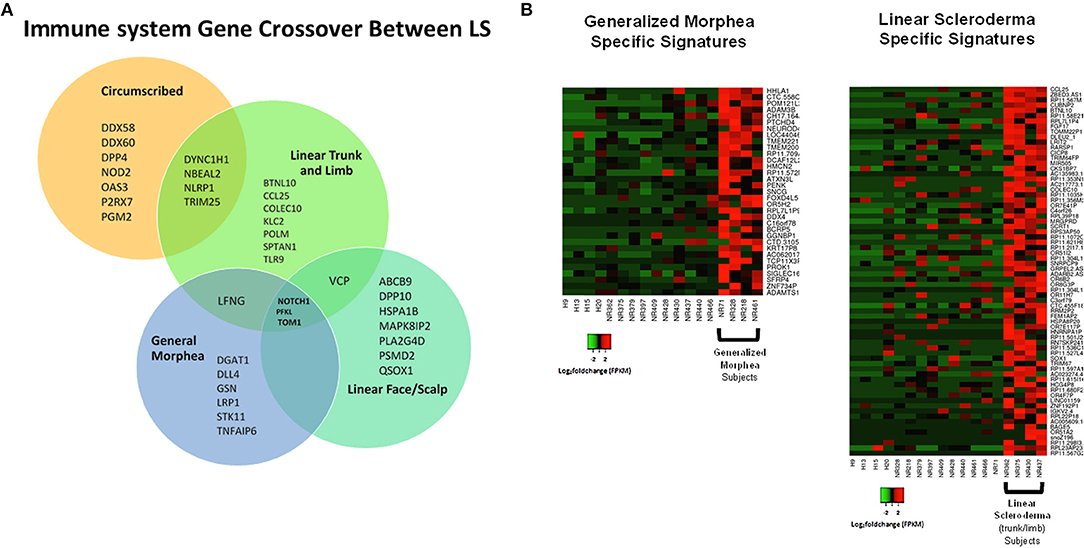
Figure 7. Shared and divergent immunological pathways among localized scleroderma subtypes. (A) Venn diagram demonstrates genetic immunological overlap occurring between subtypes, while having some uniquely different gene expression. (B) The two main LS subtypes, generalized morphea and linear scleroderma, display some areas of unique gene expression profiles that correspond to unique enrichment groups.
Gene expression profiling has traditionally used RNA extracted from fresh frozen (FF) tissue that is typically flash frozen or frozen in RNA stabilization solution such as RNAlater® (14). Unfortunately, there are many difficulties associated with the collection of FF specimens mostly pertaining to availability of enough tissue (29). However, abundant archival FFPE samples, retained from diagnostic histopathology procedures, offer an alternative to the scarcer FF sample (30). In pediatric research, the use of paraffin blocks stored from diagnostic procedures also reduces the need for children to undergo additional research biopsies. The potential for gene expression analysis lying within FFPE archives presents researchers with the opportunity to pursue retrospective studies focusing on correlations between gene expression patterns and disease states or phenotypes (31). As the storage of biopsy samples customarily involves formalin fixation and paraffin embedding, the optimization of methods for performing gene expression analysis on FFPE tissues will significantly expand the preexisting tissue resources from which researchers can draw data (18). Already, studies have demonstrated that FFPE samples may serve as a comparable alternative to FF samples for gene expression analysis in glioblastoma, endometrial, lung, and breast cancer tissues (18–22). In 2014, Hedegaard et al. investigated the potential applications of RNA-Seq and DNA Exome-Seq procedures on FFPE samples of colon, prostate, and bladder carcinomas, ultimately finding that after mapping analysis, clear correlations and similar sequence variants existed between RNA-Seq and DNA Exome-Seq results, respectively (19). Iddawella et al. performed correlational analysis on RNA profiles of matched FF and FFPE breast cancer samples and found correlations ranging from 0.83 to 0.89 which improved to 0.96 to 0.98 upon gene selection, thus solidifying that FFPE tissues could serve as reliable sources for expression profiling (32).
Sequencing of paired RNAlater and FFPE pediatric skin tissue samples has not been performed to date, and our data being presented yielded comparable results in quality and mapped genes between healthy and disease (LS) pediatric skin, with correlation analyses of RNA profiles of matched samples similar to those recently reported in breast cancer and other tissue sources (18–21, 32, 33). A high correlation of RNAlater and FFPE tissue found in this investigation indicates that FFPE tissue can be utilized for bulk RNA-seq in place of fresh or FF tissue in both healthy and disease state of the skin. This may open the door for several autoimmune and other skin diseases to be able to take advantage of stored tissue, especially in pediatrics in which repeat fresh biopsies are not tolerated well. These results supported our decision to use FFPE skin samples for RNA sequencing of 14 additional skin samples to evaluate the difference in transcriptomic expression between LS and healthy controls.
