 Yang Li
Yang Li Lijuan Fan
Lijuan Fan Rong Luo
Rong Luo Zuozhen Yang
Zuozhen Yang Meng Yuan
Meng Yuan Jinxiu Zhang
Jinxiu Zhang Jing Gan
Jing Gan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 18 February 2021
Sec. Pediatric Neurology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.641841
Introduction: O'Donnell-Luria-Rodan syndrome was recently identified as an autosomal dominant systemic disorder caused by variants in KMT2E. It is characterized by global developmental delay, some patients also exhibit autism, seizures, hypotonia, and/or feeding difficulties.
Methods: Whole-exome sequencing of family trios were performed for two independent children with unexplained recurrent seizures and developmental delay. Both cases were identified as having de novo variants in KMT2E. We also collected and summarized the clinical data and diagnosed them with O'Donnell-Luria-Rodan syndrome. Structural-prediction programs were used to draw the variants' locations.
Results: A 186 G>A synonymous variant [NM_182931.3:exon4: c.186G>A (p.Ala62=)] was found in one family, resulting in alternative splicing acid. A 5417 C>T transition variant [NM_182931.3:exon27: c.5417C>T (p.Pro1806Leu)] was found in another family, resulting in 1806 Pro-to-Leu substitution. Both variants were classified as likely pathogenic according to the ACMG (American College of Medical Genetics and Genomics) guidelines and verified by Sanger sequencing.
Conclusion: To date, three studies of O'Donnell-Luria-Rodan syndrome have been reported with heterogeneous clinical manifestations. As a newly recognized inherited systemic disorder, O'Donnell-Luria-Rodan syndrome needs to be paid more attention, especially in gene testing.
KMT2E (GenBank: NM_182931.2, MIM: 608444) (1) is also known as MLL5 and is located in the 2.5-Mb region at chromosome 7q22.1. It was first recognized in leukemia and encoded a functional protein belonging to the lysine N-methyltransferase 2 (KMT2) family (2). The enzymes of the KMT2 family modulate transcription and promote chromatin accessibility by catalyzing the methylation of histone H3 on lysine 4 (H3K4) with their highly conversed SET domain (2, 3). However, SET domains in KMT2E were absent from important motifs and were proven to lack intrinsic methyltransferase activity in vitro. Unlike a traditional histone methyltransferase, KMT2E has been thought to be a transcriptional regulator involved in cell cycle progression (4, 5), genomic stability (6), hematopoiesis, and spermatogenesis (7, 8). However, more studies are needed to explore the detailed molecular mechanisms. In previous research, the KMT2E variant was found to contribute to schizophrenia and autism (9). However, in 2019, O'Donnell-Luria et al. reported a case series of KMT2E related neurodevelopmental disorder characterized by global developmental delay and named it O'Donnell-Luria-Rodan (ODLURO) syndrome (10).
Because the disease was only recently recognized and only a limited number of cases have been reported, the genotypes and phenotypes have not been fully elucidated. Here, we present two new cases of neurodevelopmental disorder caused by a KMT2E variant to enrich the phenotypes and variants of ODLURO syndrome. We also reviewed previously reported cases and attempted to summarize the clinical features and genetic characteristics of KMT2E related neurodevelopmental disorder.
Informed consent was obtained from the parents and their families. This study was approved by the institutional review board of the West China Second University Hospital. Their clinical manifestations, electroencephalogram (EEG), brain magnetic resonance imaging (MRI), malformations, investigations of other organs, and gene variations were analyzed. We also combined the 43 KMT2E variant-related cases reported previously in our analysis. Additional phenotype data and genetic findings for individuals are summarized in Table 1. We attempted to explore the relationship between different phenotypes and genotypes by statistical analysis.
