 Zehong Lin
Zehong Lin Jinliang Li
Jinliang Li Taoyun Ji
Taoyun Ji Ye Wu
Ye Wu Yuwu Jiang
Yuwu Jiang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 21 April 2021
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.657256
Background: ATP1A1 encodes an α1 isoform of Na+/K+-ATPase, which is expressed abundantly in kidneys and central nervous system. ATP1A1 variants may cause Na+/K+-ATPase loss of function and lead to a wide spectrum of phenotypes. This study aims to summarize the clinical and genetic features of ATP1A1 de novo mutation-related disorders and explore the potential correlations between phenotypes and genotypes.
Methods: We analyzed two new cases harboring novel de novo ATP1A1 variants and reviewed all reported cases.
Results: Both our probands had developmental delay, patient 1 accompanied with sleep disorders, irritability, and patient 2 with refractory seizures. They each had a novel de novo heterozygous missense variant, c.2797G>A[p.Asp933Asn] (NM_000701) and c.2590G>A[p.Gly864Arg] (NM_000701) respectively. Four patients with de novo ATP1A1 variants have been reported in two previous papers. Among them, three patients had refractory seizures and one patient had complex hereditary spastic paraplegia (HSP). Therefore, all six patients had developmental delay, and four of them had epilepsy. All variants located in the transmembrane regions M3, M4, M7, and M8 of ATP1A1 protein. Four patients with mutations in M3 and M7 had more severe phenotypes, including developmental delay and epileptic encephalopathy, three of them with hypomagnesemia, whereas two patients with mutations in M4 and M8 had milder phenotypes, only with mild developmental delay, without seizures or hypomagnesemia. Correcting hypomagnesemia had not controlled those seizures.
Conclusions: Two novel de novo ATP1A1 variants identified in two patients here enriched the genotypic and phenotypic spectrum of ATP1A1 mutation-related disorder. Our findings suggest that hypomagnesemia in this disorder might relate to more severe phenotype and indicate more severe Na+/K+-ATPase dysfunction. Variations in M3 and M7 transmembrane regions were related to more severe phenotype than those in M4 and M8, which suggested that variations in M3 and M7 might cause more severe ATP1A1 functional defect.
Na+/K+-ATPase is an integral membrane protein of a heterodimeric enzyme containing four isoforms of an α subunit (α1–α4) and three isoforms of a β subunit (β1–β3) (1). It can extrude three Na+ ions out of the cell and import two K+ ions into the cell using energy from the hydrolysis of one ATP in each pump cycle (2). It plays an essential role in maintaining the sodium and potassium ion concentration gradients at the plasma membrane of a cell, which are necessary for osmotic regulation, sodium-coupled transport, and the electrical excitability of nerves and muscles (1, 3).
ATP1A1 encodes a transmembrane protein of 1,023 amino acids which is an α1 isoform of Na+/K+-ATPase. It locates in the human chromosome 1 region 1p2l → cen and practically ubiquitously expressed in body, especially in the kidneys and central nervous system (CNS) (1, 4). ATP1A1 protein plays an important role in cation transport and responsible for establishing and maintaining the ion electrochemical gradients of Na+/K+ across the cell membrane. ATP1A1 variants may cause Na+/K+-ATPase loss of function and abnormal cation permeability, leading to membrane depolarization and impairment of the activity of the physiological pump and thus to diseases (5). Previous studies have reported a range of diseases related to ATP1A1 mutations, including renal hypomagnesemia, refractory epilepsy, intellectual disability, Charcot-Marie-Tooth disease (CMT), HSP, and aldosterone adenoma (3, 6–12). However, the phenotypes related to ATP1A1 de novo mutations were different from those with inherited mutations and have not been well-analyzed.
This study aimed to summarize the clinical and genetic features of ATP1A1 de novo mutation-related disorders and reveal the potential correlations between phenotypes and genotypes, including the two new cases we diagnosed and four previously reported cases.
We evaluated two Chinese patients harboring de novo ATP1A1 pathogenic variants. Their clinical manifestations, serum Mg+ concentrations, brain magnetic resonance imaging (MRI), electroencephalogram (EEG), and ATP1A1 variants were analyzed. Furthermore, all four previously reported cases with de novo ATP1A1 mutations were also analyzed together in this study.
