 Yiming Qi
Yiming Qi Hongke Ding1,2
Hongke Ding1,2 Aihua Yin
Aihua Yin- 1Prenatal Diagnosis Centre, Guangdong Women and Children Hospital, Guangzhou, China
- 2Maternal and Children Metabolic-Genetic Key Laboratory, Guangdong Women and Children Hospital, Guangzhou, China
The long-term prognosis of a fetus with cardiac rhabdomyoma (CR) depends on the correlation with tuberous sclerosis complex (TSC). In recent years, the numerous variations of uncertain significance (VUS) of TSC genes produced by high-throughput sequencing have made counseling challenging, studies until now have tended to side-step the tricky topics. Here, we integrated detailed parental phenotype, echocardiography, neuro MRI, and genetic information to conduct a comprehensive evaluation of 61 CR fetuses. As a result, multiple CRs and cerebral lesions appeared in 90 and 80%, respectively of fetuses with pathogenic (P)/likely pathogenic (LP) TSC1/TSC2 variations. Overall, 85.7% of the live-born infants with P/LP presented with TSC-associated signs. While, 85.7% of VUS without nervous findings had good prognoses. Genetic evidence and cerebral MRI findings are the most sensitive index to assess long-term prognosis, which complement and confirm each other for a TSC diagnosis. In total, 68.9% of fetuses with CR could benefit from this multidisciplinary approach, which turned out to be potentially clinically actionable with precise clinical/genetic diagnosis or had a foreseeable outcome. Our practice provides a practical and feasible solution for perinatal management and prognostic guidance for fetuses with CR.
Introduction
Cardiac rhabdomyoma (CR) is one of the most common primary cardiac tumors which occurs predominately in infants and children. The reported incidence varies from 0.02 to 0.17% in live-born infants, and 0.12% in prenatal fetal studies (1, 2). An unprecedented increase in CR has benefited from the rapid development of fetal echocardiography in recent decades. However, it is still challenging to obtain accurate prognostic information of these newborns.
As a benign hamartoma, the prognosis of CR without severe complications is typically good (3). But CR is unpredictable and occurs anywhere in the heart, which may result in arrhythmias, effusions, fetal hydrops, and even stillbirth. It is worth noting, to some extent, that CR is a characteristic sign and an initial symptom for tuberous sclerosis complex (TSC) prenatally, which has a high possibility of neurological development disorder. Reported rates of TSC in fetuses with CR vary from 50 to 90% (4, 5). Approximately 90% of infants with TSC experience infantile spasms, seizures, intellectual disability, or autism (6). As a multisystem genetically based neurocutaneous syndrome, TSC is inherited in an autosomal dominant manner. Cortical tubers and subependymal nodules (SEN) are typical cerebral lesions with a high risk of obstructive hydrocephalus, which are the leading cause of mortality. There are few reports about the intrauterine brain damages of TSC, and the relationship with imaging phenotype and genotype is not clear yet.
TSC1/TSC2 mutations are the vital disease-causing genes and are recognized as independent and sufficient diagnostic criteria, even in the condition of no visible typical clinical signs (7). With the application of NGS technology, the number of identified variants has increased explosively, meanwhile massive VUS have not been far behind. Overall, 3446 TSC1 and 8112 TSC2 variations have been recorded in the LOVD Database (http://chromium.lovd.nl/LOVD2/TSC/home), including 268 VUS of TSC1 and 642 VUS of TSC2. They show a broad spectrum of variations without conspicuous hotspots in patients. Mutation types have almost equal coverage including insertions/deletions/duplications, frameshift, non-sense, missense, splicing, and sometimes encounter deep intron mutations (8). This implies that no typical mutation type was well-tolerated. Disproportionately, limited understanding of the role and lack of awareness of genotype-phenotype correlations excessively disturbs our judgment of the pathogenesis with these VUS, which is hugely critical for fetal prognosis, informed reproductive choices, and evidence-based perinatal management.
The unpredictability of the fetal CR and the uncertainty of variations in TSC1/TSC2 gene makes made phenotype and prognosis prediction of TSC clinically challenging. Here, we revealed complete imaging and the molecular portrait of a fetus with CR(s) to date, adding multi-characteristic information for a comprehensive evaluation and proposing the clinical workflow for clinical management.
Materials and Methods
Patient Recruitment and Sample Collection
The study was approved by the institutional review board of the ethics committee in Guangdong Women and Children Hospital (201601046). Pregnant women with suspected fetal CR by the second-trimester screening were recruited in our center from March 2014 to November 2019. All fetuses underwent electrocardiography to evaluate the properties of tumors, and part of them performed brain magnetic resonance imaging (MRI) scans to gather more clinical evidence. A comprehensive clinical examination was concurrently performed in pedigree. The diagnostic criteria we referred to are the international recommendations of TSC, revised in 2013 (7).
Fetal samples were collected from amniocentesis (16–24 weeks) or cordocentesis (more than 24 weeks). Karyotype and SNP-array were performed simultaneously with NGS or before. All the fetuses accompanied with chromosome aneuploidy or copy number variations (CNVs) were excluded. For each relevant family member, 5 ml peripheral blood samples were draw and stored in ethylenediaminetetraacetic acid (EDTA) anti-coagulation vacuum tubes. Skin or cardiac tissues from aborted fetuses were collected after signed informed consent was obtained from the enrolled families.
Fetal Echocardiography and Brain MRI Examinations
According to the “ISUOG Practice Guide: Fetal Heart Ultrasound Screening (Updated Version),” we comprehensively used two-dimensional ultrasound and echocardiography by the GE VoIuson E8 color Doppler ultrasound system (GE Medical Systems, Zipf, Austria) which was equipped with a 2–5 MHz two-dimensional convex array transducer and a 4–8.5 MHz three-dimensional transducer. The tumor number, location, size, echo, relationship with surrounding tissues, heart rhythm, and hemodynamic changes in details, as well as fetal brain, spine, chest, abdomen, limbs and other structures were recorded.
Fetal brain MRI was performed on a Sonata scanner (Siemens, Erlangen, Germany) using a body phased-array coil. Breath-hold imaging was performed optionally without sedation in the supine position. Images were acquired using a T2-W HASTE (half-acquisition single-shot turbo-spin-echo) sequence (TR/TE 1,260/74 ms and matrix 256) and a T1-W gradient-echo sequence (TR/TE 130–160 ms/5–10 ms, matrix 128 and α70). Slice thickness was 4 mm in the axial plane.
