 Jaewon Kim
Jaewon Kim Dong-Woo Lee
Dong-Woo Lee Ja-Hyun Jang
Ja-Hyun Jang Myungshin Kim3
Myungshin Kim3 Jisook Yim
Jisook Yim Dae-Hyun Jang
Dae-Hyun Jang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 26 February 2021
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.628190
This article is part of the Research Topic NGS Technologies of Rare Diseases Diagnosis View all 34 articles
Herein, we present a rare case of co-occurring Duchenne muscular dystrophy (DMD) and frontometaphyseal dysplasia 1 (FMD1), two different X-linked diseases, in a 7-year-old boy. He presented with proximal muscle weakness and elevated creatine phosphokinase levels. A multiplex ligation-dependent probe amplification study of DMD revealed the de novo duplications of exons 2–37, thereby confirming the diagnosis of DMD. Initial evaluation revealed atypical features, such as facial dysmorphism, multiple joint contractures, and severe scoliosis, at an early age. However, these were overlooked and were assumed to be atypical manifestations of DMD. Then, the patient's maternal cousin was diagnosed with FMD1 with pathogenic missense variant in FLNA (NM_001110556.2: c.3557C>T/p.Ser1186Leu). A family genetic test revealed that the patient and his mother had the same pathogenic variant in FLNA. The patient's atypical manifestations were considered symptoms of FMD1. Therefore, if one disease does not fully explain the patient's clinical features, an expanded genetic study is needed to detect coincidental disease.
Duchenne muscular dystrophy (DMD) is characterized by progressive muscle weakness and atrophy, and it affects ~1 in 3,500−5,000 males (1). DMD is caused by pathogenic variants in DMD on chromosome Xp21, and over 2,000 pathogenic variants have been determined. Approximately 55–65% of cases are caused by exon deletions, 30% by single-base variants or small deletions or insertions, and 5–15% by exon duplications (2–5). Individuals with this condition usually experience progressive wasting of the skeletal, respiratory, or cardiac muscles, resulting in death due to cardiac or respiratory compromise (6).
Frontometaphyseal dysplasia 1 (FMD1) is a rare genetic disorder caused by pathogenic variants in FLNA on chromosome Xq28 and is characterized by various skeletal development abnormalities. Worldwide, only a few dozen patients have been diagnosed with FMD1 to date. This condition has >10 pathogenic variants, with most being single-base variants (7). Herein, we describe a rare case of co-occurring DMD and FMD1, two different X-linked genetic disorders, coincidently diagnosed in one patient.
A 7-year-old boy visited the neuromuscular disease clinic of our institution with proximal muscle weakness and multiple contractures of the upper and lower limb joints. There was no significant family history. The patient presented with a waddling gait and Gower's sign. However, he could walk independently and go up the stairs with assistance. The manual muscle test showed grade 3/5 for proximal muscles and 4/5 for distal muscles. Whole-spine radiography revealed scoliosis with a Cobb's angle of 25°. According to laboratory tests, the patient's creatine phosphokinase (CPK) level increased to ~10,000 U/L (reference range, <250 U/L). To check for DMD, a multiplex ligation-dependent probe amplification study of DMD was conducted using a kit (MRC Holland, the Netherlands) with two probes (P034-A2 and P035-A1). Results revealed duplications of exons 2–37, which is expected to be out-of-frame (Figures 1A,B). Hence, the proband was diagnosed with DMD. The carrier test result of his mother revealed normal DMD without any pathogenic variants (Figure 1C).