Five milliliters of peripheral venous blood from each of the probands and their parents was collected, and genomic DNA was extracted for whole exome sequencing (WES) of family trios. The in silico tools PolyPhen-2, SIFT, and Mutation Taster and American College of Medical Genetics and Genomics (ACMG) guidelines were used to assess the pathogenicity of the two candidate variants (13).
Patient 1 was a girl aged 9 years and 5 months, with a de novo ATP1A1 c.2797G>A [p.Asp933Asn] variant. Her motor development was normal. Her Wechsler Intelligence Scale for Children showed a Full Scale IQ (FSIQ) score of 71 at 7.5 years old, in the borderline range. She had poor executive functioning, and her learning ability, logical ability, and memory were significantly inferior to those of her peers. She began to experience sleep disorders when she was 2 years old, which manifested as waking up suddenly and crying after 3–4 h of sleep every night and the sleep disorder has relieved since her 8 years of age. In addition, she had poor emotional control, as easy to emotional collapse and cry constantly, but no epilepsy. Her brain MRI and serum Mg2+ concentrations were normal. Her birth history and family history were normal.
Patient 2 was a boy aged 2 years and 10 months, with a de novo ATP1A1 c.2590 G>A[p.Gly864Arg] variant. He had severe developmental delay, manifesting as sitting independently but unstably, he couldn't say any words and only made a babbling sound by the time of our follow-up. He began to develop epileptic seizures 5 months after birth, characterized by a tonic seizure in one limb and, later, evolving into generalized tonic–clonic seizures. EEG showed widespread slow waves and spike slow waves during sleep and wide 3–4 Hz slow waves, especially in para-midline and midline regions, during waking and sleeping stages (Figure 1). He was diagnosed as developmental and epileptic encephalopathy. His brain MRI and serum Mg2+ concentrations were normal. The child has taken a total of three antiepileptic drugs including diazepam, levetiracetam, and topiramate, the latter two are currently used, but the seizures have not been controlled. He was born at 36 6/7 weeks of gestation with normal delivery. His family history was unremarkable.
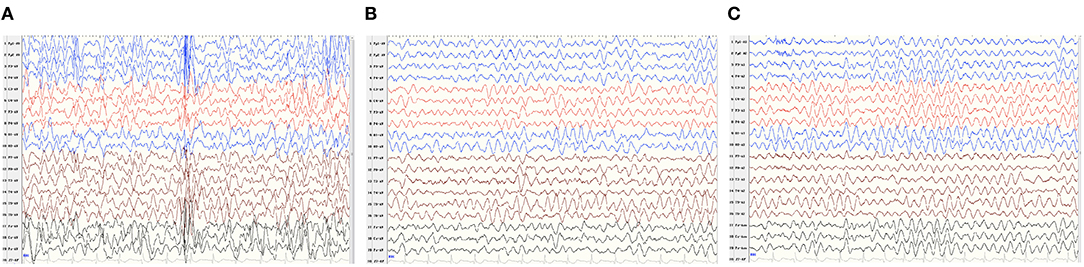
Figure 1. V-EEG: Widespread slow waves and spike slow waves during sleep and 3–4 Hz slow waves, especially in para-midline and midline regions, during waking, and sleeping periods. (A–C) EEG features described as the caption of Patient 2.
Both patients were identified de novo variants of ATP1A1. Patient 1 was found to have c.2797G>A[p.Asp933Asn] and patient 2 was found to have c.2590 G>A[p.Gly864Arg]; variants were de novo in both cases (Figure 1). Sequence alignment among multiple species suggested that p.Asp933Asn and p.Gly864Arg were located in highly conserved sites (Figure 2). Variants were predicted to be pathogenic by multiple bioinformatic tools (e.g., PolyPhen-2, SIFT, and Mutant Taster. In addition, they were not included in genomAD, ExAC, and 1000 genomes. According to ACMG guidelines, we confirmed that both variants are likely pathogenic (see Table 1 for details). Furthermore, we did not find other pathogenic variants of genes known to be associated with CNS-related diseases in the two probands using Trio-WES.