Amniocentesis and Cordocentesis
The procedure was performed using an aseptic technique with infiltration of local anesthesia, under ultrasound guidance, using a real-time ultrasound machine equipped with a 2–5-MHz transabdominal probe. For amniocentesis, the ultrasound probe may be tilted up to 45°, with respect to the mid-sagittal plane of the maternal abdomen, away from the intended side of needle entry, whilst remaining in the same transverse plane. Once the needle was located correctly in the uterine cavity, the stylet was removed and the syringe, or a Luer adapter attached to a Vacutainer® holder, was connected. Finally, ~10–20 ml of amniotic fluid was drawn. For cordocentesis, a 22- or 23-gauge spinal needle was used. The puncture site, either near the cord insertion or in a free-floating loop, was chosen according to the accessibility and quality of visualization, regardless of location. Then, about 4 ml of cord blood was collected, ideally without contamination by maternal blood cells.
Follow-Up
The patient flow diagram is depicted in Figure 1. Continuing pregnancies were observed, and infants with CRs were born at the author's center or the referral center. All children born were followed up by a pediatric expert team which included a pediatric cardiologist, a pediatric neurologist, an imaging expert, and a genetic counselor. Termination of pregnancy (TOP) or obtaining a diagnosis (clinically or/and genetically) was the endpoint of clinical observation. Postnatal examinations included clinical examination, echocardiography, and MRI scanning if suspicious extra cardiac complications were found.
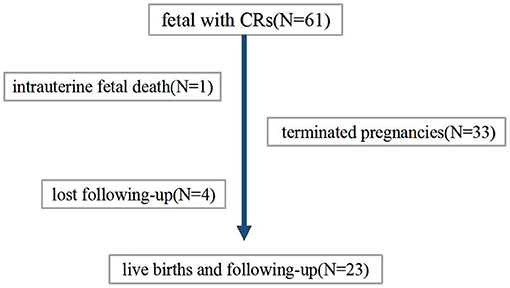
Figure 1. Patient flow diagram.
Next-Generation Sequencing
Genomic DNA samples were extracted using the Qiagen DNA Blood Midi/Mini kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's instructions. Sequencing of the TSC1 gene and TSC2 gene in Trios was performed with a custom-designed Roche Nimblegen (Madison, WI) (SeqCap EZ Choice Library) chip capturing the TSC1 (NM_000368, exon 3-23, exons 1 and 2 are UTRs) and TSC2 (NM_000548, exon 2-42, exons 1 UTRs) genes following the manufacturer's protocols (JIAJIAN Clinical Laboratories, BEIJING, China). The found variants were further verified by Sanger sequencing in fetuses and parents. Analysis of genetic results was based on the genomic variation database (http://dgv.tcag.ca/dgv/app/home), DECIPHER database (https://decipher.sanger.ac.uk/), and OMIM database (http://www.ncbi.nlm.nih.gov/omim). Variant classification was based on the guidelines of the ACMG and divided into five categories: “I: pathogenic (P),” “II: likely pathogenic (LP),” “III: variants of uncertain significance (VUS),” “IV: likely benign (LB),” and “V: benign (B).”
Results
General Information and Imaging Results Prenatally
A total of 61 fetuses with CR were included in the study. The median age of pregnant women was 28 (range 22–38) years. The average GA of CRs detected during routine scans was 25 ± 3.5 weeks (range 16–34). There were 59 single pregnancies and two dichorionic twin pregnancies, in which one of the fetuses presented with CR. Sonographic images showed round, hyperechogenic, homogenous masses with clear boundaries and absence of color in the Doppler flow inner. Solitary and multiple rhabdomyomas were detected in 18 fetuses (29.5%) and 43 fetuses (70.5%), respectively (Supplementary Figures 1A,B). All CRs were distributed in the ventricle, interventricular septum, and the atrium; the most common areas were the left ventricle and ventricular septum. Tumor size was recorded in 59 cases, and, of these, the median diameter was 8.1 mm (range 2.2–31.8). Forty-nine fetuses had more than one echocardiography monitoring record. Throughout the pregnancy, in 41% of the 49 fetuses tumors increased, seven cases kept stable, and only one had somehow subsided. Echocardiographic findings are summarized in Table 1.
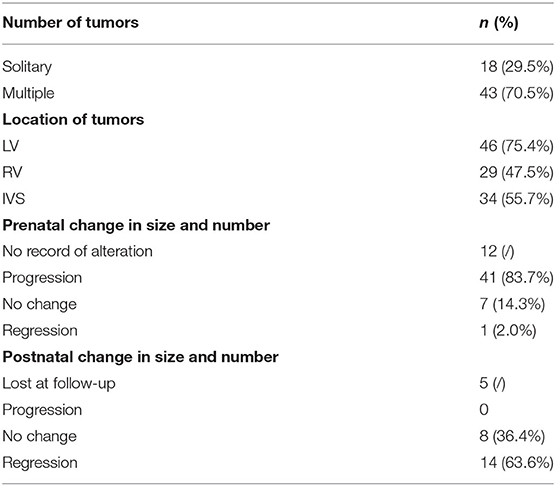
Table 1. Summary of echocardiographic findings in fetuses with CR.
Severe hemodynamic changes occurred in 6.6% (4/61) of the fetuses. Six fetuses presented left/right ventricular outflow tract obstruction, and three of them progressed to pericardial and peritoneal effusion (Supplementary Figure 1C). Moderate tricuspid regurgitation with or without pulmonary hypertension was observed in two fetuses (Supplementary Figure 1D). In case 6, the gravida had been diagnosed as TSC clinically, she underwent sonographic screening ahead of schedule at 16 weeks of GA. The fetus appeared to have a ventricular hyper echo accompanied by severe congenital cardiac structural deformities, which were directly related to a poor perinatal outcome rather than a TSC-associated prognosis. All subjects did not detect arrhythmia throughout pregnancy. MRI was performed from 20 to 36 weeks of gestation in 53 fetuses, 25 of them found SENs and cortical tubers. Three cases showed lateral or bilateral ventriculomegaly. Detailed information is contained in Table 2.

Table 2. Characteristics of 61 cases of prenatally diagnosed cardiac rhabdomyoma (CR).
Detection of TSC1 and TSC2 Variants
In general, TSC1 and TSC2 variants were detected in 45 (Figure 2G) of 61 fetuses (73.8%), leaving 16 with no variant identified (NVI) (Figure 2A). Two-thirds of these variants were de novo (Figure 2B). Roughly 50% of these variants had not been reported previously (Figure 2C). Following ACMG Standards, we identified 30 variants of P/LP and 16 VUS (Figure 2D). The P/LP variant spectrum in TSC1/TSC2 was 16 (34%) non-sense, 11 (23.4%) missense, 7 (14.9%) frameshift, 5 (10.6%) splice, 4 (8.5%) indels, and 2 (4.3%) introns (Figure 2E). When we focused on VUS, poor prognosis presented in all the truncating variant fetuses. Half of the missense/indel variants and all the intron variants tended to have a good outcome (Figure 2F).