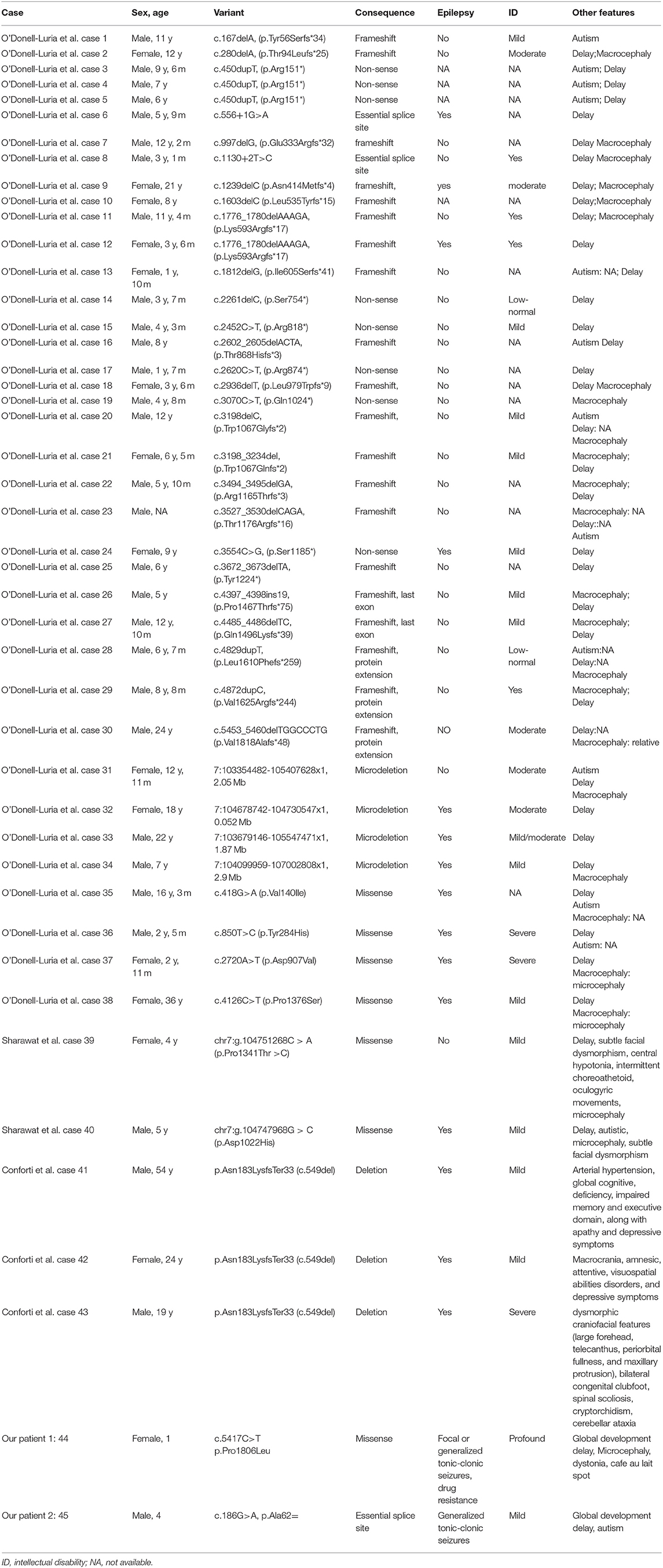
Table 1. Clinical information of cases with KMT2E mutations in our study.
Peripheral blood samples were collected from the probands and their families. Genomic DNA was extracted. A total amount of 1.0 μg genomic DNA per sample was used as input material for the DNA sample preparation. Sequencing libraries were generated using xGen Exome Research Panel probes (IDT, USA) following the manufacturer's recommendations. Sequencing was performed on the Illumina Novaseq 6000 platform.
Burrows-Wheeler Aligner (BWA) (11) was utilized to map the paired-end clean reads to the human reference genome (hg19). GATK (12) was utilized for the recalibration base quality score, SNP, and short indel calling. The variations were annotated using ANNOVAR (13). Variants were picked up in exonic and splicing regions with a minor allele frequency of ≤0.005 in the SNP database (ExAC_EAS, ExAC_ALL, 1000Genomes, gnomAD). The identified variants were both de novo variants that were absent in the parents. These were further validated by Sanger sequencing. The ACMG guideline was used to judge the degree of pathogenicity of both variants to evaluate their pathogenicity.
To understand the biophysical consequences of KMT2E protein sequence changes, structural-prediction programs (HMMER, PHYRE2, InterProScan, and NetPhos) were applied to evaluate the presence of protein domains and major secondary structure elements (e.g., helices, strands, loops, disorder, and post-translational modification sites).