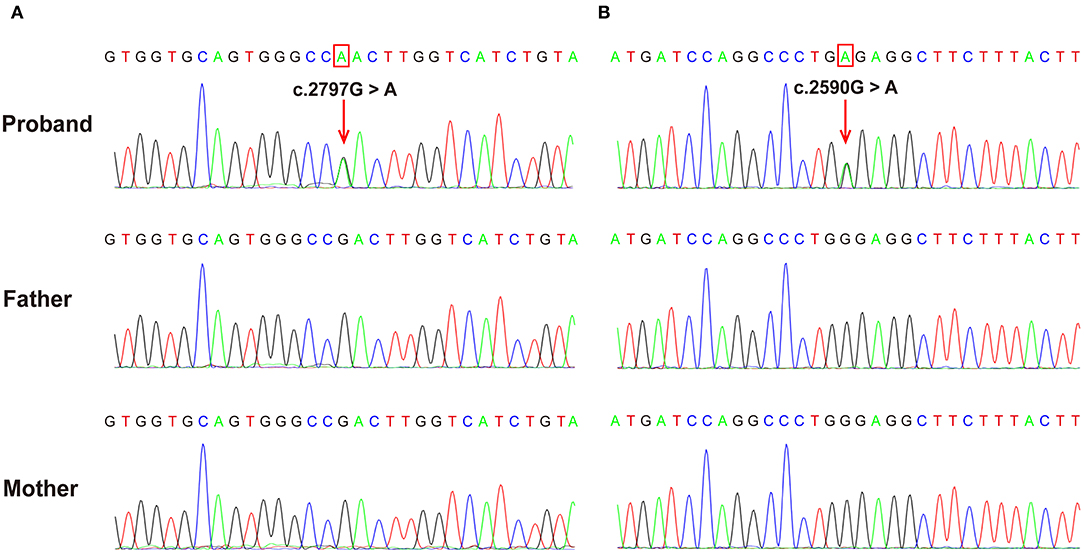
Figure 2. The de novo ATP1A1 variants were confirmed using Sanger sequencing. (A) c.2797G>A variant in case 1 resulting in a p.Asp933Asn substitution. (B) c.2590G>A variant in case 2 resulting in a p.Gly864Arg substitution. Verified as de novo variants using Trio-WES and Sanger sequencing.

Table 1. Pathogenic prediction in ATP1A1 variants.
We searched PubMed with the keywords “ATP1A1” and “de novo.” A total of four children with de novo variants in ATP1A1 were identified.
Schlingmann et al. (9) first described three patients harboring the de novo variations c.905T>C[p.Leu302Arg], c.907G>C[p.Gly303Arg], and c.2576T>G[p.Met859Arg] (NM_000701) in ATP1A1, involving renal hypomagnesemia, refractory epilepsy, and intellectual disability[HOMGSMR2, MIM618314] (Table 2, cases 3, 4, and 5). The onset ages of the three patients ranged from 6 days to 6 months. The first symptoms were generalized seizures and severe hypomagnesemia. All patients had received antiepileptic drugs and magnesium supplementation, but seizures continued. Moreover, all three patients involved severe generalized developmental delay. Among them, two patients (Table 2, cases 4 and 5) also showed additional clinical features consistent with autism spectrum disorders. Two patients underwent brain MRI, which both showed cerebral atrophy. None of the children's parents had any related clinical manifestations or hypomagnesemia.
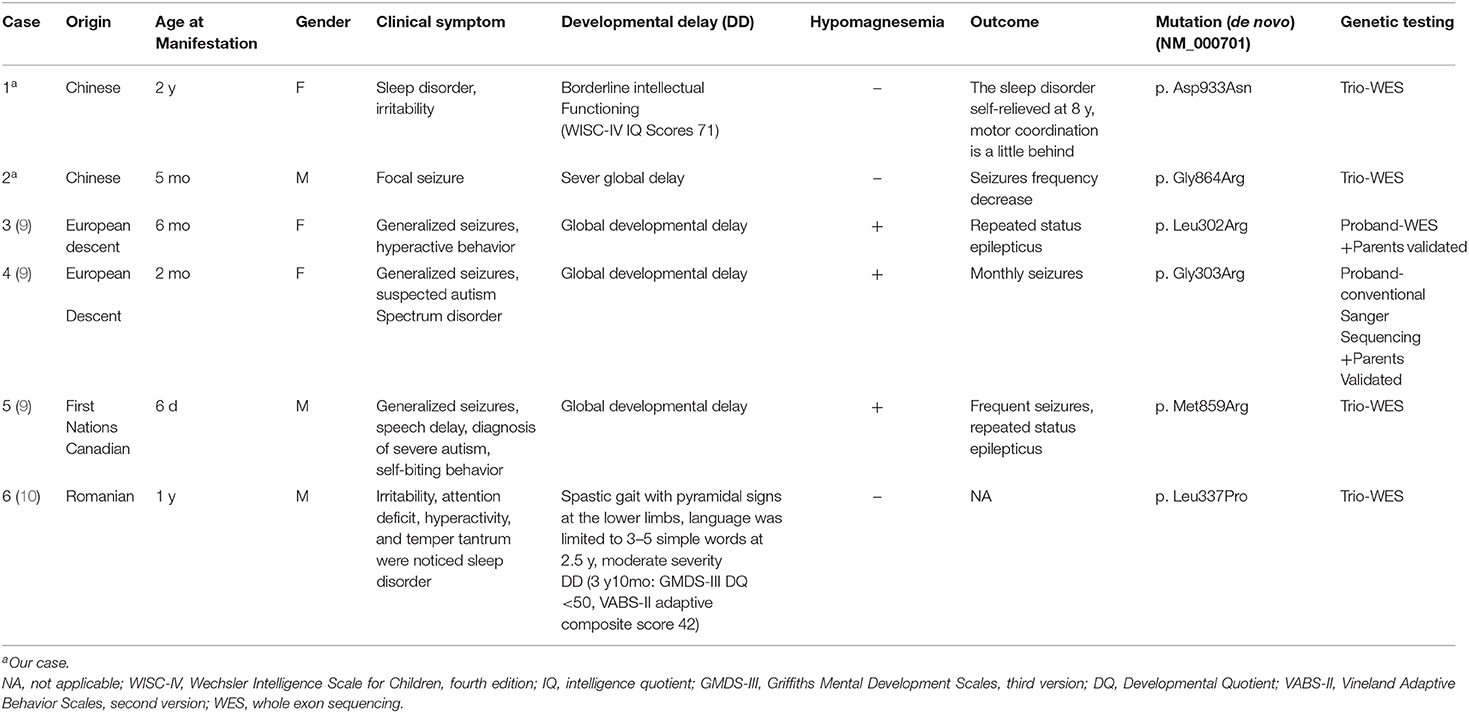
Table 2. Clinical features of patients with ATP1A1 mutations.
Stregapede et al. (10) reported a de novo ATP1A1 mutation-related HSP, with the c.1010 T>C[p.Leu337Pro] variant (NM_000701) (Table 2, case 6). The patient was 4 years and 6 months old. He did not have epilepsy, but exhibited motor developmental delay in his first year of life. He could sit independently at the age of 12 months, walk at 18 months with an abnormal