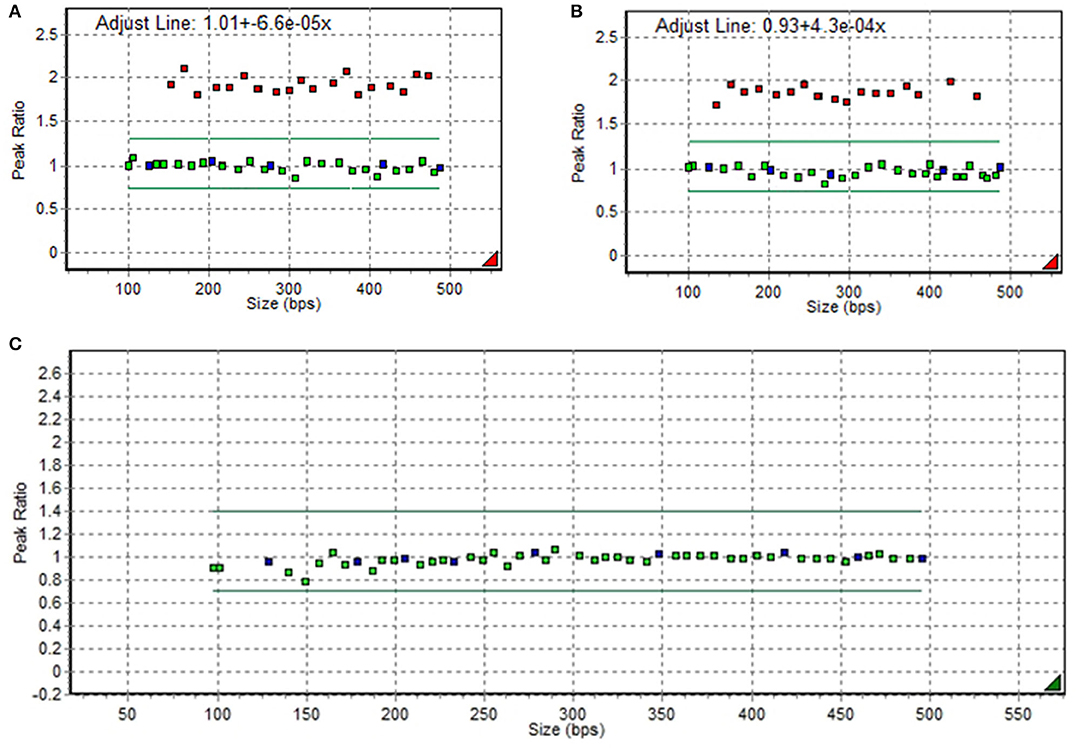
Figure 1. Duplications of exons 2−37 in DMD on multiplex ligation-dependent probe amplification electropherogram in the proband (A,B) and in his mother (C).
Based on physical examination, the patient showed facial dysmorphism with hypodontia, prominent supraorbital ridge, broad and depressed nasal ridge, and micrognathia. Other than muscular dystrophy, various features, such as multiple joint contractures and facial dysmorphism, were atypical manifestations of DMD. The patient received regular steroid and physical therapy. However, multiple joint contractures worsened over time, and 4 years after diagnosis, at 11 years of age, he started using a wheelchair. The patient was subsequently lost to follow-up.
After 3 years, another 13-year-old boy with an appearance similar to that of the current patient visited the neuromuscular disease clinic of our institution due to multiple contractures and facial dysmorphism. Using multigene panel sequencing for skeletal dysplasia, the patient was diagnosed with FMD1 caused by FLNA located at chromosome Xq28. The missense FLNA variant, which was previously reported as pathogenic variant was identified (NM_001110556.2: c.3557C>T /p.Ser1186Leu) (8, 9). Based on family history, the two children were identified as maternal cousins.
When the DMD patient was 15 years old, we called up the patient and requested him to visit our neuromuscular disease clinic to assess the pathogenic variant of FLNA. His joint contractures and scoliosis had worsened (Cobb's angle increased from 25° to 51°), and he was wheelchair-bound due to progressive weakness, joint contracture, and dyspnea. The strength of both shoulder girdles was graded 2/5; upper limbs, 3/5; proximal lower limbs, 2/5; and distal lower limbs, 3/5, and CPK level was 3,156 U/L.
The patient underwent a Sanger sequencing study for FLNA. This study revealed that he had a hemizygous missense variant (NM_001110556.2: c.3557C>T), which is the same variant observed in his cousin. Finally, the patient was diagnosed with FMD1 caused by FLNA missense variant (c.3557C>T) inherited from the asymptomatic maternal carrier and, coincidently, DMD caused by de novo DMD exons 2–37 duplications.
Through family testing, we found that the proband's mother, aunt, and female cousin also had the same pathogenic heterozygous variant of the FLNA. None of them showed definite phenotype of FMD1 (Figures 2A,B, 3).