To date, studies examining the pathogenesis of LS have consisted of reports of circulating chemokine profiles or antibodies, flow cytometry of peripheral blood, and immunostaining often in a limited number of samples or without controls (34–37). Most of these studies have observed increased serum CXCL9 and CXCL10 levels that are associated with increased clinical measures of disease activity (2, 11, 38–41). Despite these studies, the pathogenesis of LS is largely unknown with transcriptional profiling by microarray or RNA bulk sequencing limited to two available studies.
Little is known about the transcriptional profile of localized scleroderma (LS) and even less about pediatric LS. Only a few studies have investigated the gene expression of LS through NGS methods, and our study is the first to examine a wide range of subtypes from pediatric LS subjects. In SSc, transcriptional investigation has been successful at classifying samples based on genetic differences that could indicate or predict disease progression and potential future treatment plans. In the landmark SSc microarray skin expression studies defining these classifications by Milano et al. there were three adult LS subjects included with the 24 SSc subjects. The skin expression patterns classified the SSc subjects by four distinct genetic signatures: diffuse proliferation, inflammatory, limited, and normal-like (42). All three LS subjects' microarray expression aligned with the inflammatory signature on the DEG heat map, expressing primarily T-lymphocyte- and IFNγ-related genes and associating with the early diffuse cutaneous SSc subjects (42).
The other available NGS transcriptional study of LS skin was conducted in pediatric patients but was limited to those with craniofacial scleroderma subtype, termed Parry Romberg Syndrome. RNAseq analyses demonstrated that LS samples (n = 16) had increased enrichment groups pertaining to inflammation driven by chemokines/cytokines and interleukins as well as apoptotic signaling which includes genes such as IL24, PROK2, CSF3, RTL1, DPP2, WISP2, and SCARA5 (43).
In our dataset, the comparison of LS to healthy samples clearly identified a distinct transcriptomic difference between disease and healthy skin. Similar enrichment groups related to inflammation as seen in both transcriptional studies performed to date on LS skin were reflected; however, the specific genetic profile of the pediatric LS samples more closely matches the samples within the inflammatory signature that were analyzed with the SSc samples described by Milano et al. Furthermore, within the diverse group of samples included in this study, a distinct subset of patients expressed similar inflammatory genes including interferon-inducible chemokines such as CXCL9, CXCL10, CXCL11, and IFNγ itself. Similar to SSc, subgroupings of LS patients are expected to be present as these inflammatory genes were more highly expressed in LS patients with more inflammatory or active lesions, with associated higher clinical activity scores of mLoSSI and PGA-A. Smaller less distinct subgroupings based on fibrotic and transitioning characteristics are less distinct in this dataset, but clear disease progression in genetic profile is observed.
A defining gene characteristic in the DEG analysis between all LS patients compared to controls is the inflammatory signaling enrichment groups. A specific interest was taken in the presence of the KRAS signaling pathway, which has previously been defined as a key component of the MAPK/ERK signaling pathway for modulating ERK activity and was highly enriched in the dataset. This pathway plays an important role in cell proliferation, migration, differentiation, and apoptosis and especially T cell infiltration to tissue, including transitioning to T regulatory and TH17 cells (44). The abundance of Tregs and TH17 cells has been contested in recent publications, but the consensus is that both of these cell types are decreased in LS but with an overall increase in interferon expression (all interferon subtypes). Overexpression of the KRAS signaling pathway has been shown to be critical to the development of Th17 cells and the Th17/Treg cell imbalance (45). The KRAS pathway is thought to induce the IRF7 signaling that leads to increased gene expression and induction of key proteins involved in expression and secretion of interferon gene expression inducing the conversion of CD4+-naïve T to Treg cells by upregulating genes that characterize Tregs, including FOXP3 (46–48). In this study, only the KRAS signaling and interferon signaling pathways were observed, including IRF7, with no regulatory component present. The lack of FOXP3 expression observed here and in our prior publication of LS circulating PBMC profile (46) indicates that while the KRAS signaling pathway is inducing interferon-related expression, as seen in the data through high expression of IRF7, IFI27, CCL2, CXCL12, and ARG1, the induction of Th17 and Treg populations is low. Additionally, KRAS and the MAPK/ERK pathway have also been linked to matrix metalloproteinase (MMP) expression (49). MMPs are antifibrotic molecules that induce the degradation of the collagens and other ECM components; unbalanced expressions of MMP and tissue inhibitor of metalloproteinases (TIMP) are involved in the fibrotic process of related autoimmune diseases (50, 51).