gait and frequent falls, and speak 3–5 simple words at 2 years and 6 months; his eye contact was normal. In addition, attention-deficit hyperactivity disorder, temper tantrums, and sleep disorders were identified in the patient. Both MRI (brain and spine) and serum Mg2+ concentrations were normal. Neurologic examination revealed pyramidal signs at the lower limbs. Motor evoked potential revealed that the central conduction time at the lower limbs was increased. EEG and nerve conduction study of the upper and lower limbs were inconclusive. Neuropsychological assessments suggested moderate to severe developmental delay at 3 years and 10 months (Griffiths Mental Development Scales, third version: developmental quotient <50 and developmental age equivalent to 29 months; Vineland Adaptive Behavior Scales, second version: adaptive composite score 42).
In brief, including the two cases we reported here, there have been six patients with de novo ATP1A1 mutations identified to date. The pathogenic ATP1A1 variants in these patients were p.Asp933Asn, p.Gly864Arg, p.Leu302Arg, p.Gly303Arg, p.Met859Arg, and p.Leu337Pro (Figure 3). All patients had different degrees of developmental delay, and four had epilepsy. Of the four patients with epilepsy, three had generalized epileptic seizures and hypomagnesemia, and one patient had focal epileptic seizure without hypomagnesemia. Antiepileptic medicine or magnesium supplementation cannot control epileptic seizures in all these patients.

Figure 3. Multiple species sequence alignment. Conservation analysis of amino acid sequences on p.Asp933Asn and p.Asp933Asn variant sites demonstrated that they were located on highly conserved sites. The bold red letters mean the amino acids that represent the same location in different species are all the same, indicated that they are highly conserved. (A) p.Asp933Asn variant in case 1, (B) p.Gly864Arg variant in case 2.