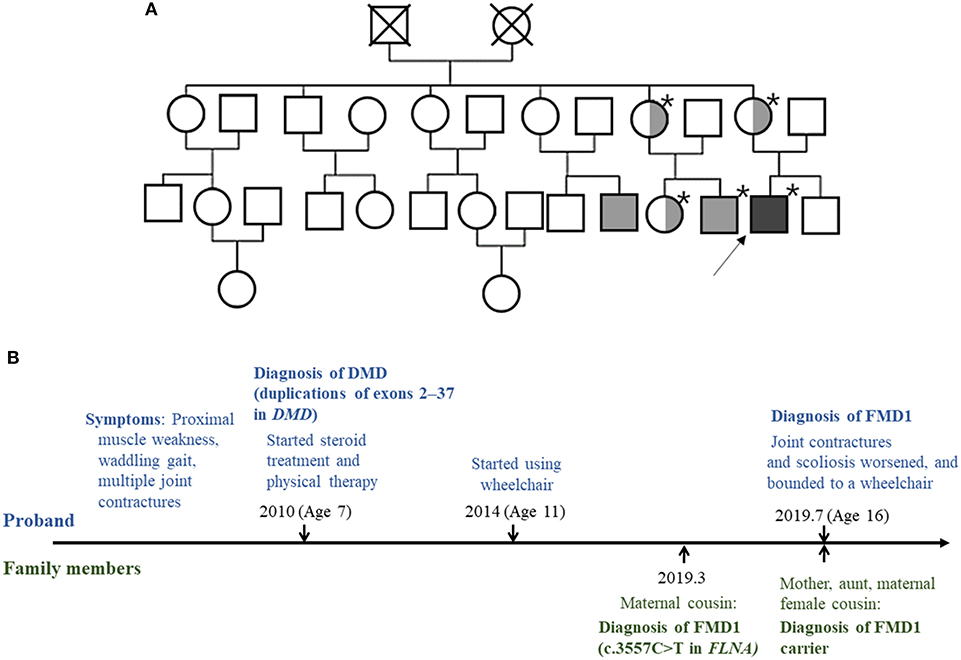
Figure 2. (A) Pedigree of the family. The squares and circles represent male and female family members, respectively. The asterisk indicates people tested for the FLNA pathogenic variant. Light gray color represents family members with the FMD1 phenotype. Symbols divided into halves indicate heterozygous carriers of FMD1. Dark gray color represents the co-occurrence of FMD1 and DMD. The arrow indicates the proband. (B) Timeline of the clinical presentation and molecular diagnosis of the proband and his family members.
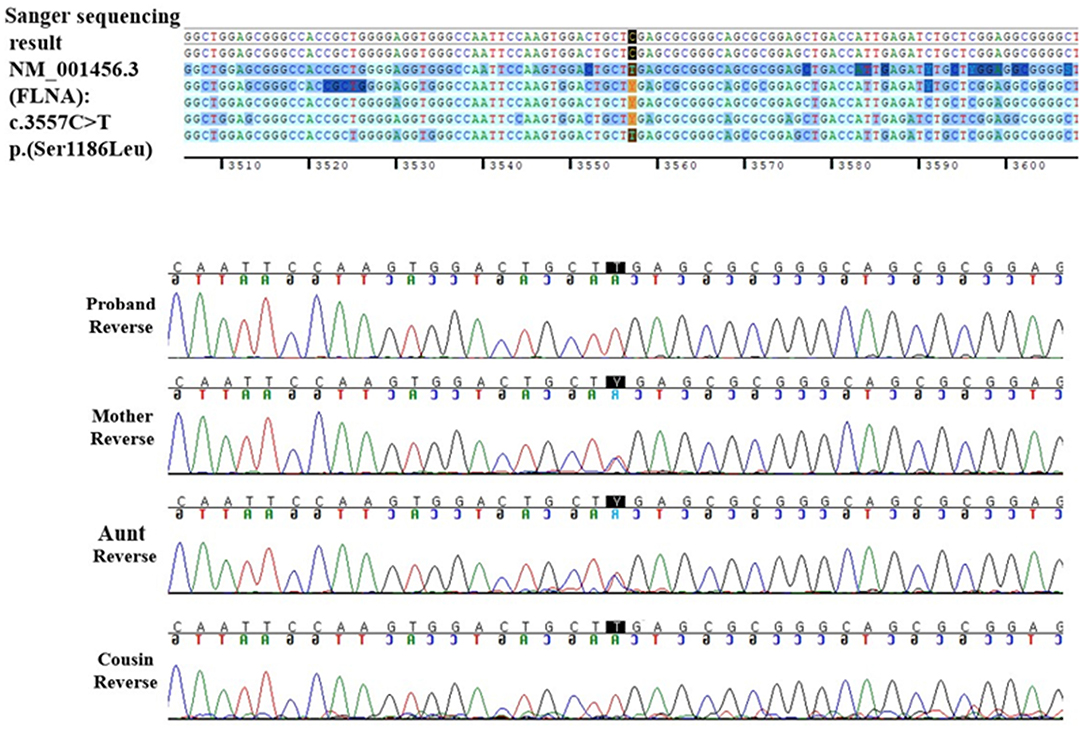
Figure 3. FLNA reverse DNA sequencing chromatogram of the proband and his family.