Case 1 was a full-term, 5-month-old test-tube girl with uncomplicated prenatal and neonatal courses. Her other family members, including her twin sister, were all healthy. This girl had seizure onset at 4 months, with focal to generalized tonic-clonic seizures. One small café au lait spot was found on her trunk and hypotonia of the extremities. She had microcephaly with a head circumference of only 38 cm (OFC <-3 SD). She also presented specific facial features, including a slightly large forehead, prominent cheeks, and long eyelashes (Supplementary Figure 1). However, investigations of other organs (heart, eye, liver, kidney, etc.) were negative. She exhibited a failure to thrive, and her development was delayed and stagnant. Upon admission at 5 months of age, she could not lift her neck or roll over and had a poor reaction to human faces, lights, and sound. The EEG showed multifocal spike waves, sharp waves, spike-slow wave complexes, and slow waves in the left posterior temporal region, while the brain MRI was normal (Supplementary Figure 2, 3). Both blood and urine metabolic screening were normal. The auditory brainstem response and visual evoked potential tests were negative. Her seizures were not effectively controlled even though different combinations of anti-epileptic drugs had been administered, often lasting for 10 min to 1 h for every episode. Oxcarbazepine (40 mg/kg.d), levetiracetam (40 mg/kg.d), valproic acid (30 mg/kg.d), topiramate (8 mg/kg.d), clonazepam (1 mg bid), and a ketogenic diet were tried. The patient is currently taking valproic acid (30 mg/kg.d), levetiracetam (40 mg/kg.d), clonazepam (1 mg bid), and a ketogenic diet. At her latest follow-up assessment at 13 months of age, the seizures were not controlled, but the frequency had decreased to 1–5 episodes per month. Repeated EEG monitoring showed improvement in the epileptiform discharge with moderate and low-amplitude sharp and slow wave discharge, obvious in the occipital region. Her occipitofrontal head circumference (OFC) was only 40 cm (OFC <-3 SD). She had profound global developmental delay, with a Gesell Developmental Scale score of 25.
Case 2 was a 5-year-old boy whose birth was uneventful. He showed global developmental delay with mild autism. He experienced two episodes of febrile seizure at 2 years old when EEG showed slow background waves. On admission, he suffered frequent generalized tonic-clonic seizures that lasted for 30 min, which fulfilled the diagnostic criteria of convulsive status epilepticus. His occipitofrontal head circumference was 50 cm. When he was 3 years old, he could walk without support, speak simple words, and follow simple instructions. Additionally, he had difficulties with social interaction and communication without any tests for intellectual disability or autism spectrum disorder before admission. On the day of admission, he was in a coma with a Glasgow Coma Scale score of nine. He had no neck stiffness, but the Babinski sign was positive. No malformations of other organs were identified. Cytological and biochemical examinations of the cerebrospinal fluid were normal, and etiological tests including herpes simplex virus, cytomegalovirus, Epstein-Barr virus, Japanese encephalitis virus, coxsackievirus, mycoplasma pneumoniae, tubercle bacillus, and fungal culture were all negative. In addition, autoimmune encephalitis and metabolic diseases were excluded by antibody and metabolic screening. The EEG showed sharp spikes in the frontal, temporal, and parietal lobes with a slow background (Supplementary Figure 4). Brain MRI showed cerebellar atrophy, as well as broadening of the lateral and third ventricles (Supplementary Figure 5). He was treated with levetiracetam (50 mg/kg/d), valproic acid (40 mg/kg/d), and clonazepam (2 mg bid). After ventilator weaning and withdrawal of sedatives, the boy still had paroxysmal hypertonia of the extremities. Finally, he was seizure-free 1 month later on a combination of levetiracetam (50 mg/kg/d), valproic acid (40 mg/kg/d), and clonazepam (1 mg bid). By the last follow-up, he had been seizure-free for 6 months. Repeated EEG monitoring showed a generalized low-amplitude wave with an absent sleeping spindle. His Wechsler Intelligence Scale score was 55. A trio whole-exome sequencing test was performed on both patients.