While signaling and fibrogenic pathways did not enrich in the active LS DEG analysis compared to controls, the overall inflammatory signal dominated the genetic landscape. Enrichment pathways for cytokine and IFNγ signaling predominate, and many genes included in the active LS DEG list correlate with clinical parameters such as disease scoring and lesion number. CXCR3-related ligands, CXCL9, CXCL10, and CXCL11, have repeatedly been presented as active indicators of LS in both protein and transcriptional expressions (11, 38–40). The appearance of these genes as DEGs in active LS expression lists provides further evidence of the importance of these cytokines in LS disease propagation. Transcript staining of these markers in pediatric skin matches immunohistochemistry (IHC) from the skin from adult subjects in the Morphea in Adult and Children (MAC) cohort in which the expressions of TH1 cell markers (CD3, CD4, CXCR3) and CXCL 9 and 10 chemokines were investigated in LS skin (52). A predominate lymphocytic infiltrate was identified in perivascular and periadnexal areas of the superficial and deep dermis of LS subjects, all of which show strong staining with increased percentages of CD3+, CD4+, and CXCR3+ cells (52). The RNAscope findings in our study corroborate the IHC staining by Walker et al., revealing that CXCL9-expressing macrophages reside close to CD4 lymphocytes expressing CXCR3 but neither cell co-expresses the other (52), and supporting our hypothesis that LS macrophage activation mediates IFNγ expression and subsequent T cell CXCR3 receptor binding to overexpress CXCL9 chemokines. Fusiform cells with fibroblast morphology were also observed as CXCL9 expressers in the dermis of LS patients (52). This observation provides some linkage to the idea that fibrosis in LS is inflammatory driven by chemokine fibroblast stimulation and expression.
Inactive LS sample DEG enrichment groups predominantly relate to ECM formation and dermal restructuring. Previous studies have identified sub-epithelial thickening and deposition of the extracellular matrix (ECM) as common features of scleroderma skin biopsies (53–55). Fibrogenetic transition states have been implicated in fibrosis, especially in autoimmune-mediated diseases, because of the large role these transition states play in connective tissue composition. Mesenchymal cells such as fibroblasts are the predominant source of many ECM proteins, which is particularly true for fibroblasts that have differentiated into a myofibroblast phenotype (56), i.e., alpha smooth muscle actin (α-SMA)-expressing fibroblasts. Myofibroblasts are known to be the primary source of type I and III collagen in fibrotic lesions, and this is thought to be a consequence of a phenotype differentiation that is dependent on stimulation by TGFβ in many fibrotic diseases (57, 58). In our LS samples, transcription factors that are important in regulating the production of factors that control epithelial–mesenchymal interactions, cellular proliferation, and extracellular matrix production (59, 60) such as WNT, ERK, PI3K- TBX, FOX, RUNX, and SRF were demonstrated to be highly expressed. However, TGFβ is distinctly absent in the LS signature in this dataset. As mentioned, TGFβ is thought to be a key regulator for wound healing and other fibrotic diseases, including SSc, a disease that shares some skin characteristic with LS (61). One of the key factors of this pediatric LS dataset is the lack of TGFβ expression in DEGs between different disease activities and subtypes, which may indicate that TGFβ is not the driving pediatric localized scleroderma fibrosis like it is in SSc skin.
The gene signature of the active samples compared to the inactive samples contains genes related to the general inflammatory response or dermal restructuring as seen in the independent active and inactive comparison to healthy samples, but to a lesser degree of expression change and more truncated gene list. Although the expression is lower, genes related to higher pathway functionality can be seen more clearly than in the other comparisons. The KRAS signaling pathway was among the highest enriched groups with increased expression seen in active samples. This supports our findings from the active samples compared to healthy controls and indicates that this signaling pathway might be directly related to active disease, potentially supporting this pathway as a biomarker of the active disease state in LS. A positive correlation of some of these genes with disease activity scores further supports this relationship.
Of the significantly upregulated pathways associating with disease activity, the JAK/STAT pathways are the most prevalent in DEG comparisons between disease and healthy controls and between active and inactive diseases. These pathways are of clinical interest, as medications inhibiting this pathway exist for autoimmune disease. Inhibitors of these pathways were shown to decrease dermal thickness in a mouse model of LS (62) and improvement symptoms of disease, such as erythema, induration, range of motion, and strength in limited human application. Two of the most used JAK inhibitors, tofacitinib (inhibits Jak1/3) and baricitinib (inhibits Jak1/2), were found to be effective in both human patients and scleroderma mouse models, although the direct mechanism is less clear. The inhibitors might directly inhibit excess collagen production by fibroblasts (62). STAT3 and STAT5 are activated in response to growth, stress, and inflammatory stimuli and are critical for the induction of immune response and pro-proliferative genes (63, 64). Additionally, a direct physical interaction between STAT3 and several NF-κB subunits has been shown to act together to regulate the expression of an overlapping group of target genes (including SERPINE1, BCL3, and BCL2), which result in both transactivation and repression depending on the cellular context (65). While this transcriptomic examination cannot pinpoint direct areas of dysregulation in these pathways, it is highly likely that an active disease in LS is directly linked to those identified which can be examined further in future studies.
These findings strongly support unique genetic profiles for active and inactive stages of LS and provide areas in which therapeutic intervention could be better targeted. The predominant inflammatory signature in active LS explains why common systemic therapies largely consisting of methotrexate and corticosteroids (66–70) are reasonably effective in the early stages of disease, although they are associated with significant side effects, limiting tolerability and compliance (71–74). The signaling pathways that increased in active disease compared to inactive disease provide further insight to possible disease propagation and potentially pathogenesis, which could help guide current therapies and provide targets for future approaches. The transcriptome classification system in SSc that was determined by Milano et al., has been used more recently to predict patient response to therapy, such as the inflammatory subset showing better response to mycophenolate mofetil and the fibroproliferative group showing better response to stem cell transplant (75, 76). A methodologically similar classification of LS using immunophenotyping of the transcriptome in a higher number of and more diverse group of patients could help to delineate immunological subtypes and determine therapeutic responses to disease.