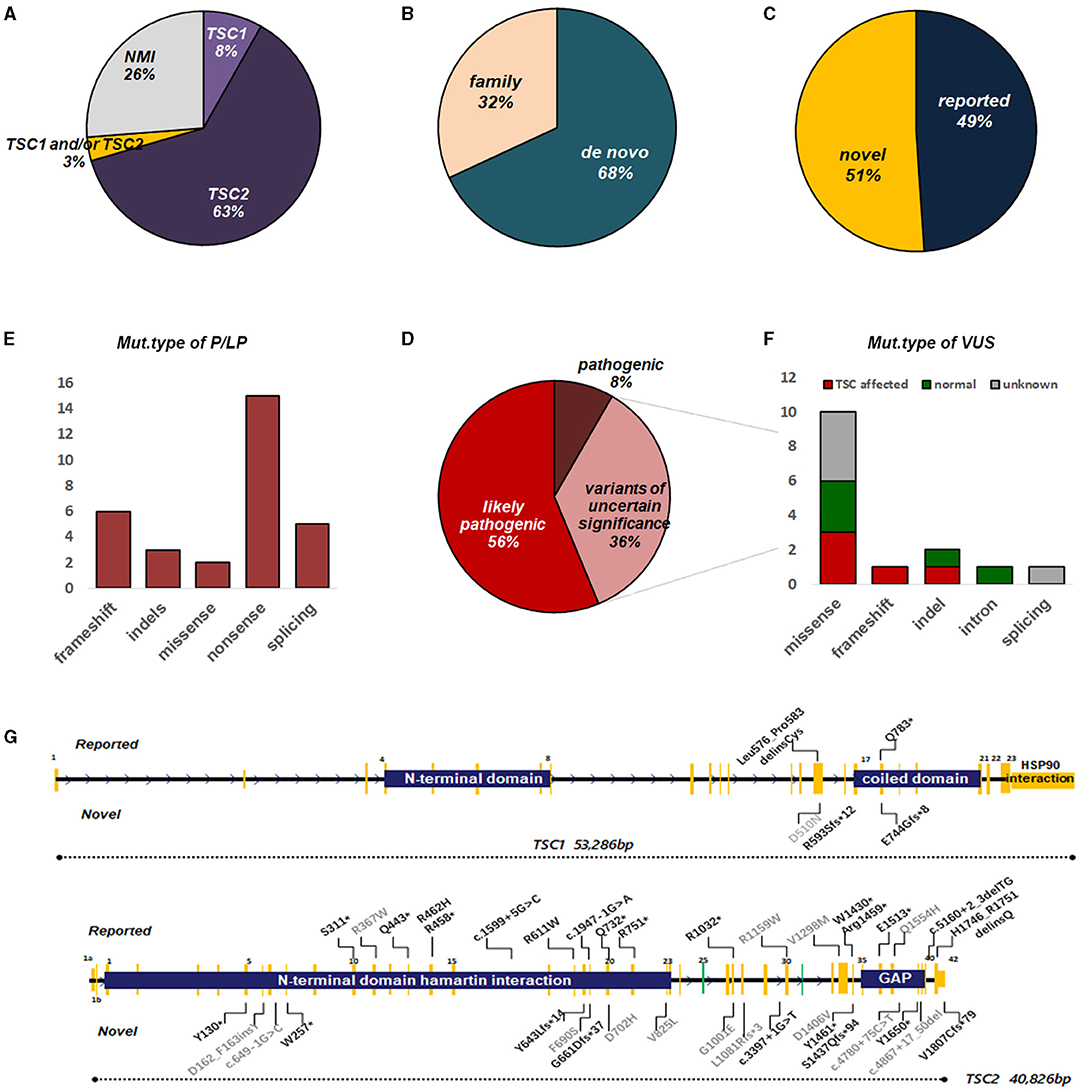
Figure 2. Overview of genetic testing results of TSC1/2 in fetuses with CR. (A) Percentages of TSC mutation found or no mutation identified (NMI) in fetuses with CR. (B) The proportion of the origin of TSC variation in fetuses with CR. (C) The ratio of the reported sites to the novel sites in TSC1 and TSC2 genes. (D) Among the identified mutations, pathogenic, likely pathogenic, or uncertain significance, TSC1/2 mutations are indicated. (E) Molecular consequences of mutations classified as pathogenic or possibly pathogenic. (F) Molecular consequences of mutations classified as VUS. Annotations include the following: (G) Distribution of mutation types in fetuses with CR(s). For TSC2, mutations were scattered throughout the gene with no enrichment in specific hotspots or domains, although there were several recurrent mutations, suggesting founder effects in certain populations. The number of mutations on the TSC1 gene was much less, mainly concentrated near the C-domain and interacting with tuberin. Annotations include the following: reported mutations (top); novel mutations (bottom); mutations of pathogenic/likely pathogenic (black); mutations of uncertain significance (gray). Splice site mutations are not shown here (see Supplementary Table 1). GAP, GTPase-activating protein.
Genotype-Phenotype Correlation
The profile of genotype and phenotype in fetuses with CR(s) is depicted in Table 3. For the 30 cases identified with variations of P/LP: 90% (27/30) had multiple CRs. Overall, 80% (20/25) were cerebral MRI positive before birth, and TSC-associated signs appeared in 85.7% (6/7) of the live-born infants. Two babies with multiple CRs but normal brain MRI were born, one of which developed epilepsy at 6 months, and SENs appeared. In the fetuses carrying VUS: the incidence rate of multiple tumors was as high as 73.3% (11/15), yet cerebral lesions observed prenatally were much lower (46.7%). In total, 85.7% (6/7) of VUS without nervous findings had the normal phenotype as far as we followed up. Postnatal brain damage and neurological symptoms onset occurred in only one infant without MRI findings in utero. As to the group with NVI, multiple CRs were found in 31.2% (5/16), and 87.5% (14/16) had normal nervous imaging. In two NVI fetuses (p36, p59), cortical nodules proved by MRI supported the diagnosis of TSC clinically. Both of them underwent TOP, and the heart tissues testing confirmed the NVI result (data are not shown). In terms of the mutation genes, TSC2 occupied a dominant position. Considering the TSC2 pathogenic variants are related to a more severe phenotype than TSC1 (9), we compared them in fetuses with CR(s). Both the frequency of TSC-causing mutations and number of CRs had no significant difference (P > 0.05). However, TSC2 variants were more related to positive neuroimaging findings than TSC1 (P < 0.05) (Table 4).