Two de novo variants in the KMT2E gene were identified: a 186 G>A synonymous variant (NM_182931.3:exon4: c.186G>A (p.Ala62=) resulting in alternative splicing acid and a 5417 C>T transition variant [NM_182931.3:exon27: c.5417C>T (p.Pro1806Leu)] resulting in 1806 Pro-to-Leu substitution. Sanger sequencing confirmed the variants in their families (Figure 1). The variant c.5417 C>T results in protein changes from proline to leucine (p.Pro1806Leu) and was predicted to be deleterious by Polyphen2, Mutation taster, and SIFT. As a synonymous variant, the 186 G>A variant was evaluated by “Ada” and “RF” scores from InterVar software (0.9984 and 0.832, respectively, Table 2; threshold: 0.6; higher score means greater probability altering the splicing). The effect of the 186 G>A variant was predicted to be splicing donor site loss, probably resulting in an abnormal isoform (Figure 2A).
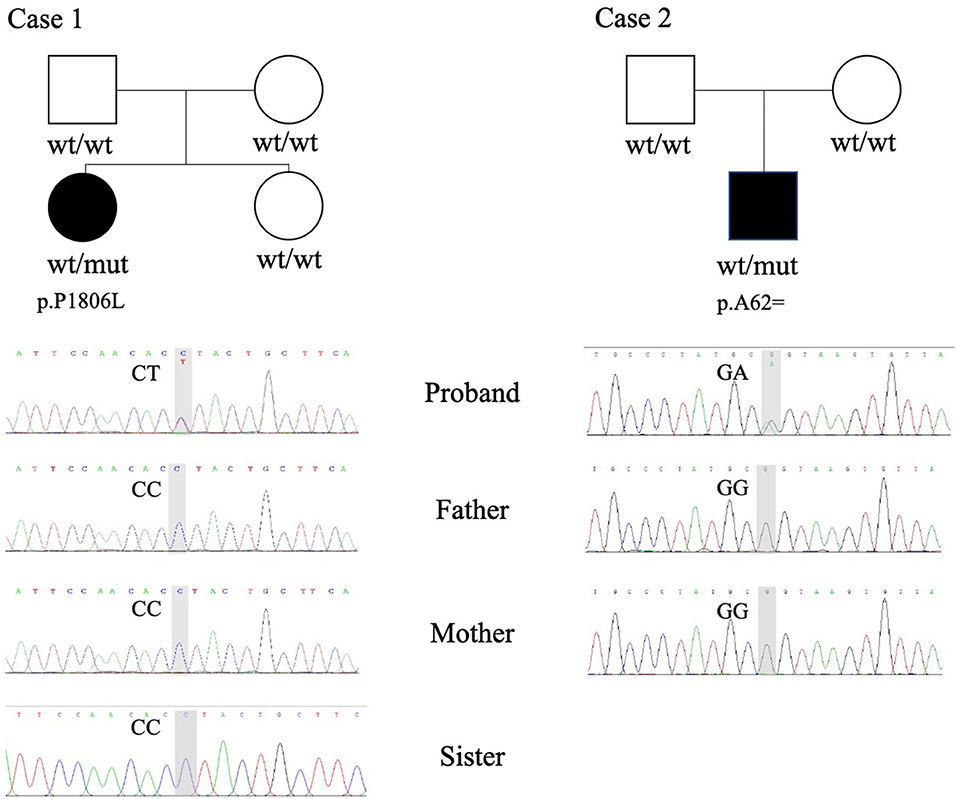
Figure 1. Whole-exome sequencing (WES) and Sanger sequencing revealed heterozygous variants in case 1 (p.P1806L) and case 2 (p.A62=). wt, wildtype; mut, mutation.
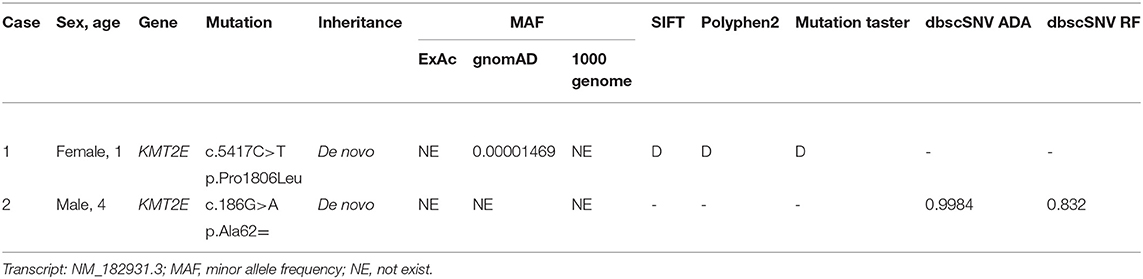
Table 2. The analysis of pathogenicity of the variants in KMT2E.