Since the patient's cousin had Chiari I malformation and syringomyelia, brain and whole-spine magnetic resonance imaging was conducted and Chiari I malformation was detected. Additionally, head computed tomography scan revealed craniosynostosis similar to his cousin.
Herein, we report a rare case of co-occurring DMD caused by DMD exons 2–37 duplications and FMD1 caused by FLNA pathogenic variant (NM_001110556.2: c.3557C>T/p.Ser1186Leu) in a patient. The patient presented with atypical DMD characteristics, such as severe scoliosis, joint contractures, facial dysmorphism, Chiari I malformation, and craniosynostosis. Joint contracture is a common complication of DMD; however, in this case, the patient had developed severe joint contracture at an early age (10).
There have been few cases in which DMD alone induced facial dysmorphism. In a previous case, one patient with DMD presented with facial dysmorphism and craniosynostosis. However, only DMD gene-targeted sequencing was conducted, and the presence of other genetic abnormalities was not validated (11). In our case, the presence of multiple contractures and facial dysmorphism, which are not indicative of DMD, was only explained after the diagnosis of FMD1.
This patient presented with two genetic diseases that cause musculoskeletal abnormalities. Hence, whether defective dystrophin and filamin A had interactive or synergic effects was not evaluated in our study but should be considered in the future.
DMD is an X-linked recessive inheritance disorder caused by pathogenic variants in DMD that encodes for dystrophin protein, which acts as a connector of the cytoskeleton of muscle fibers and extracellular matrix. These variants help in muscle fiber stabilization during muscle exertion. In the absence of normally functioning dystrophin, progressive muscular damage occurs and regeneration is restricted. Muscle fibers are subsequently transformed into fibrotic or adipose tissues, thus causing pseudohypertrophy (12). In DMD, musculoskeletal complications include progressive muscle degeneration and contracture, resulting in postural compensation or deformities including scoliosis (13). In the later stages, the involvement of cardiac and respiratory muscle becomes apparent. In general, patients require ventilation support during their 20s, and premature death usually occurs during their third or fourth decade of life (14, 15).
FMD1 is a rare genetic disease classified under otopalatodigital syndrome spectrum disorder. It is characterized by skeletal dysplasia, such as multiple joint contractures involving the fingers and/or toes, facial deformity, supraorbital hyperostosis, scoliosis, and sensorineural/conductive hearing loss. The facial features of FMD1 include prominent supraorbital ridge, hypertelorism, and micrognathia (8). In most cases, FMD1 is caused by single-base variants, and all pathogenic variants can maintain the reading frame (9). The FLNA comprises 48 exons encoding a 280 kDa filamin A protein. In humans, the filamins are a group of proteins comprising filamins A, B, and C. These proteins are involved in the internal networks of cytoskeleton and filamentous actin rearrangements in the networks in response to mechanical stress. It anchors various transmembrane proteins to the actin cytoskeleton and mediates the signaling processes. Furthermore, filamin A interacts with the glycoprotein-Ibα subunit of the von Willebrand factor receptor, which is associated with cytoskeletal rearrangements, and plays a role in blood vessel and blood clotting (16, 17). Further, filamin C and dystrophin protein were found to have an indirect interaction with each other. However, whether filamin A and dystrophin are correlated or have an interaction with each other is not elucidated (18). Therefore, from a molecular biologic aspect, the presence of these two diseases in one patient is not expected to exert a synergic effect. However, from a clinical aspect, the patient presented with an accelerated deterioration of function, such as gait disturbance, early wheelchair-bound status, severe joint contracture, and scoliosis. If the patient was diagnosed earlier, intensive rehabilitative intervention could have had a positive effect in this case.
The current patient presented with duplications of exons 2–37 in DMD, which is an out-of-frame variant, and might have caused DMD (19). In addition, a hemizygous missense variant (NM_001110556.2: c.3557C>T /p.Ser1186Leu) in FLNA was previously considered pathogenic, thereby leading to a diagnosis of FMD1.
Until now, there are only a few cases of DMD wherein two or more pathogenic variants occurred simultaneously in the X chromosome. Previous studies have reported Fabry disease (Xq22.1), hemophilia A (Xq28), X-linked myotubular myopathy (Xq28), and oculofaciocardiodental syndrome (Xp11.4) (20–23) (Table 1). DMD is the largest human gene known to date, of which one of three pathogenic variants occurs as de novo. Hence, in some cases, DMD and other pathogenic variants can coexist in the X chromosome (24).