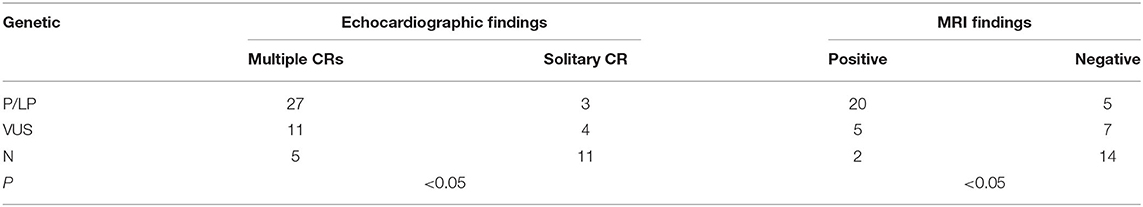
Table 3. The profile of genotype and phenotype in fetuses with CR(s).

Table 4. Phenotype differences between TSC1 and TSC2 in fetuses with CR(s).
Clinical Outcome
Finally, 34 families decided to terminate the pregnancy. Among these, case 25 experienced intrauterine fetal death at GA of 26 weeks for severe hemodynamic alteration. A considerable tumor (28 × 28 mm) obstructed the left ventricular outflow tract and was complicated with pericardial effusion/peritoneal effusion. In the remaining 27 cases, postnatal data were available for 23 live births; four were lost at follow-up (Table 5).
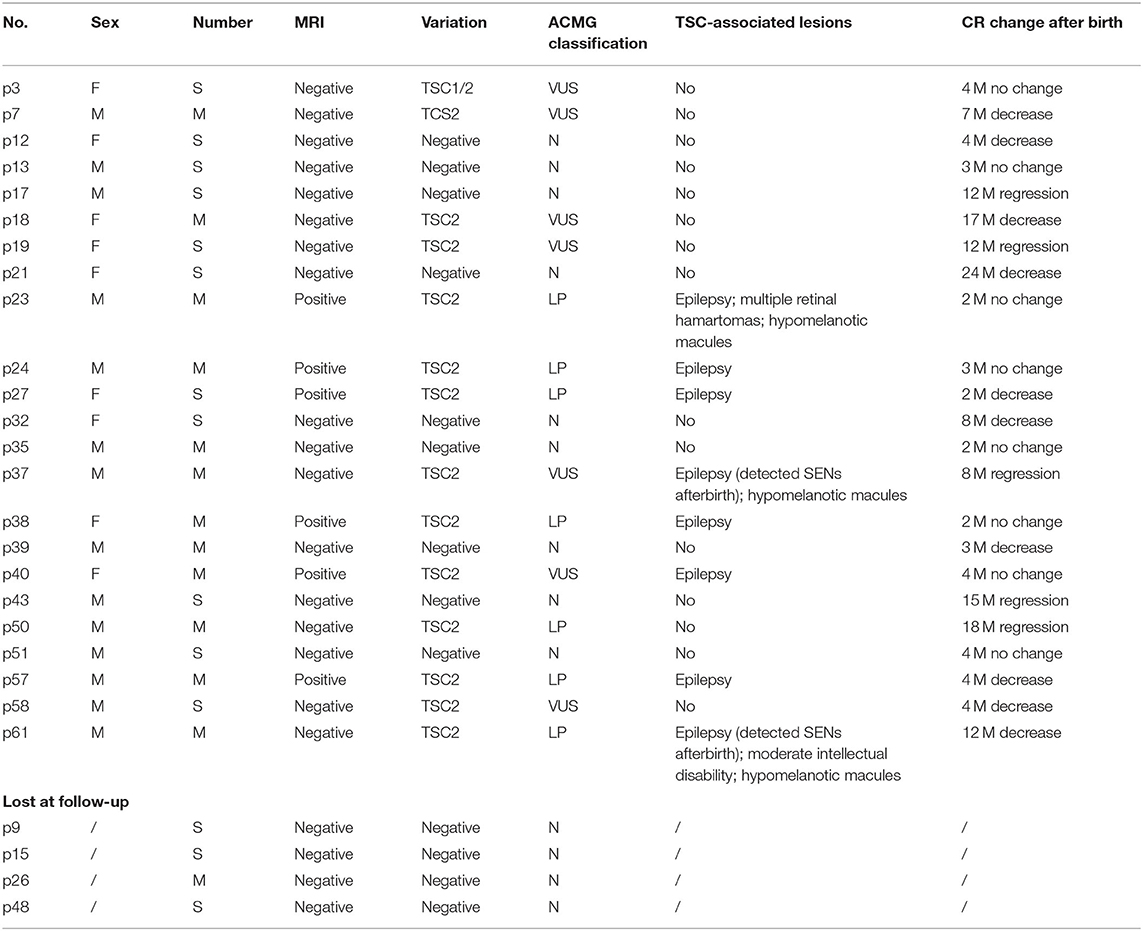
Table 5. Prognosis information for live-born fetuses or those lost at follow-up.
Among the 23 newborns (15 boys, eight girls), the tumor decreased or regressed in 15 of them (10 boys, five girls) no <3 months after birth. Overall, nine of the 23 newborns were negative for TSC1/2 mutation, seven for VUS, and seven for LP. All the brain MRI positive fetuses presented neurological symptoms within 1 year after birth. MRI imaging was standard in p37 and p61 before delivery, for those who carried a VUS and an LP mutation, respectively. While new SENs were found in both when epilepsy occurred at about half a year old. All the fetuses with LP variants were finally proved to have brain lesions except one (case 50). Case 50 showed a known TSC2 missense variant (c.1385G>A; p.R462H), which had already been classified as a likely pathogenic marker for TSC (clinvar). NGS-trio analysis of the family confirmed the father also carried this mutation without the TSC-associated phenotype. What is more, we also detected c.1385G>A in a healthy sibling, aunt, and grandfather (Figure 3A).