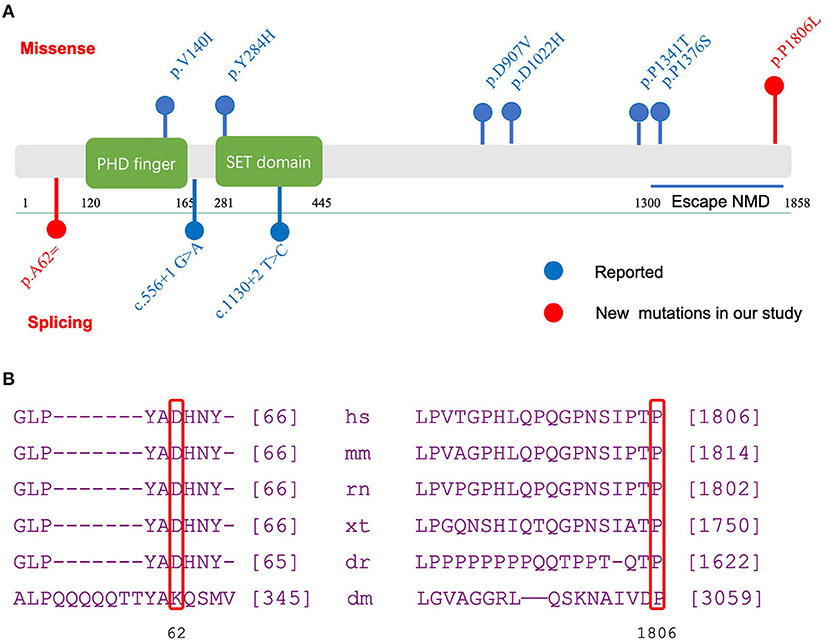
Figure 2. Functional domains of KMT2E and evolutionary conservation analysis of amino acid residue. (A) Functional domains and regions in KMT2E gene as indicated. Red dots: new mutations in our study; Blue dots: reported mutations; Above bar: Missense mutation; below bar: splicing mutation. (B) Evolutionary conservation of amino acid residue at position 62 and 1806 in the KMT2E gene among species.
Further conservation analysis showed that the locus was highly conserved (Figure 2B). The 5417 C>T transition variant was reported once in the gnomAD database. The 186 G>A variant was not found in the public databases (gnomAD, Exome Aggregation Consortium, or 1000 Genomes; Table 2). These two variants were classified as likely pathogenic according to the ACMG guideline (Table 3). Other pathogenic variants of genes known to be associated with epilepsy or intellectual disability were not found in the two probands.
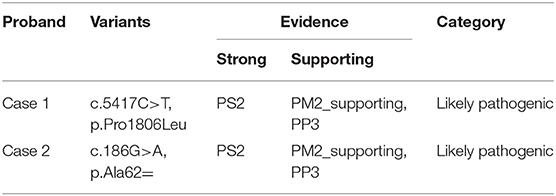
Table 3. Pathogenicity classification referred to ACMG guideline.
We combined all genetic variants and phenotypes from the published cases [43 cases: 38 individuals in 36 families from O'Donell-Luria et al. (10); two individuals in two families from Sharawat et al. (14); three individuals in one family from Conforti et al. (15)] and two cases from two families in our study (Table 1) to explore the relationship between different phenotypes and variants. For each phenotype, we defined the overall incidence of ODLURO syndrome as background and compared each specific phenotype incidence rate in the sub-variant type group to the overall incidence. We found that the incidence rates of ID (intellectual disability) (100 vs. 86.7%, p < 0.001), epilepsy (85.7 vs. 41.5%, p < 0.001), and microcephaly (66.7 vs. 9.3%, p < 0.001) in the missense group were significantly higher than their overall incidence (Table 4). However, no macrocephaly (0 vs. 46.5%, p < 0.001) was observed in the missense group (Table 4).