This study provides the initial foundation demonstrating different immunophenotypes in LS more based on disease activity status rather than disease subtype as a stronger clustering motif. Although all subtypes and activity levels of LS were captured in our study, which is novel to the existing RNA seq literature, sequencing of additional LS sample numbers is underway, which will help to further define these immunophenotypes.
The results of this study provide two novel aspects: (1) an initial dive into the understanding of the pathways promoting and/or sustaining disease in LS by providing initial skin transcriptomic data across disease states and subtypes in LS and (2) demonstration of the utility of RNA seq in paraffinized skin, opening the door to the study of numerous skin conditions using clinically obtained stored specimens, especially helpful in pediatric skin conditions in which a repeat skin biopsy for fresh tissue for research purposes is usually not accepted well by patients and parents.
The datasets presented in this study can be found online at the NIH GEO repository under GSE 166861.
The studies involving human participants were reviewed and approved by University of Pittsburgh IRB. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
KT obtained funding to perform the study, was in charge of oversight of the study from initiation to completion, reviewed the data and assisted in interpretation, and contributed to manuscript drafting and review. EM contributed to the majority of complex data analyses and summary of results, as well as drafting and editing of the manuscript. JW and RM contributed to additional data analyses and manuscript review. QY, XW, and WC contributed to computational data analysis guidance, experimental design, and manuscript editing. CL and KS contributed to manuscript editing and results presentation. LK provided materials and oversight of RNAScope analyses and manuscript review. All authors contributed to manuscript review and read and approved the final manuscript.
This work was supported by The Nancy Taylor Foundation for Chronic Diseases, Inc. Role: supporting clinical research personnel to analyze the complex RNA sequencing data and shipment charges for skin specimens. The Scleroderma Foundation: Established Investigator Award, The Stephen J Katz, MD Memorial Grant. Role: supporting clinical research personnel to analyze the complex single-cell sequencing data.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to acknowledge the clinical coordinators and the patients who helped provide samples for the analyses in this study. The authors would also like to thank the plastic surgeon, Lorelei Grunwaldt, and plastic surgery physician assistant, Megan Natali, for assistance with skin biopsy procedures and specimens.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.669116/full#supplementary-material
LS, localized scleroderma; SSc, systemic sclerosis; RNAseq, RNA sequencing; IFNγ, interferon γ; IRGS, inflammatory response gene signature; CARRA, Childhood Arthritis and Rheumatology Research Alliance; mLoSSI, modified Localized Scleroderma Skin Severity Index; PGA-A, Physician Global Assessment of Activity; TH1, CD4+ IFNγ + T cells; Tregs, FOXP3+ regulatory T cells; TH17, IL17+ T cells; CXCL9, monokine induced by gamma interferon [MIG]; CXCL10, interferon gamma-induced protein 10 [IP-10]; NGS, next-generation sequencing; FF, fresh-frozen; FFPE, formalin-fixed; paraffin-embedded; NRCOS, National Registry for Childhood Onset Scleroderma; GSEA, gene set enrichment; MMP, matrix metalloproteinases; TIMP, tissue inhibitor of metalloproteinases; IHC, immunohistochemistry; MAC, Morphea in Adult and Children; ECM, extracellular matrix; α-SMA, alpha smooth muscle actin; TGFβ, transforming growth factor beta.
1. Peterson LS, Nelson AM, Su WP, Mason T, O'Fallon WM, Gabriel ES. The epidemiology of morphea (localized scleroderma) in Olmsted County 1960-1993. J Rheumatol. (1997) 24:73–80.
2. Mirizio E, Marathi A, Hershey N, Ross C, Schollaert K, Salgado C, et al. Identifying the signature immune phenotypes present in pediatric localized scleroderma. J Invest Dermatol. (2019) 139:715–8. doi: 10.1016/j.jid.2018.09.025
3. Arkachaisri T, Fertig N, Pino S, Medsger TA Jr. Serum autoantibodies and their clinical associations in patients with childhood- and adult-onset linear scleroderma. A single-center study. J Rheumatol. (2008) 35:2439–44. doi: 10.3899/jrheum.080098
4. Arkachaisri T, Vilaiyuk S, Li S, O'Neil KM, Pope E, Higgins GC, et al. The localized scleroderma skin severity index and physician global assessment of disease activity: a work in progress toward development of localized scleroderma outcome measures. J Rheumatol. (2009) 36:2819–29. doi: 10.3899/jrheum.081284
5. Kelsey CE, Torok SK. The localized scleroderma cutaneous assessment tool: responsiveness to change in a pediatric clinical population. J Am Acad Dermatol. (2013) 69:214–20. doi: 10.1016/j.jaad.2013.02.007
6. Marzano AV, Menni S, Parodi A, Borghi A, Fuligni A, Fabbri P, et al. Localized scleroderma in adults and children. Clinical and laboratory investigations on 239 cases. Eur J Dermatol. (2003) 13:171–6.