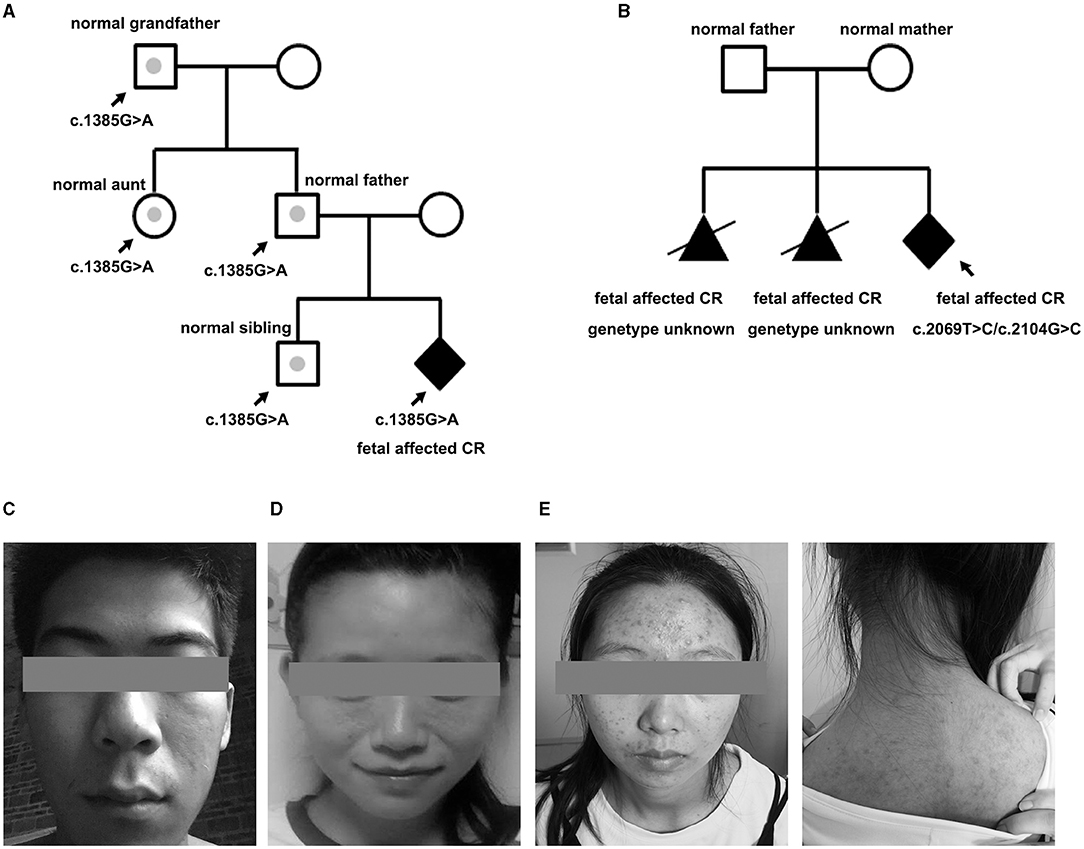
Figure 3. Pedigree of the family variation in TSC2 in a particular family and photographs. (A) The CR-affected fetus had a heterozygote LP variant, which had also been confirmed in the normal relatives (case 50). Gray dots indicate carrying the LP variant but without a TSC phenotype. (B) Pedigree of case 33 is shown with potential gonadal mosaicism. The black triangle indicates the aborted fetuses suffering from CR but without a genetic test. (C) Father of fetus 61 who was identified with 12% mosaicism of the TSC-causing mutation, only having subtle signs of facial angiofibroma. (D) Mother of case 47 who carried a TSC1 mutation but only showed subtle signs of facial lesions. (E) Face and back of the mother of case 20. The facial angiofibroma was occasionally accompanied by infection, but she thought it was intractable acne and never received standardized diagnosis and treatment.
For case 33, it was the third time they had fallen pregnant with a fetus with CR. NGS-trio identified two de novo missense TSC2 mutations in the fetus (Figure 3B). The couple decided to TOP immediately without further evaluation of the neurological examination. We had no more information to clarify the pathogenicity of the two mutants. Considering the uncertain significance of these variants and the possibility of gonadal mosaicism, risk of recurrence is still high.
Work-Flow Suggestion
Thinking over the fact that most intrauterine CRs were first observed coincidentally through a level III ultrasound during 22-26 GA except for the one who had detailed family history, we proposed a simplified clinical work-flow for this situation based on our experience (Figure 4). When cardiac tumors are detected in the mid-trimester, a detailed fetal echocardiography examination is recommended firstly to assess the cardiac structure and function and to give proper treatment. Then, an MRI for the nervous system should be performed as well as family history collection. For CRs in fetuses whose family history is ambiguous or mutation unknown, targeted NGS-Trios could be the most effective and time-saving path when searching the molecular etiology. Genetic counseling should be provided after a comprehensive analysis of clinical symptoms, imaging, and molecular results, which are essential for pregnancy decision or clinical management. Follow-up before and after birth are both indispensable. Benefiting from the multidisciplinary approach, 68.9% of fetuses (42/61) with CRs could receive a precise clinical/genetic diagnosis, or a known outcome, which turned out to be potentially clinically actionable.
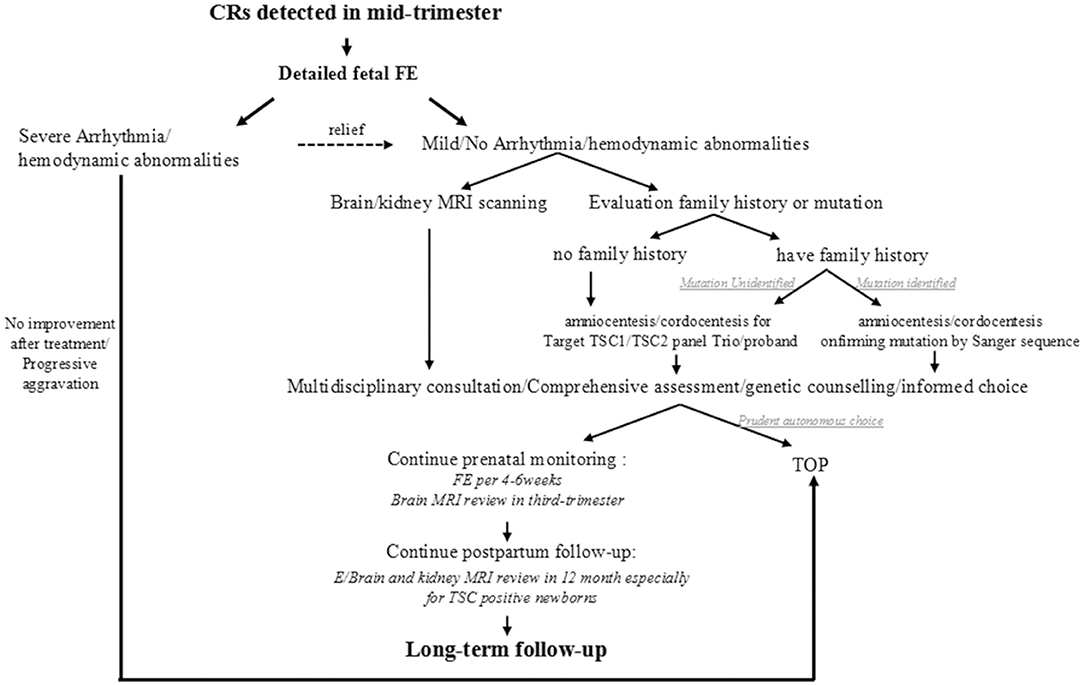
Figure 4. Proposed clinical workflow following CR(s) for other clues to TSC.
Discussion
When cardiac rhabdomyomas are detected in a fetus, both the perinatal outcome dominated by tumor space occupation and the long-term prognosis related to TSC are critical, but tricky. We tried to assess perinatal risk and prenatal diagnosis of TSC with CR as the initial symptom through a multidisciplinary approach, which opens up the possibility of relatively reliable assessment for numerous TSC VUS.