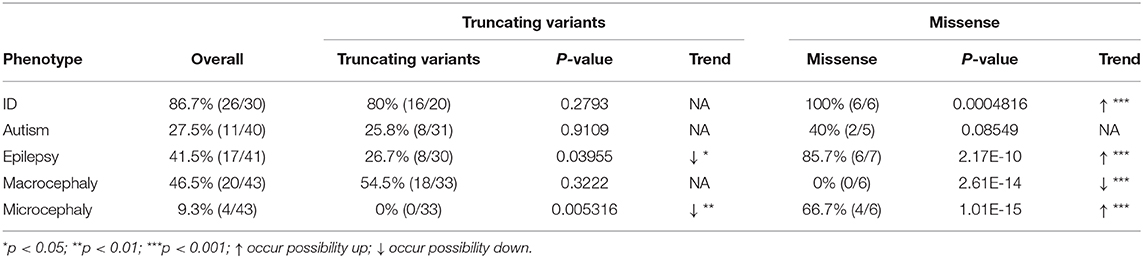
Table 4. Phenotype related to variant type.
ODLURO syndrome is a recently reported genetic disease caused by heterozygous variants in the KMT2E (the lysine N-methyltransferase 2E) gene (10). To date, 43 cases of ODLURO syndrome have been reported. The clinical characteristics are developmental delay, but macrosomia, cerebellar ataxia, leukoencephalopathy, hypotonia, speech difficulties, memory dysfunction, autism, and other behavioral concerns such as attention-deficit/hyperactivity disorder and depression can also occur (10, 14–17). Many individuals with hypotonia were reported to have feeding difficulties, including reflux, vomiting, or bowel motility issues (10). Patients may have dysmorphic features including a large forehead, full cheeks, prominent nasolabial folds, deep-set eyes with down-slanting palpebral fissures, macrocephaly, bilateral congenital clubfoot, spinal scoliosis, and cryptorchidism (10).
This study described the clinical manifestations of two Chinese patients with ODLURO syndrome from two unrelated families. The cases we reported share the characteristics of other affected patients. Both patients had epilepsy. Case 1 presented with dystonia, profound global developmental delay, and drug-resistant focal seizures or generalized tonic-clonic seizures. Case 1 also had microcephaly which is not a common feature of ODLURO syndrome (4 out of 43 according to reported cases). Case 2 presented with global developmental delay, autism, and generalized tonic-clonic seizures. Of the 43 cases of KMT2E variants with neuropathic disorder reported, only 12 had epilepsy, 5 of which were treatment-resistant epilepsy. The main forms of seizures were tonic seizures, clonic seizures, focal or generalized tonic-clonic seizures, myoclonic seizures, epileptic spasms, and absence seizures. There seem to be phenotypic differences between individuals with protein-truncating and missense variants in KMT2E. Patients carrying the missense variant might present with a more severe phenotype and are more likely to have seizures than patients with the truncating variant (10, 15). O'Donell-Luria et al. (10) reported that only 5 of 30 patients with the KMT2E protein-truncating variant had epilepsy, of whom only one was treatment-resistant, while all four patients with missense variants had epilepsy, of whom three were treatment-resistant. In addition, patients with a missense variant presented with more severe intellectual disability, and half had microcephaly. The reason is not yet understood but might result from the pathogenic mechanism of haploinsufficiency vs. gain-of-function or dominant-negative effects. More research is required to elucidate this.
The MRI abnormalities of ODLURO syndrome were varied and unspecific. They included thinning or partial agenesis of the corpus callosum, widening of the subarachnoid space, ventricle enlargement, various cysts, increased white matter signal, a hyperintense signal in the basal ganglia, decreased volume, delayed myelination, small areas of heterotopia, cerebellar dysplasia, and Chiari I malformation. Among them, thinning of the corpus callosum may be common and was seen in 7 of 41 patients (although MRI data were limited) (10, 15). In our study, case 1 had normal MRI findings, while the MRI of case 2 demonstrated cerebellar atrophy and broadening of the lateral and third ventricles.