The ATP1A1 protein is composed of ten transmembrane helices and three cytoplasmic domains: A (actuator), N (nucleotide-binding), and P (phosphorylation) (14). All de novo ATP1A1 variants located in the regions encoding transmembrane regions M3, M4, M7, and M8 of the ATP1A1 protein (Figure 4), which suggests that these regions may be susceptible to de novo ATP1A1 mutation. Interestingly, the amino acid residues in M3 and M7 (two cases each: M3: p.Leu302Arg, p.Gly303Arg; M7: p.Gly864Arg, p.Met859Arg) were all changed to arginine. If a mutation introduces an arginine into the transmembrane region, this positively charged residue may interfere with the negatively charged ion binding sites of the pump, and generally, a charged residue is unfavorable in the transmembrane region. Such mutations are therefore more likely to result in pump dysfunction, either because they directly disrupt binding and transport of sodium and/or potassium, or because they disturb the overall structure.
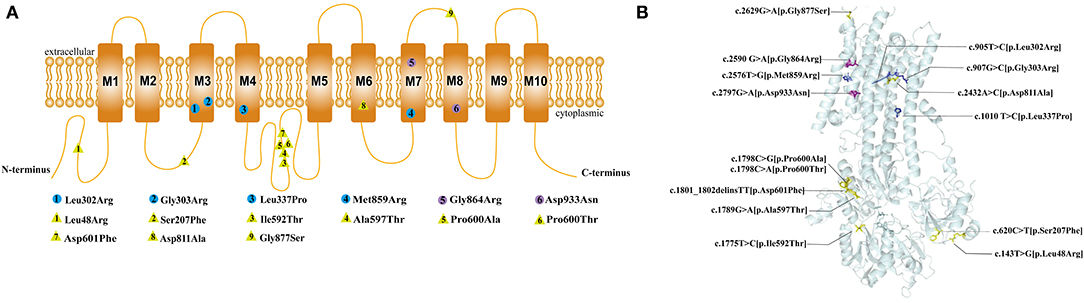
Figure 4. Location of the pathogenic variants in ATP1A1 protein. (A) 2D structure: The circles represent the de novo variants, with purple filled representing the novel variants that we found and blue filled representing the previously reported variants. The yellow triangles represent inherited variants associated with CMT previously reported. (B) 3Dstructure: Location of all the pathogenic variants in ATP1A1 protein. All the de novo mutations were concentrated in the transmembrane region, whereas 88.9% (8/9) of inherited variants concentrated in the topological regions, and 11.1% (1/9) locate in the transmembrane regions.
To date, the phenotype spectrum of de novo ATP1A1 pathogenic variants has included neurodevelopmental disorders (including developmental delay, autism spectrum disorders, and epilepsy), emotional disorders, sleep disorders, and HSP. It is suggested that ATP1A1 mutation-related disorders may have a wide spectrum of phenotypes, which may be related to the extensive expression and important functions of ATP1A1 in the nervous system (1, 4, 5). Among neurodevelopmental disorders, four patients (age from 2 months to 10 years) had generalized developmental delay, and one patient with HSP had significant language retardation. Four patients (4/6) were diagnosed with epilepsy, including three patients with generalized epileptic seizure and one patient with focal seizure; the generalized seizure patients featured hypomagnesemia, while the focal seizure patient did not. Seizures were not controlled after combined treatments of several antiepileptic drugs and magnesium supplementation. EEG features of ATP1A1 mutation related epilepsy were not described in previous reports. Patient 2 exhibited focal epileptic seizure with severe developmental delay, and MRI showed normal brain structure. His EEG showed the features of genetic developmental and epileptic encephalopathy.
The mechanism of hypomagnesemia caused by ATP1A1 variants is associated with dysfunction of renal tubular Na+/K+-ATPase. The Na+/K+-ATPase expressed on the outside of the renal tubule basement membrane establishes a favorable electrochemical gradient for the influx of cations through the magnesium ion channel and provides an exit mechanism for reabsorbed sodium ions. The distal convoluted tubule mediates active transcellular magnesium transport and has the highest Na+/K+-ATPase density and activity; ATP1A1 encodes the most prevalent form of Na+/K+-ATPase in the distal convoluted tubule (9). Therefore, ATP1A1 variants may lead to Na+ and K+ transport dysfunction and interfere with the active reabsorption of magnesium ions, resulting in hypomagnesemia. In our cases, we found that seizures were not controlled after magnesium supplementation. This supported that epilepsy in these patients might be a direct result of neuronal energy dysfunction caused by Na+/K+-ATPase dysfunction in the CNS rather than a secondary feature of hypomagnesemia (9).