7. Wu EY, Li SC, Torok KS, Virkud YV, Fuhlbrigge RC, Rabinovich CE, et al. Baseline description of the juvenile localized scleroderma subgroup from the childhood arthritis and rheumatology research alliance legacy registry. ACR Open Rheumatol. (2019) 1:119–24. doi: 10.1002/acr2.1019
8. Falanga V, Medsger TA Jr, Reichlin M, Rodnan PG. Linear scleroderma. Clinical spectrum, prognosis, laboratory abnormalities. Ann Intern Med. (1986) 104:849–57. doi: 10.7326/0003-4819-104-6-849
9. Christen-Zaech S, Hakim MD, Afsar FS, Paller SA. Pediatric morphea (localized scleroderma): review of 136 patients. J Am Acad Dermatol. (2008) 59:385–96. doi: 10.1016/j.jaad.2008.05.005
10. Torok KS, Li SC, Jacobe HM, Taber SF, Stevens AM, Zulian F, et al. Immunopathogenesis of pediatric localized scleroderma. Front Immunol. (2019) 10:908. doi: 10.3389/fimmu.2019.00908
11. O'Brien JC, Rainwater YB, Malviya N, Cyrus N, Auer-Hackenberg L, Hynan LS, et al. Transcriptional and cytokine profiles identify CXCL9 as a biomarker of disease activity in morphea. J Invest Dermatol. (2017) 137:1663–70. doi: 10.1016/j.jid.2017.04.008
12. Jones W, Greytak S, Odeh H, Guan P, Powers J, Bavarva J, et al. Deleterious effects of formalin-fixation and delays to fixation on RNA and miRNA-Seq profiles. Sci Rep. (2019) 9:6980. doi: 10.1038/s41598-019-43282-8
13. Li J, Fu C, Speed TP, Wang W, Symmans WF. Accurate RNA sequencing from formalin-fixed cancer tissue to represent high-quality transcriptome from frozen tissue. JCO Precis Oncol. (2018) 2018:PO.17.00091. doi: 10.1200/PO.17.00091
14. Jovanovic B, Sheng Q, Seitz RS, Lawrence KD, Morris SW, Thomas LR, et al. Comparison of triple-negative breast cancer molecular subtyping using RNA from matched fresh-frozen versus formalin-fixed paraffin-embedded tissue. BMC Cancer. (2017) 17:241. doi: 10.1186/s12885-017-3237-1
15. Cieslik M, Chugh R, Wu YM, Wu M, Brennan C, Lonigro R, et al. The use of exome capture RNA-seq for highly degraded RNA with application to clinical cancer sequencing. Genome Res. (2015) 25:1372–81. doi: 10.1101/gr.189621.115
16. Conroy JM, Pabla S, Glenn ST, Burgher B, Nesline M, Papanicolau-Sengos A, et al. Analytical validation of a next-generation sequencing assay to monitor immune responses in solid tumors. J Mol Diagn. (2018) 20:95–109. doi: 10.1016/j.jmoldx.2017.10.001
17. Abramovitz M, Ordanic-Kodani M, Wang Y, Li Z, Catzavelos C, Bouzyk M, et al. Optimization of RNA extraction from FFPE tissues for expression profiling in the DASL assay. Biotechniques. (2008) 44:417–23. doi: 10.2144/000112703
18. Esteve-Codina A, Arpi O, Martinez-Garcia M, Pineda E, Mallo M, Gut M, et al. A comparison of RNA-Seq results from paired formalin-fixed paraffin-embedded and fresh-frozen glioblastoma tissue samples. PLoS ONE. (2017) 12:e0170632. doi: 10.1371/journal.pone.0170632
19. Hedegaard J, Thorsen K, Lund MK, Hein AM, Hamilton-Dutoit SJ, Vang S, et al. Next-generation sequencing of RNA and DNA isolated from paired fresh-frozen and formalin-fixed paraffin-embedded samples of human cancer and normal tissue. PLoS ONE. (2014) 9:e98187. doi: 10.1371/journal.pone.0098187
20. Kissel HD, Paulson TG, Liu K, Li X, Swisher E, Garcia R, et al. Feasibility of RNA and DNA extraction from fresh pipelle and archival endometrial tissues for use in gene expression and SNP arrays. Obstet Gynecol Int. (2013) 2013:576842. doi: 10.1155/2013/576842
21. Vukmirovic M, Herazo-Maya JD, Blackmon J, Skodric-Trifunovic V, Jovanovic D, Pavlovic S, et al. Identification and validation of differentially expressed transcripts by RNA-sequencing of formalin-fixed, paraffin-embedded (FFPE) lung tissue from patients with idiopathic pulmonary fibrosis. BMC Pulm Med. (2017) 17:15. doi: 10.1186/s12890-016-0356-4
22. Zhao W, He X, Hoadley KA, Parker JS, Hayes DN, Perou MC. Comparison of RNA-Seq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genomics. (2014) 15:419. doi: 10.1186/1471-2164-15-419
23. Carrick DM, Mehaffey MG, Sachs MC, Altekruse S, Camalier C, Chuaqui R, et al. Robustness of next generation sequencing on older formalin-fixed paraffin-embedded tissue. PLoS ONE. (2015) 10:e0127353. doi: 10.1371/journal.pone.0127353