So far, most of the KMT2E related ODLURO syndrome cases reported had a de novo variant (10). These variants are distributed throughout the gene with no hotspot variant. Most reported pathogenic variants were frameshift and non-sense, and only six missense and two essential splice sites were reported previously (10, 14, 15). Synonymous variants are generally considered non-pathogenic and are not expected to change the function of the protein. However, recent studies have shown that synonymous changes in codon usage may alter the efficiency and speed of translation, ultimately affecting protein folding (18, 19). “Ada” and “RF” scores have been widely used for evaluating potential splicing variants predicted by dbscSNV (13). Both are ensemble scores, which are derived from the outputs of several machine learning algorithms. The score is scaled from 0 to 1, with higher values indicating a greater probability that the variant will alter the splicing of the gene. Moreover, 0.6 was regarded as a threshold value for dichotomous effects. In our study, the Ada and RF scores were 0.9984 and 0.832, respectively. Thus, these scores demonstrate a highly likely splicing result for KMT2E.
Our study reported two KMT2E variants from two children with de novo variants and evaluated the impact of this variant on protein structure. The variant c.186G>A [p.Ala62=] was predicted to influence splicing. The effect of the 186 G>A variant was predicted to be splicing donor site loss, which probably results in an abnormal isoform. The missense variant (c.5417C>T, p.Pro1806Leu) falls in the terminal exon of the gene. Thus, it potentially escapes non-sense-mediated decay and extends the open reading frame. It is predicted to be functionally damaging by several bioinformatics software programs. An evolutionary analysis showed a conserved result at 62 and 1806 sites among multiple species (Figure 2B), indicating a crucial role in gene function. In summary, the variants c.5417C>T [p.Pro1806Leu] and c.186G>A [p.Ala62=] in KMT2E can be considered pathogenic and the cause of ODLURO syndrome in these patients.
Our study adds two new patients to the ODLURO syndrome case series that were genetically, clinically, and radiologically evaluated. Two novel variants, c.5417C>T [p.Pro1806Leu] and c.186G>A [p.Ala62=] were reported, both of which were de novo variations. Individuals with missense variants may have a more severe phenotype than individuals with truncating variants. The pathological mechanism of KMT2E variants in neurological diseases and the relationship between different genotypes and phenotypes have not been entirely established. Our study adds to the limited information that has been published on ODLURO syndrome and promotes further research.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: NCBI BioProject, accession no: PRJNA690597.
The studies involving human participants were reviewed and approved by West China Second University Hospital, Sichuan University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
YL: conceptualization, methodology, and data mining. LF, MY, and JZ: writing (original draft preparation). RL: writing (original draft preparation) and supervision. ZY: software, data mining, and investigation. JG: supervision and writing (reviewing and editing). All authors contributed to the article and approved the submitted version.
This work was supported by the National Science Foundation of China: (No. 82071686, 81501301), a Grant from the Science and Technology Bureau of Sichuan province to Jing Gan (No. 2021YFS0093), a Grant from the Clinical Research Fund of West China Second University Hospital (No. KL072), and a Grant from the Yun epilepsy Genetics Research and Applicant Fund.