Hypomagnesemia in the patients with ATP1A1 mutations may indicate more severe Na+/K+-ATPase dysfunction, because the epileptic phenotype occurs more often in individuals with hypomagnesemia. All three patients with hypomagnesemia had refractory epilepsy, whereas among the three patients without hypomagnesemia, only one had refractory focal epilepsy, and the other two did not have seizures. Four patients with epilepsy had significant language delay. Among them, three patients (age from 4 to 10 years) could only speak a single word; one patient, aged 2 years and 10 months, could only make a babbling sound. The two patients without epilepsy had less developmental delay, one of them had mild language delay, and the other had normal language development.
Four patients with mutations in M3 and M7 (two cases each) had more severe phenotypes, which manifested as developmental delay and epileptic encephalopathy; three of these patients featured hypomagnesemia. However, two other patients with mutations in M4 and M8 had less severe phenotypes, which mainly manifested as mild developmental delay, sleep disorders, emotional disorders, HSP, and a lack of seizures or hypomagnesemia. This suggests that variations in M3 and M7 may have a greater detrimental impact on ATP1A1 function and lead to more severe phenotypes.
In contrast to patients with de novo variants, patients with inherited variants are mainly presented CMT. Eight patients had familiar ATP1A1 mutation-related autosomal dominant CMT, one of them inherited the mutation from his mother with mosaic mutation (3, 8). The inheritance mode of one case with CMT could not be confirmed due to lacking of the father's genetic test (3). The age of onset ranged from 8 to 50 years old, and the mutations mainly caused weakness and atrophy of the distal extremities and sensory disturbances rather than seizures. No information about the blood magnesium test were found in those previous reports. These mutations were mainly concentrated in the topological regions of ATP1A1 (88.9%, 8/9), and one (11.1%, 1/9) located at the transmembrane region, suggesting that topological regions may have a specificity for peripheral neuropathy. Sweadner et al. (15) reported that the putatively pathogenic mutations of ATP1A1 found in CMT2 axonal sensorimotor neuropathy were mainly in the P domain, N domain, or extracellular surface which may affect only biosynthesis or activity. But the pathogenic mutations of ATP1A1 found in patients with epilepsy were all in the transmembrane regions which significantly reduced the K+ affinity and even cooperativity of K+ binding (9). In the CNS, constant Na+/K+-ATPase activity is required for generating the resting membrane potential and buffering and clearance of extracellular K+ transients during neuronal activity. It suggested that the decreases in Na+/K+-ATPase activity from these mutations might cause more sever phenotypes.
In summary, two novel de novo ATP1A1 variants were identified in two cases in this study, enriching the phenotypic spectrum of ATP1A1 mutation-related genetic diseases. Together with four previously reported cases, we summarized the clinical characteristics of all six patients with ATP1A1 de novo mutation-related diseases. The most common phenotypes are developmental delay and refractory epilepsy. ATP1A1 variants can cause dysfunction of renal tubular reabsorption, leading to hypomagnesemia, but seizures were not controlled by magnesium supplementation. Our findings suggest that seizures are not secondary to hypomagnesemia but rather caused directly by Na+/K+-ATPase dysfunction in the CNS. However, hypomagnesemia might indicate more severe Na+/K+-ATPase dysfunction, particularly in the CNS, because patients with hypomagnesemia are more likely to have epilepsy. Moreover, the transmembrane regions M3, M4, M7, and M8 might be susceptible to de novo ATP1A1 mutations. The ATP1A1 variations located in M3 and M7 might have a greater detrimental impact on ATP1A1 function and lead to more severe phenotypes. However, the case number were too limited to clarify the complete features for this disease, and further large-sample studies are needed to confirm our findings.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by Institutional Review Board of Peking University First Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
YJ and KG: study design, analysis, and revision of the manuscript. ZL: follow-up of patient's information and draft preparation. ZL, JL, TJ, YW, KG, and YJ: collection of clinical and WES data. All authors contributed to the article and approved the submitted version.