24. Condie D, Grabell D, Jacobe H. Comparison of outcomes in adults with pediatric-onset morphea and those with adult-onset morphea: a cross-sectional study from the morphea in adults and children cohort. Arthritis Rheumatol. (2014) 66:3496–504. doi: 10.1002/art.38853
25. Leitenberger JJ, Cayce RL, Haley RW, Adams-Huet B, Bergstresser PR, Jacobe TH. Distinct autoimmune syndromes in morphea: a review of 245 adult and pediatric cases. Arch Dermatol. (2009) 145:545–550. doi: 10.1001/archdermatol.2009.79
26. Lis-Swiety A, Skrzypek-Salamon A, Ranosz-Janicka I, Brzezinska-Wcislo L. Localized scleroderma: clinical and epidemiological features with emphasis on adulthood- versus childhood-onset disease differences. J Eur Acad Dermatol Venereol. (2017) 31:1595–603. doi: 10.1111/jdv.14197
27. Matsubara T, Soh J, Morita M, Uwabo T, Tomida S, Fujiwara T, et al. DV200 index for assessing RNA integrity in next-generation sequencing. Biomed Res Int. (2020) 2020:9349132. doi: 10.1155/2020/9349132
28. Sommer A, Gambichler T, Bacharach-Buhles M, von Rothenburg T, Altmeyer P, Kreuter A. Clinical and serological characteristics of progressive facial hemiatrophy: a case series of 12 patients. J Am Acad Dermatol. (2006) 54:227–33. doi: 10.1016/j.jaad.2005.10.020
29. Kalmar A, Wichmann B, Galamb O, Spisak S, Toth K, Leiszter K, et al. Gene-expression analysis of a colorectal cancer-specific discriminatory transcript set on formalin-fixed, paraffin-embedded (FFPE) tissue samples. Diagn Pathol. (2015) 10:126. doi: 10.1186/s13000-015-0363-4
30. Xie R, Chung JY, Ylaya K, Williams RL, Guerrero N, Nakatsuka N, et al. Factors influencing the degradation of archival formalin-fixed paraffin-embedded tissue sections. J Histochem Cytochem. (2011) 59:356–65. doi: 10.1369/0022155411398488
31. Bibikova M, Talantov D, Chudin E, Yeakley JM, Chen J, Doucet D, et al. Quantitative gene expression profiling in formalin-fixed, paraffin-embedded tissues using universal bead arrays. Am J Pathol. (2004) 165:1799–807. doi: 10.1016/S0002-9440(10)63435-9
32. Iddawela M, Rueda OM, Klarqvist M, Graf S, Earl HM, Caldas C. Reliable gene expression profiling of formalin-fixed paraffin-embedded breast cancer tissue (FFPE) using cDNA-mediated annealing, extension, selection, and ligation whole-genome (DASL WG) assay. BMC Med Genomics. (2016) 9:54. doi: 10.1186/s12920-016-0215-4
33. Byrum S, Avaritt NL, Mackintosh SG, Munkberg JM, Badgwell BD, Cheung WL, et al. A quantitative proteomic analysis of FFPE melanoma. J Cutan Pathol. (2011) 38:933–6. doi: 10.1111/j.1600-0560.2011.01761.x
34. Danczak-Pazdrowska A, Polanska A, Synakiewicz J, Gurgul E, Molinska-Glura M, Ruchala M. Morphea and antithyroid antibodies. Postepy Dermatol Alergol. (2018) 35:470–3. doi: 10.5114/ada.2018.75839
35. Budzynska-Wlodarczyk J, Michalska-Jakubus MM, Kowal M, Krasowska D. Evaluation of serum concentrations of the selected cytokines in patients with localized scleroderma. Postepy Dermatol Alergol. (2016) 33:47–51. doi: 10.5114/pdia.2015.48044
36. Ihn H, Sato S, Fujimoto M, Kikuchi K, Takehara K. Demonstration of interleukin-2, interleukin-4 and interleukin-6 in sera from patients with localized scleroderma. Arch Dermatol Res. (1995) 287:193–7. doi: 10.1007/BF01262331
37. Uziel Y, Feldman BM, Krafchik BR, Yeung RS, Laxer MR. Methotrexate and corticosteroid therapy for pediatric localized scleroderma. J Pediatr. (2000) 136:91–5. doi: 10.1016/S0022-3476(00)90056-8
38. Kurzinski K, Torok SK. Cytokine profiles in localized scleroderma and relationship to clinical features. Cytokine. (2011) 55:157–64. doi: 10.1016/j.cyto.2011.04.001
39. Magee KE, Kelsey CE, Kurzinski KL, Ho J, Mlakar LR, Feghali-Bostwick CA, et al. Interferon-gamma inducible protein-10 as a potential biomarker in localized scleroderma. Arthritis Res Ther. (2013) 15:R188. doi: 10.1186/ar4378