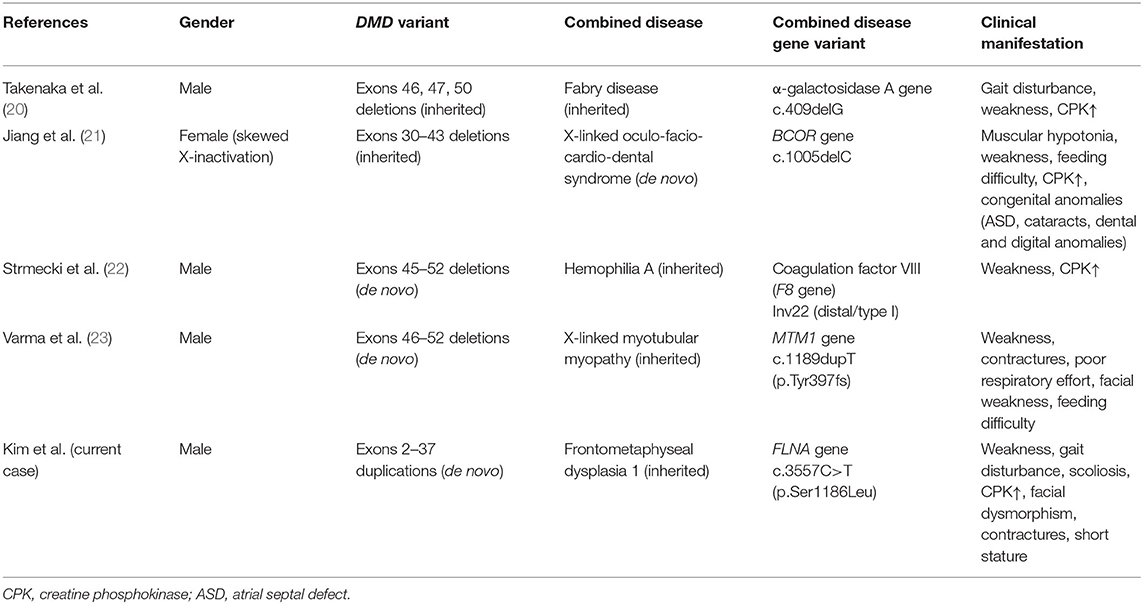
Table 1. Pathogenic variants simultaneously found in the X chromosome in patients with DMD.
Here we report the co-occurrence of DMD and FMD1 in patient, which has not been previously reported to date. Both diseases showed musculoskeletal involvement and X-linked recessive inheritance. DMD occurred de novo, and it was incidentally found that FMD1 was a maternal inheritance. In rare cases, two or more genetic disorders can occur concurrently. Therefore, if one disease does not fully explain the patient's clinical features, an expanded genetic study is required to detect coincidental diseases.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Institutional Review Board, Incheon St. Mary's Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
JK: acquisition of data, analysis and interpretation of data, and writing. D-WL: acquisition of data and critical revision of manuscript. J-HJ, MK, and JY: analysis and interpretation of data. D-HJ: study concept and design, acquisition of data, analysis and interpretation of data, study supervision, and critical revision of manuscript for intellectual content. All authors: contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.628190/full#supplementary-material
1. Emery AE. Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul Disord. (1991) 1:19–29. doi: 10.1016/0960-8966(91)90039-U
2. Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. (1987) 50:509–17. doi: 10.1016/0092-8674(87)90504-6
3. Roberts RG, Gardner RJ, Bobrow M. Searching for the 1 in 2,400,000: a review of dystrophin gene point mutations. Hum Mutat. (1994) 4:1–11. doi: 10.1002/humu.1380040102
4. Sitnik R, Campiotto S, Vainzof M, Pavanello RC, Takata RI, Zatz M, et al. Novel point mutations in the dystrophin gene. Hum Mutat. (1997) 10:217–22. doi: 10.1002/(SICI)1098-1004(1997)10:3<217::AID-HUMU7>3.0.CO;2-F
5. Hu X, Ray PN, Murphy EG, Thompson M, Worton R. Duplicational mutation at the Duchenne muscular dystrophy locus: its frequency, distribution, origin, and phenotypegenotype correlation. Am. J. Hum. Genet. (1990) 46:682
6. Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. (2018) 17:347–61. doi: 10.1016/S1474-4422(18)30025-5