The Influence of Tumor on Perinatal Outcome
Outflow tract obstruction was not rare as it occurred in ~10% of this study. But severe hemodynamics disorders were scattered partly because multiple CRs, which accounted for 70% in this study, tended to be smaller (10) and chamber-located (11). Although 83.7% of tumors increased by the stimulation of maternal estrogen prenatally (12), the increment reduced after the 32nd week (13). So the masses were at their most significant in the mid/last trimester and therefore required more rigorous and careful monitoring. The earliest ultrasonic observable time of CR was reported at 15 weeks of GA (14), but the exact time of tumorigenesis is difficult to deduce. Case 6 provided some clues for early events in tumor formation. We conjectured that early-onset CRs might be manifested as the diffuse ventricular hyperechoic type and could affect the normal remodeling process in the heart. Thus, the diagnosis of TSC in some complex cardiac structural malformation fetuses might be underestimated or ignored. Although the proliferation of CRs seems self-limiting and independent of TSC gene alteration, tumorigenesis might result in gene instability, such as a loss of heterozygosity or a second variant (15). Supporting evidence from targeted-deep sequencing in TSC-associated hamartoma samples found that roughly 2/3 of hamartomas from TSC individuals harbored two TSC hits (16). We tested CR tissues in two aborted TSC fetuses, and both the mutations were consistent with their amniotic fluid results (data are not shown), suggesting a germline origin of mutations. Therefore, strengthening echocardiography monitoring and reassessing tumors in middle pregnancy is indispensable.
Some postpartum studies revealed that up to 23% of children with CRs may present with arrhythmia (17), which may be related to abnormal conduction of electrical excitement in tumor tissues near the atrioventricular junction (18). Tetsuya et al. reviewed 20 fetuses with CRs and speculated that, regardless of number and location, tumor diameter >30 mm is associated with postnatal arrhythmia (19). We observed no arrhythmia in all the subjects including the one who suffered a single huge tumor located in the LV which exceeded 30 mm.
The Influence of Tumor-Related TSC on Long-Term Prognosis
The influence of CR-related TSC is a decisive factor for long-term prognosis, although this is contested. NGS facilitates the detection process but leaves the problem of variant recognition. We amplified the detection yield to 73.8% (including mutations of P, LP, and VUS), which was much higher than 10 years before (20). Chen et al. (11) identified TSC1/TSC2 P/LP variants in 69.8% of fetuses with CR which seemed more instructive. We speculate that there was a selection bias for the inclusion criteria. Rhabdomyomas could not be fully confirmed pathologically until fetus tissues were obtained. Thence the specificity of ultrasound for the diagnosis of CR is the main reason for the difference in the detection rate between different cohorts. Some other benign cardiac tumors, such as teratomas, fibroids, hemangiomas, or hamartomas, might pull down the variants detection rate. In order to avoid this selection bias as much as possible, we differentiated these tumors by rigorous fetal echocardiogram selection according to various characteristics before inclusion (21). The majority of teratomas arise from the pericardium and are of mixed echogenicity with cystic or calcified structures. Almost all are associated with pericardial effusion. Fibroid and hemangioma were often higher in size and grew fast, with or without calcification. Myxomas may attach to the atria and swing with the rhythm; also, malignant tumors are rare (2). Słowińska et al. confirmed the feasibility of a similar algorithm in early diagnosis, before seizure onset, of CR children (22). The cohort composed of newborns or infants within the first 16 weeks of age, so the algorithm was mostly based on postpartum clinical signs, supplemented by brain MRI, echocardiography, skin examination, and genetic testing. Compared with our study, whose objects were all fetuses, antenatally observable phenotypes were extremely limited. Therefore, we weighted the results from genetic assessments in prenatal diagnosis. The accuracy of the algorithm for VUS is vital information that affects family decision-making.
Family phenotype collection is in the top priority of genetic counseling. About 32% of mutations identified in this cohort were inherited from parents without a typical phenotype. Clinical expression is hugely variable. Some subtle signs, mostly skin manifestations, were easily overlooked or misdiagnosed, and 3–6 outpatient visits were needed to obtain a preliminary diagnosis in China. p20 and p23 had both suffered from “acne” for decades, which were actually atypical facial angiofibromas for TSC (Figures 3D,E). Mosaicism is another essential factor affecting phenotype, especially of mild cases with NVI by conventional testing, which afforded 7.5% of patients with a clinical TSC diagnosis (23). Variant allele fraction (VAF) in the blood was positively correlated with the number of major features (23, 24). Interestingly, low-level mosaicism (0–10%, median 1.7%), which likely arises from a later postzygotic variant, had a milder and distinct clinical phenotype in comparison with other TSC series, with similar facial angiofibromas (92%) and kidney angiomyolipomas (83%) (25). VAF of the p61 husband was 12%, he showed an unnoticeable asymmetric facial angiofibroma (Figure 3C). However, the p61 infant, who carried the heterozygous variant, not only had facial lesions but also had intellectual disability, and epilepsy presented at 6 months. The difference in performance is not only closely related to the mosaic level, but may also be affected by factors such as the structure of the variant, the tissue distribution, and the penetrance rate. Cutaneous signs are the most common signs in TSC phenotype series, and vice versa, the variant of TSC1/TSC2 was the most frequently detected in skin lesions (26). Thus, dermatological consultations or even skin biopsy sequencing are necessary for asymptomatic parents whose variants are accidentally discovered by NGS-trio to confirm the diagnosis, especially when encountering a VUS.
Penetrance is high but not absolute, which also influences the assessment of pathogenicity of variants. The TSC2 p.R462H substitution has been annotated as likely pathogenic by LOVD. In vitro experimental studies showed that the missense mutation had an effect on TORC1 activity and interfered with the formation of the TSC1-TSC2 complex, and resulted in an unstable protein and impaired protein function (27). The mutant was not recorded in population databases (rs45494392, ExAC no frequency) but uniquely presented in the p50 pedigree (Figure 3A). The fetus was negative for neuroimaging findings during the whole pregnancy. All indexes point to benign prognosis, which had been confirmed in follow-up. Integrated genetic information in trios, and imaging results may enable early implementation of further diagnostic investigations, perinatal surveillance, and family counseling, especially for couples with no signs of TSC phenotype on clinical examination.