40. Torok KS, Kurzinski K, Kelsey C, Yabes J, Magee K, Vallejo AN, et al. Peripheral blood cytokine and chemokine profiles in juvenile localized scleroderma: T-helper cell-associated cytokine profiles. Semin Arthritis Rheum. (2015) 45:284–93. doi: 10.1016/j.semarthrit.2015.06.006
41. Florez-Pollack S, Kunzler E, Jacobe TH. Morphea: current concepts. Clin Dermatol. (2018) 36:475–86. doi: 10.1016/j.clindermatol.2018.04.005
42. Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS ONE. (2008) 3:e2696. doi: 10.1371/journal.pone.0002696
43. Chen JT, Eisinger B, Esquibel C, Poore SO, Eliceiri K, Siebert WJ. Changes in cutaneous gene expression after microvascular free tissue transfer in parry-romberg syndrome. Plast Reconstr Surg. (2018) 142:303e–9e. doi: 10.1097/PRS.0000000000004638
44. Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou FH. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res. (2015) 35:600–4. doi: 10.3109/10799893.2015.1030412
45. Zayoud M, Marcu-Malina V, Vax E, Jacob-Hirsch J, Elad-Sfadia G, Barshack I, et al. Ras signaling inhibitors attenuate disease in adjuvant-induced arthritis via targeting pathogenic antigen-specific Th17-type cells. Front Immunol. (2017) 8:799. doi: 10.3389/fimmu.2017.00799
46. Lee SE, Li X, Kim JC, Lee J, Gonzalez-Navajas JM, Hong SH, et al. Type I interferons maintain Foxp3 expression and T-regulatory cell functions under inflammatory conditions in mice. Gastroenterology. (2012) 143:145–54. doi: 10.1053/j.gastro.2012.03.042
47. Wang Z, Hong J, Sun W, Xu G, Li N, Chen X, et al. Role of IFN-gamma in induction of Foxp3 and conversion of CD4+ CD25- T cells to CD4+ Tregs. J Clin Invest. (2006) 116:2434–41. doi: 10.1172/JCI25826
48. Wang Z, Zheng Y, Hou C, Yang L, Li X, Lin J, et al. DNA methylation impairs TLR9 induced Foxp3 expression by attenuating IRF-7 binding activity in fulminant type 1 diabetes. J Autoimmun. (2013) 41:50–9. doi: 10.1016/j.jaut.2013.01.009
49. Lu N, Malemud JC. Extracellular signal-regulated kinase: a regulator of cell growth, inflammation, chondrocyte and bone cell receptor-mediated gene expression. Int J Mol Sci. (2019) 20:3792. doi: 10.3390/ijms20153792
50. Sohara N, Trojanowska M, Reuben A. Oncostatin M stimulates tissue inhibitor of metalloproteinase-1 via a MEK-sensitive mechanism in human myofibroblasts. J Hepatol. (2002) 36:191–9. doi: 10.1016/S0168-8278(01)00265-3
51. Trojanowska M. Molecular aspects of scleroderma. Front Biosci. (2002) 7:d608–18. doi: 10.2741/A798
52. Walker D, Susa JS, Currimbhoy S, Jacobe H. Histopathological changes in morphea and their clinical correlates: results from the morphea in adults and children cohort V. J Am Acad Dermatol. (2017) 76:1124–30. doi: 10.1016/j.jaad.2016.12.020
53. Gilbane AJ, Denton CP, Holmes MA. Scleroderma pathogenesis: a pivotal role for fibroblasts as effector cells. Arthritis Res Ther. (2013) 15:215. doi: 10.1186/ar4230
54. Burgess JK, Mauad T, Tjin G, Karlsson JC, Westergren-Thorsson G. The extracellular matrix - the under-recognized element in lung disease? J Pathol. (2016) 240:397–409. doi: 10.1002/path.4808
55. Yang L, Serada S, Fujimoto M, Terao M, Kotobuki Y, Kitaba S, et al. Periostin facilitates skin sclerosis via PI3K/Akt dependent mechanism in a mouse model of scleroderma. PLoS ONE. (2012) 7:e41994. doi: 10.1371/journal.pone.0041994
56. Kis K, Liu X, Hagood SJ. Myofibroblast differentiation and survival in fibrotic disease. Expert Rev Mol Med. (2011) 13:e27. doi: 10.1017/S1462399411001967
57. Phan SH. Biology of fibroblasts and myofibroblasts. Proc Am Thorac Soc. (2008) 5:334–37. doi: 10.1513/pats.200708-146DR
58. Petrov VV, Fagard RH, Lijnen JP. Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension. (2002) 39:258–63. doi: 10.1161/hy0202.103268
59. Angel P, Szabowski A. Function of AP-1 target genes in mesenchymal-epithelial cross-talk in skin. Biochem Pharmacol. (2002) 64:949–56. doi: 10.1016/S0006-2952(02)01158-9
60. Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. (2002) 4:E131–6. doi: 10.1038/ncb0502-e131
61. Frangogiannis N. Transforming growth factor-beta in tissue fibrosis. J Exp Med. (2020) 217:e20190103. doi: 10.1084/jem.20190103