This work was supported by the National Key Research and Development Program of China (grant numbers: 2016YFC1306201, 2020YFA0804003, and 2016YFC0901505), the National Natural Science Foundation of China (grant numbers: 81971211 and 81601131), the Beijing Key Laboratory of Molecular Diagnosis and Study on Pediatric Genetic Diseases (grant number: BZ0317), and the Fundamental Research Funds for the Central Universities (grant numbers: BMU2017JI002, BMU2018XY006, and PKU2017LCX06).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We sincerely thank the patients' families for their kind support during the study.
1. Blanco G. Na,K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin Nephrol. (2005) 25:292–303. doi: 10.1016/j.semnephrol.2005.03.004
2. Holm TH, Lykke-Hartmann K. Insights into the pathology of the alpha3 Na(+)/K(+)-ATPase ion pump in neurological disorders; lessons from animal models. Front Physiol. (2016) 7:209. doi: 10.3389/fphys.2016.00209
3. Lassuthova P, Rebelo AP, Ravenscroft G, Lamont PJ, Davis MR, Manganelli F, et al. Mutations in ATP1A1 cause dominant charcot-marie-tooth type 2. Am J Hum Genet. (2018) 102:505–14. doi: 10.1016/j.ajhg.2018.01.023
4. Yang-Feng TL, Schneider JW, Lindgren V, Shull MM, Benz EJ Jr, Lingrel JB, et al. Chromosomal localization of human Na+, K+-ATPase alpha- and beta-subunit genes. Genomics. (1988) 2:128–38. doi: 10.1016/0888-7543(88)90094-8
5. Cechova P, Berka K, Kubala M. Ion pathways in the Na(+)/K(+)-ATPase. J Chem Inf Model. (2016) 56:2434–44. doi: 10.1021/acs.jcim.6b00353
6. Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet. (2013) 45:1055–60. doi: 10.1038/ng.2716
7. Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet. (2013) 45:440–4. doi: 10.1038/ng.2550
8. He J, Guo L, Lin S, Chen W, Xu G, Cai B, et al. ATP1A1 mutations cause intermediate Charcot-Marie-Tooth disease. Hum Mutat. (2019) 40:2334–43. doi: 10.1002/humu.23886
9. Schlingmann KP, Bandulik S, Mammen C, Tarailo-Graovac M, Holm R, Baumann M, et al. Germline de novo mutations in ATP1A1 cause renal hypomagnesemia, refractory seizures, and intellectual disability. Am J Hum Genet. (2018) 103:808–16. doi: 10.1016/j.ajhg.2018.10.004
10. Stregapede F, Travaglini L, Rebelo AP, Cintra VP, Bellacchio E, Bosco L, et al. Hereditary spastic paraplegia is a novel phenotype for germline de novo ATP1A1 mutation. Clin Genet. (2020) 97:521–6. doi: 10.1111/cge.13668
11. Williams TA, Monticone S, Schack VR, Stindl J, Burrello J, Buffolo F, et al. Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertension. (2014) 63:188–95. doi: 10.1161/HYPERTENSIONAHA.113.01733
12. Seidel E, Schewe J, Scholl UI. Genetic causes of primary aldosteronism. Exp Mol Med. (2019) 51:1-12. doi: 10.1038/s12276-019-0337-9
13. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
14. Kanai R, Ogawa H, Vilsen B, Cornelius F, Toyoshima C. Crystal structure of a Na+-bound Na+,K+-ATPase preceding the E1P state. Nature. (2013) 502:201–6. doi: 10.1038/nature12578
Keywords: ATP1A1 gene, developmental delay, epilepsy, hypomagnesemia, genotype-phenotype correlations
Citation: Lin Z, Li J, Ji T, Wu Y, Gao K and Jiang Y (2021) ATP1A1 de novo Mutation-Related Disorders: Clinical and Genetic Features. Front. Pediatr. 9:657256. doi: 10.3389/fped.2021.657256
Received: 22 January 2021; Accepted: 25 March 2021;
Published: 21 April 2021.
Edited by:
Loredana Bury, University of Perugia, ItalyReviewed by:
Francesco Nicita, Bambino Gesù Children Hospital (IRCCS), ItalyCopyright © 2021 Lin, Li, Ji, Wu, Gao and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuwu Jiang, amlhbmd5dXd1QGJqbXUuZWR1LmNu; Kai Gao, Z2Fva2FpX2dsaWFAMTYzLmNvbQ==
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.