ZY was employed by Cipher Gene LLC.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the probands and their families for their kind cooperation and Cipher Gene LCC for data mining.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.641841/full#supplementary-material
1. Yun H, Damm F, Yap D, Schwarzer A, Chaturvedi A, Jyotsana N, et al. Impact of MLL5 expression on decitabine efficacy and DNA methylation in acute myeloid leukemia. Haematologica. (2014) 99:1456–64. doi: 10.3324/haematol.2013.101386
2. Zhang X, Novera W, Zhang Y, Deng LW. MLL5 (KMT2E): structure, function, clinical relevance. Cell Mol Life Sci. (2017) 74:2333–44. doi: 10.1007/s00018-017-2470-8
3. Park K, Kim JA, Kim J. Transcriptional regulation by the KMT2 histone H3K4 methyltransferases. Biochim Biophys Acta Gene Regul Mech. (2020) 1863:194545. doi: 10.1016/j.bbagrm.2020.194545
4. Ali M, Rincon-Arano H, Zhao W, Rothbart SB, Tong Q, Parkhurst SM, et al. Molecular basis for chromatin binding and regulation of MLL5. Proc Natl Acad Sci USA. (2013) 110:11296–301. doi: 10.1073/pnas.1310156110
5. Deng L-W, Chiu I, Strominger JL. MLL 5 protein forms intranuclear foci, and overexpression inhibits cell cycle progression. Proc Natl Acad Sci USA. (2004) 101:757–62. doi: 10.1073/pnas.2036345100
6. Liu J, Cheng F, Deng LW. MLL5 maintains genomic integrity by regulating the stability of the chromosomal passenger complex through a functional interaction with Borealin. J Cell Sci. (2012) 125(Pt 19):4676–85. doi: 10.1242/jcs.110411
7. Heuser M, Yap DB, Leung M, de Algara TR, Tafech A, McKinney S, et al. Loss of MLL5 results in pleiotropic hematopoietic defects, reduced neutrophil immune function, and extreme sensitivity to DNA demethylation. Blood. (2009) 113:1432–43. doi: 10.1182/blood-2008-06-162263
8. Yap DB, Walker DC, Prentice LM, McKinney S, Turashvili G, Mooslehner-Allen K, et al. Mll5 is required for normal spermatogenesis. PLoS ONE. (2011) 6:e27127. doi: 10.1371/journal.pone.0027127
9. Shen E, Shulha H, Weng Z, Akbarian S. Regulation of histone H3K4 methylation in brain development and disease. Philos Trans R Soc Lond B Biol Sci. (2014) 369:1652. doi: 10.1098/rstb.2013.0514
10. O'Donnell-Luria AH, Pais LS, Faundes V, Wood JC, Sveden A, Luria V, et al. Heterozygous variants in KMT2E cause a spectrum of neurodevelopmental disorders and epilepsy. Am J Hum Genet. (2019) 104:1210–22. doi: 10.1016/j.ajhg.2019.03.021
11. Abuin JM, Pichel JC, Pena TF, Amigo J. BigBWA: approaching the Burrows-Wheeler aligner to Big Data technologies. Bioinformatics. (2015) 31:4003–5. doi: 10.1093/bioinformatics/btv506
12. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
13. Li Q, Wang K. InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. (2017) 100:267–80. doi: 10.1016/j.ajhg.2017.01.004
14. Sharawat IK, Panda PK, Dawman L. Clinical characteristics and genotype-phenotype correlation in children with KMT2E gene-related neurodevelopmental disorders: report of two new cases and review of published literature. Neuropediatrics. (2020). doi: 10.1055/s-0040-1715629
15. Conforti R, Iovine S, Santangelo G, Capasso R, Cirillo M, Fratta M, et al. ODLURO syndrome: personal experience and review of the literature. Radiol Med. (2020). doi: 10.1007/s11547-020-01255-2
16. Dong S, Walker MF, Carriero NJ, DiCola M, Willsey AJ, Ye AY, et al. De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep. (2014) 9:16–23. doi: 10.1016/j.celrep.2014.08.068
17. Abdelhalim AN, Alberico RA, Barczykowski AL, Duffner PK. Patterns of magnetic resonance imaging abnormalities in symptomatic patients with Krabbe disease correspond to phenotype. Pediatr Neurol. (2014) 50:127–34. doi: 10.1016/j.pediatrneurol.2013.10.001
18. Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. (2007) 315:525–8. doi: 10.1126/science.1135308
Keywords: KMT2E, O'Donnell-Luria-Rodan syndrome, epilepsy, whole-exome sequencing, neurodevelopmental disorder
Citation: Li Y, Fan L, Luo R, Yang Z, Yuan M, Zhang J and Gan J (2021) Case Report: De novo Variants of KMT2E Cause O'Donnell-Luria-Rodan Syndrome: Additional Cases and Literature Review. Front. Pediatr. 9:641841. doi: 10.3389/fped.2021.641841
Received: 15 December 2020; Accepted: 26 January 2021;
Published: 18 February 2021.
Edited by:
Pasquale Striano, University of Genoa, ItalyReviewed by:
Antonietta Coppola, University of Naples Federico II, ItalyCopyright © 2021 Li, Fan, Luo, Yang, Yuan, Zhang and Gan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Gan, Z29yZG9ucmFjaGVsQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.