62. Damsky W, Patel D, Garelli CJ, Garg M, Wang A, Dresser K, et al. Jak inhibition prevents bleomycin-induced fibrosis in mice and is effective in patients with morphea. J Invest Dermatol. (2020) 140:1446–9.e4. doi: 10.1016/j.jid.2019.12.019
63. Seif F, Khoshmirsafa M, Aazami H, Mohsenzadegan M, Sedighi G, Bahar M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signal. (2017) 15:23. doi: 10.1186/s12964-017-0177-y
64. O'Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. (2012) 36:542–50. doi: 10.1016/j.immuni.2012.03.014
65. Martincuks A, Andryka K, Kuster A, Schmitz-Van de Leur H, Komorowski M, Muller-Newen G. Nuclear translocation of STAT3 and NF-kappaB are independent of each other but NF-kappaB supports expression and activation of STAT3. Cell Signal. (2017) 32:36–47. doi: 10.1016/j.cellsig.2017.01.006
66. Kreuter A, Gambichler T, Breuckmann F, Rotterdam S, Freitag M, Stuecker M, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol. (2005) 141:847–52. doi: 10.1001/archderm.141.7.847
67. Weibel L, Sampaio MC, Visentin MT, Howell KJ, Woo P, Harper IJ. Evaluation of methotrexate and corticosteroids for the treatment of localized scleroderma (morphoea) in children. Br J Dermatol. (2006) 155:1013–20. doi: 10.1111/j.1365-2133.2006.07497.x
68. Torok KS, Arkachaisri T. Methotrexate and corticosteroids in the treatment of localized scleroderma: a standardized prospective longitudinal single-center study. J Rheumatol. (2012) 39:286–94. doi: 10.3899/jrheum.110210
69. Zulian F, Athreya BH, Laxer R, Nelson AM, Feitosa de Oliveira SK, Punaro MG, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. An international study. Rheumatology. (2006) 45:614–20. doi: 10.1093/rheumatology/kei251
70. Zulian F, Vallongo C, Patrizi A, Belloni-Fortina A, Cutrone M, Alessio M, et al. A long-term follow-up study of methotrexate in juvenile localized scleroderma (morphea). J Am Acad Dermatol. (2012) 67:1151–56. doi: 10.1016/j.jaad.2012.03.036
71. Zulian F, Martini G, Vallongo C, Vittadello F, Falcini F, Patrizi A, et al. Methotrexate treatment in juvenile localized scleroderma: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. (2011) 63:1998–2006. doi: 10.1002/art.30264
72. Bulatovic M, Heijstek MW, Verkaaik M, van Dijkhuizen EH, Armbrust W, Hoppenreijs EP, et al. High prevalence of methotrexate intolerance in juvenile idiopathic arthritis: development and validation of a methotrexate intolerance severity score. Arthritis Rheum. (2011) 63:2007–13. doi: 10.1002/art.30367
73. van Dijkhuizen EH, Bulatovic Calasan M, Pluijm SM, de Rotte MC, Vastert SJ, Kamphuis S, et al. Prediction of methotrexate intolerance in juvenile idiopathic arthritis: a prospective, observational cohort study. Pediatr Rheumatol Online J. (2015) 13:5. doi: 10.1186/s12969-015-0002-3
74. Fatimah N, Salim B, Nasim A, Hussain K, Gul H, Niazi S. Frequency of methotrexate intolerance in rheumatoid arthritis patients using methotrexate intolerance severity score (MISS questionnaire). Clin Rheumatol. (2016) 35:1341–5. doi: 10.1007/s10067-016-3243-8
75. Franks JM, Martyanov V, Wang Y, Wood TA, Pinckney A, Crofford LJ, et al. Machine learning predicts stem cell transplant response in severe scleroderma. Ann Rheum Dis. (2020) 79:1608–15. doi: 10.1136/annrheumdis-2020-217033
Keywords: localized scleroderma, morphea, pediatric rheumatology, bulk RNA sequencing, inflammation
Citation: Mirizio E, Liu C, Yan Q, Waltermire J, Mandel R, Schollaert KL, Konnikova L, Wang X, Chen W and Torok KS (2021) Genetic Signatures From RNA Sequencing of Pediatric Localized Scleroderma Skin. Front. Pediatr. 9:669116. doi: 10.3389/fped.2021.669116
Received: 18 February 2021; Accepted: 12 April 2021;
Published: 07 June 2021.
Edited by:
Erkan Demirkaya, Western University, CanadaReviewed by:
Ozgur Kasapcopur, Istanbul University-Cerrahpasa, TurkeyCopyright © 2021 Mirizio, Liu, Yan, Waltermire, Mandel, Schollaert, Konnikova, Wang, Chen and Torok. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathryn S. Torok, S2F0aHJ5bi50b3Jva0BjaHAuZWR1; orcid/org.0000-0002-1662-143X
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.