7. FLNA Gene. Available online at: https://ghr.nlm.nih.gov/gene/FLNA (accessed July 13, 2020).
8. Robertson SP, Jenkins ZA, Morgan T, Ades L, Aftimos S, Boute O, et al. Frontometaphyseal dysplasia: mutations in FLNA and phenotypic diversity. Am J Med Genet A. (2006) 140:1726–36. doi: 10.1002/ajmg.a.31549
9. Robertson SP, Twigg SR, Sutherland-Smith AJ, Biancalana V, Gorlin RJ, Horn D, et al. Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat Genet. (2003) 33:487–91. doi: 10.1038/ng1119
10. Emery AE, Muntoni F, Quinlivan RC. Duchenne Muscular Dystrophy. 4th ed. New York, NY: OUP Oxford (2015). p. 29–51.
11. Xu Y, Wang H, Xiao B, Wei W, Liu Y, Ye H, et al. Novel noncontiguous duplications identified with a comprehensive mutation analysis in the DMD gene by DMD gene-targeted sequencing. Gene. (2018) 645:113–8. doi: 10.1016/j.gene.2017.12.037
12. Verhaart IEC, Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol. (2019) 15:373–86. doi: 10.1038/s41582-019-0203-3
13. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. (2010) 9:177–89. doi: 10.1016/S1474-4422(09)70272-8
14. Emery AE. The muscular dystrophies. Lancet. (2002) 359:687–95. doi: 10.1016/S0140-6736(02)07815-7
15. Van Ruiten H, Bushby K, Guglieri M. State of the art advances in Duchenne muscular dystrophy. Eur. Med. J. (2017) 2:90–9.
16. van der Flier A, Sonnenberg A. Structural and functional aspects of filamins. Biochim Biophys Acta. (2001) 1538:99–117. doi: 10.1016/S0167-4889(01)00072-6
17. Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, et al. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. (2001) 2:138–45. doi: 10.1038/35052082
18. Dalkilic I, Kunkel LM. Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev. (2003) 13:231–8. doi: 10.1016/S0959-437X(03)00048-0
19. Ling C, Dai Y, Fang L, Yao F, Liu Z, Qiu Z, et al. Exonic rearrangements in DMD in Chinese Han individuals affected with Duchenne and Becker muscular dystrophies. Hum Mutat. (2020) 41:668–77. doi: 10.1002/humu.23953
20. Takenaka T, Sakuraba H, Hashimoto K, Fujino O, Fujita T, Tanaka H, et al. Coexistence of gene mutations causing Fabry disease and Duchenne muscular dystrophy in a Japanese boy. Clin Genet. (1996) 49:255–60. doi: 10.1111/j.1399-0004.1996.tb03783.x
21. Jiang YH, Fang P, Adesina AM, Furman P, Johnston JJ, Biesecker LG, et al. Molecular characterization of co-occurring Duchenne muscular dystrophy and X-linked oculo-facio-cardio-dental syndrome in a girl. Am J Med Genet A. (2009) 149A:1249–52. doi: 10.1002/ajmg.a.32863
22. Strmecki L, Hudler P, Benedik-Dolnicar M, Komel R. De novo mutation in DMD gene in a patient with combined hemophilia A and Duchenne muscular dystrophy. Int J Hematol. (2014) 99:184–7. doi: 10.1007/s12185-013-1488-4
23. Varma U, Mukherjee D, Hughes I, Sethuraman C, Kamupira S. X-linked myotubular myopathy and Duchenne muscular dystrophy in a preterm infant: a rare combination. Pediatrics. (2020) 146:e20182879. doi: 10.1542/peds.2018-2879
Keywords: Duchenne muscular dystrophy, frontometaphyseal dysplasia 1, X-linked genetic diseases, FLNA gene mutation, genetic disease
Citation: Kim J, Lee D-W, Jang J-H, Kim M, Yim J and Jang D-H (2021) Case Report: Co-occurrence of Duchenne Muscular Dystrophy and Frontometaphyseal Dysplasia 1. Front. Pediatr. 9:628190. doi: 10.3389/fped.2021.628190
Received: 11 November 2020; Accepted: 08 February 2021;
Published: 26 February 2021.
Edited by:
Emiliano González Vioque, University Clinical Hospital of Santiago, SpainReviewed by:
Muhammad Jawad Hassan, National University of Medical Sciences (NUMS), PakistanCopyright © 2021 Kim, Lee, Jang, Kim, Yim and Jang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dae-Hyun Jang, ZGhqYW5nbWRAbmF2ZXIuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.