For post-test analysis in the NGS era, it was common for phenotypic fetuses to show VUS of TSC1/2 despite over 10,000 recorded variants. Some other angles of view need to be introduced to aid further evaluation. Pathological changes in the CNS are observed in almost all TSC individuals, and ~80–90% of them present with cortical tubers and/or SENs, and seems equally familiar in TSC1 and TSC2 pathogenic mutations (26). However, antenatal neurological manifestations are easily overlooked and mostly checked after CR detection during the second-trimester scanning when the pathological changes in the CNS might have gone a long way. Saada et al. (14) indicated that the characteristic cerebral lesions of TSC may form as early as 10–20 weeks of embryonic development. As recently as 2007, Mühler et al. preliminarily explored the application of fetal cerebral MRI in sonographically proven CR (28). Here, we disclosed that neuroimaging finding was a much more specific indication of genetic TSC and was proportional to poor prognosis. TSC2 mutations seemed more likely to have neuropathy than TSC1 in prenatal fetuses. In combination with fetal MRI, neurological lesions were found in two NVI cases and five TSC2 VUS cases, for whom a clinical diagnosis of TSC was made. Exceptionally, in one case with VUS, neurological symptoms occurred at 16 weeks after birth. Benefiting from prenatal MRI scanning, some VUS, or even NVI turned out to be potentially clinically actionable. Neuro MRI should be included in the clinical diagnosis process to achieve regular monitoring in a fetus with CR.
Besides, the possibility of parental gonadal mosaicism cannot be overlooked in a typical CR fetus with more than one proband sibling. The reported incidence of germ-line mosaicism ranges from 2 to 5% (29). Case 33 here was a family with two fetuses with CR(s). For this pregnancy, two missense variations of TSC2 were detected in the fetus but absent in both parents. Although both of the mutations were pathogenically ambiguous, it was strongly suspected to be germ-line mosaicism in the couple. It implied that the recurrence risk of unaffected parents who have had an affected child would increase because of germ-line mosaicism, and families with high suspicion of this should be advised to undergo prenatal diagnosis for each pregnancy.
Conclusion
We recommended strengthening fetal heart examination during middle and late pregnancy, especially for pregnancy with a family history of TSC. Fetal echocardiography is the preferred method for detecting fetuses with CR. Neuroimaging findings are a more sensitive indicator compared to the number of CR to imply the pathogenicity of TSC1/2 variants and a poor prognosis. Combined with neuro MRI and genetic testing, it facilitates early detection of TSC and is of great significance for perinatal management and prognostic guidance. The multidisciplinary approach could allow nearly 70% of fetuses with CR(s) to be potentially clinically actionable.
Limitation
Our study still has some limitations that warrant further consideration. First, the pathological information of these cardiac tumors is insufficient and lacks autopsy information. Second, a limited number of cases identified TSC1 mutations, making it difficult to assess the relationship between CR, genotype, and neuroimaging changes for this subpopulation of TSC. Third, the clinical manifestations of TSC can be mimicked by mutations in many genes. Sequence coverage was restricted to exons and adjacent intronic mutations, which perhaps missed mutations deep within introns or in other unknown regulatory regions.
Data Availability Statement
The data presented in the study are deposited in the NCBI repository (https://www.ncbi.nlm.nih.gov/sra/PRJNA742183), accession number (PRJNA742183).
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Guangdong Women and Children Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
YQ carried out all the data analyses, participated in the design of the work, and wrote the draft. HD and YZh collected all clinical data. YH, YZe, LY, and LL participated in molecular genetic test work. AY designed the work and revised the manuscript. All authors have materially participated in the study and manuscript preparation, critically reviewed the manuscript and provided final approval for submission, and agreed to be accountable for all aspects of the work, ensuring the accuracy and integrity of the publication.
Funding
The study was financially supported by the National Key Research and Development Program of China (2016YFC1000700, 2016YFC1000703) and the Medical Scientific Research Foundation of Guangdong Province of China (A2020257). This funding supported us in completing the experiments, sequencing, data analysis, and publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the patients and family members who participated in this study, and all medical staff members who contributed to the collection of clinical information.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.628238/full#supplementary-material
Supplementary Figure 1. Fetal echocardiography. (A) Four-chamber view of p21 at 19 weeks' gestation, a single giant rhabdomyoma (32.2 × 22.5 mm) presented on the left ventricular, with clear border, uniform internal echo, and no obvious relationship with the posterior leaflet of the mitral valve. (B) Multiple rhabdomyomas scattered in LV, RV, and IVS at 32 weeks' gestation of p29, the diameter ranged from 2.2 to 9.0 mm. (C) In the four-chamber view, a large rhabdomyoma was visible in LV, and a small amount of fluid can be seen in the pericardial cavity, about 6.0 mm deep. The p25 fetus was concurrent with peritoneal effusion and stillbirth at 26 weeks' gestation. (D) Four-chamber view of p38 at 35 weeks' gestation, the tricuspid valve is well-open, while moderate regurgitation can be detected when closed.
Supplementary Table 1. Genotypic and phenotypic features of patients with germline mutation of TSC1/2.
Abbreviations
CR, cardiac rhabdomyoma; TSC, tuberous sclerosis complex; MRI, magnetic resonance imaging; VUS, variations of uncertain significance; P, pathogenic; LP, likely pathogenic; SENs, subependymal nodules; CNVs, copy number variations; TOP, termination of pregnancy; NVI, no variant identified; NGS, next generation sequence.
References
1. Yuan SM. Fetal primary cardiac tumors during perinatal period. Pediatr Neonatol. (2017) 58:205–10. doi: 10.1016/j.pedneo.2016.07.004
2. Isaacs H Jr. Fetal and neonatal cardiac tumors. Pediatr Cardiol. (2004) 25:252–73. doi: 10.1007/s00246-003-0590-4
3. Yuan SM. Fetal cardiac tumors: clinical features, management and prognosis. J Perinat Med. (2018) 46:115–21. doi: 10.1515/jpm-2016-0311
4. Altmann J, Kiver V, Henrich W, Weichert A. Clinical outcome of prenatally suspected cardiac rhabdomyomas of the fetus. J Perinat Med. (2019) 48:74–81. doi: 10.1515/jpm-2019-0246
5. Dragoumi P, O'Callaghan F, Zafeiriou DI. Diagnosis of tuberous sclerosis complex in the fetus. Eur J Paediatr Neurol. (2018) 22:1027–34. doi: 10.1016/j.ejpn.2018.08.005
6. Henske EP, Józwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. (2016) 2:16035. doi: 10.1038/nrdp.2016.35
7. Northrup H, Krueger DA, on behalf of the International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr Neurol. (2013) 49:243e54. doi: 10.1016/j.pediatrneurol.2013.08.001
8. Hasbani DM, Crino PB. Crino. Chapter 52: Tuberous sclerosis complex. Handb Clin Neurol. (2018) 148:813–22. doi: 10.1016/B978-0-444-64076-5.00052-1
9. Ogórek B, Hamieh L, Hulshof HM, Lasseter K, Klonowska K, Kuijf H, et al. TSC2 pathogenic variants are predictive of severe clinical manifestations in TSC infants: results of the EPISTOP study. Genet Med. (2020) 22:1489–97. doi: 10.1038/s41436-020-0823-4
10. Chao AS, Chao A, Wang TH, Chang YC, Chang YL, Hsieh CC, et al. Outcome of antenatally diagnosed cardiac rhabdomyoma: case series and a meta-analysis. Ultrasound Obstet Gynecol. (2008) 31:289–95. doi: 10.1002/uog.5264
11. Chen J, Wang J, Sun H, Gu X, Hao X, Fu Y, et al. Fetal cardiac tumor: echocardiography, clinical outcome and genetic analysis in 53 cases. Ultrasound Obstet Gynecol. (2019) 54:103–9. doi: 10.1002/uog.19108
12. Barnes BT, Procaccini D, Crino J, Blakemore K, Sekar P, Sagaser KG, et al. Maternal sirolimus therapy for fetal cardiac rhabdomyomas. N Engl J Med. (2018) 378:1844–5. doi: 10.1056/NEJMc1800352
13. Ekmekci E, Ozkan BO, Yildiz MS, Kocakaya B. Prenatal diagnosis of fetal cardiac rhabdomyoma associated with tuberous sclerosis: a case report. Case Rep Womens Health. (2018) 19:e00070. doi: 10.1016/j.crwh.2018.e00070
14. Saada J, Hadj Rabia S, Fermont L, Le Bidois J, Bernardes LS, Martinovic J, et al. Prenatal diagnosis of cardiac rhabdomyomas: incidence of associated cerebral lesions of tuberous sclerosis complex. Ultrasound Obstet Gynecol. (2009) 34:155–9. doi: 10.1002/uog.6367
15. Karim F, Reza H. Effect of induced hypothermia and rewarming on renal hemodynamics in anesthetized dogs. Life Sci I. (1970) 9:115–63. doi: 10.1016/0024-3205(70)90148-7
16. Martin KR, Zhou W, Bowman MJ, Shih J, Au KS, Dittenhafer-Reed KE, et al. The genomic landscape of tuberous sclerosis complex. Nat Commun. (2017) 8:15816. doi: 10.1038/ncomms15816
17. Józwiak S, Kotulska K, Kasprzyk-Obara J, Domańska-Pakieła D, Tomyn-Drabik M, Roberts P, et al. Clinical and genotype studies of cardiac tumors in 154 patients with tuberous sclerosis complex. Pediatrics. (2006) 118:e1146–51. doi: 10.1542/peds.2006-0504
18. Kocabaş A, Ekici F, Cetin II, Emir S, Demir HA, Ari ME, et al. Cardiac rhabdomyomas associated with tuberous sclerosis complex in 11 children: presentation to outcome. Pediatr Hematol Oncol. (2013) 30:71–9. doi: 10.3109/08880018.2012.734896
19. Ide T, Miyoshi T, Katsuragi S, Neki R, Kurosaki KI, Shiraishi I, et al. Prediction of postnatal arrhythmia in fetuses with cardiac rhabdomyoma. J Matern Fetal Neonatal Med. (2019) 32:2463–8. doi: 10.1080/14767058.2018.1438402
20. Milunsky A, Ito M, Maher TA, Flynn M, Milunsky JM. Prenatal molecular diagnosis of tuberous sclerosis complex. Am J Obstet Gynecol. (2009) 200:321.e1-6. doi: 10.1016/j.ajog.2008.11.004
21. Zhou QC, Fan P, Peng QH, Zhang M, Fu Z, Wang CH. Prenatal echocardiographic differential diagnosis of fetal cardiac tumors. Ultrasound Obstet Gynecol. (2004) 23:165–71. doi: 10.1002/uog.979
22. Słowińska M, Józwiak S, Peron A, Borkowska J, Chmielewski D, Sadowski K, et al. Early diagnosis of tuberous sclerosis complex: a race against time. How to make the diagnosis before seizures? Orphanet J Rare Dis. (2018) 13:25. doi: 10.1186/s13023-018-0764-z
23. Tyburczy ME, Dies KA, Glass J, Camposano S, Chekaluk Y, Thorner AR, et al. Mosaic and intronic mutations in TSC1/TSC2 explain the majority of TSC patients with no mutation identified by conventional testing. PLoS Genet. (2015) 11:e1005637. doi: 10.1371/journal.pgen.1005637
24. Treichel AM, Hamieh L, Nathan NR, Tyburczy ME, Wang JA, Oyerinde O, et al. Phenotypic distinctions between mosaic forms of tuberous sclerosis complex. Genet Med. (2019) 21:2594–604. doi: 10.1038/s41436-019-0520-3
25. Giannikou K, Lasseter KD, Grevelink JM, Tyburczy ME, Dies KA, Zhu Z, et al. Low-level mosaicism in tuberous sclerosis complex: prevalence, clinical features, and risk of disease transmission. Genet Med. (2019) 21:2639–43. doi: 10.1038/s41436-019-0562-6
26. Cotter JA. An update on the central nervous system manifestations of tuberous sclerosis complex. Acta Neuropathol. (2020) 139:613–24. doi: 10.1007/s00401-019-02003-1
27. Hoogeveen-Westerveld M, Wentink M, van den Heuvel D, Mozaffari M, Ekong R, Povey S, et al. Functional assessment of variants in the TSC1 and TSC2 genes identified in individuals with tuberous sclerosis complex. Hum Mutat. (2011) 32:424–35. doi: 10.1002/humu.21451
28. Mühler MR, Rake A, Schwabe M, Schmidt S, Kivelitz D, Chaoui R, et al. Value of fetal cerebral MRI in sonographically proven cardiac rhabdomyoma. Pediatr Radiol. (2007) 37:467–74. doi: 10.1007/s00247-007-0436-y
Keywords: cardiac rhabdomyoma, prenatal diagnoses, tuberous sclerosis (TSC), fetal tumor, targeted exome capture sequencing
Citation: Qi Y, Ding H, Huang Y, Zeng Y, Yu L, Liu L, Zhang Y and Yin A (2021) A Multidisciplinary Approach in Prenatal Diagnosis of TSC With Cardiac Rhabdomyoma as the Initial Symptom. Front. Pediatr. 9:628238. doi: 10.3389/fped.2021.628238
Received: 12 February 2021; Accepted: 21 June 2021;
Published: 27 August 2021.
Edited by:
Umberto Morbiducci, Politecnico di Torino, ItalyReviewed by:
Sergiusz Jozwiak, Warszawski Uniwersytet Medyczny, PolandElizabeth Martina Bebin, University of Alabama at Birmingham, United States
Kang Zeng, Southern Medical University, China
Copyright © 2021 Qi, Ding, Huang, Zeng, Yu, Liu, Zhang and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aihua Yin, eWluYWl3YUB2aXAuMTI2LmNvbQ==