 Tashunka Taylor-Miller
Tashunka Taylor-Miller Jeremy Allgrove
Jeremy Allgrove- Department of Endocrinology and Metabolic Medicine, Great Ormond Street Hospital for Children NHS Foundation Trust, London, United Kingdom
The physiology and regulation of bone minerals in the fetus and the newborn is significantly different from children and adults. The bone minerals calcium, phosphate and magnesium are all maintained at higher concentrations in utero to achieve adequate bone accretion. This is an integral component of normal fetal development which facilitates safe neonatal transition to post-natal life. When deciphering the cause of bone mineral disorders in newborns, the potential differential diagnosis list is broad and complex, including several extremely rare conditions. Also, significant discoveries including new embryological molecular genetic transcription factors, the role of active placental mineral transport, and hormone regulation factors have changed the understanding of calcium and phosphate homeostasis in the fetus and the newborn. This article will guide clinicians through an updated review of calcium and phosphate physiology, then review specific conditions pertinent to successful neonatal care. Furthermore, with the advancement of increasingly rapid molecular genetic testing, genomics will continue to play a greater role in this area of fetal diagnostics and prognostication.
Take Home Points:
1. Fetal and neonatal mineral metabolism differs significantly from that in later life.
2. The regulation of sodium/phosphate cotransporter activity in the renal tubules is the primary mechanism by which phosphate homeostasis is maintained. Major phosphaturic hormones that regulate renal phosphate handling are PTH and FGF23.
3. Advances in genetics have identified new gene mutations in which have clarified the causes of several conditions previously thought to be “idiopathic.”
4. A thorough understanding of the topic is essential to correct diagnosis and treatment of disorders of calcium and phosphate in the newborn.
Introduction
Over the past 35 years there have been significant advances in the understanding of materno-fetal mineral homeostatic mechanisms. Parathyroid hormone related peptide (PTHrP) was first described in 1985 as a new compound with parathyroid hormone (PTH)-like bioactivity that accounted for the discrepancy between human umbilical cord and maternal PTH levels (1). This discovery provided new insight as to why fetal PTH levels were so low, yet fetal calcium levels were maintained higher than and independent of maternal calcium concentrations. Another important novel finding was made in 2000, when bone-derived hormone Fibroblast Growth Factor-23 (FGF23) was found to cause autosomal dominant hypophosphataemic rickets (ADHR), which provided the underlying mechanism for the previously unknown “phosphaturic factor” causing hypophosphataemia (2, 3). Genomic discoveries have continued to provide new insights into the mechanisms facilitating transplacental bone mineral transport and unveil the causation of conditions previously thought to be idiopathic. As the fetus accumulates 80% of its bone mineral content in the third gestational trimester (4), this time is critical to achieve normal skeletal mineralisation by 40 weeks gestation and support successful transition to post-natal life. Passive and active transport of bone-minerals occurs across the placenta to achieve higher fetal concentration of calcium, phosphate, and magnesium compared to maternal levels. Once the baby is born, loss of placental delivery of minerals causes a sudden drop in serum concentrations of these bone minerals which triggers a rise in regulating factors such as PTH, 1,25-dihydroxyvitamin D [1,25(OH)2D, calcitriol] and FGF23 to maintain postnatal homeostasis. This article will first examine current understanding of fetal-to-neonatal mineral homeostasis mechanisms, and then review specific conditions pertinent to successful neonatal care. Magnesium and vitamin D homeostasis will also be briefly discussed.
Fetal Calcium Homeostasis
Fetal blood calcium concentrations are maintained ~0.3–0.5 mmol/L higher than in maternal circulation, with the placenta transporting 100–150 mg/kg/day of calcium during the third trimester (4–6). To achieve this, active materno-fetal transplacental transport is facilitated by transmembrane calcium-selective channel TRPV6, calbindin D9k and plasma membrane calcium-ATPase. Once calcium has been delivered to the fetus, concentrations are tightly regulated by the calcium sensing receptor (CaSR) which is primarily expressed in the fetal parathyroid glands and kidneys. The CaSR activates magnesium-dependent G-protein coupled downstream signalling cascades to control PTH secretion and renal calcium handling. Mutations in CASR result in distinct phenotypes causing either hyper- or hypocalcaemia.
PTH is integral for achieving normal bone mineralisation and maintaining fetal calcium homeostasis by regulating expression of calciotropic genes and other solute transporters within the placenta (7). By the 10th week of gestation PTH is synthesised from fetal parathyroid glands, but circulating concentrations are kept low during fetal life due to relative hypercalcaemia dictated by the CaSR. Fetal parathyroid glands differentiate from endoderm cells in the third and fourth pharyngeal pouches, and mutations in any of the involved genes or transcription factors results in several genetic hypoparathyroidism conditions (8, 9). Both PTH and PTHrP, acting on PTH1 receptor to increase resorption of calcium from bone and kidney and expression of 1a-hydroxylase enzyme, play a critical role in endochrondral bone formation and stimulation of placental calcium transport (4, 10, 11).
Birth causes disruption of the maternal-fetal calcium supply and rapid 30% drop in serum calcium concentrations (4). This triggers a 2–5-fold increase in PTH secretion to stimulate calcitriol synthesis, resorption of calcium from renal tubules, and mobilisation of calcium from skeletal stores to maintain normocalcaemia in the first 48 postnatal hours (4, 6). Hypocalcaemia can be much more pronounced in premature infants due to lack of third trimester bone mineral accretion and gestational unresponsiveness of the parathyroid glands (6). The gastrointestinal tract then becomes the main source of calcium for the newborn. As such, the feeding mode and volume determines calcium availability. For example, exclusively breast-fed infants receive ~200 mg/day calcium (12). Active calcium absorption is driven by calcitriol. The PTH surge drives upregulation of calcitriol synthesis which increases serum total calcium to adult values within 48 h which are then strictly maintained between 2.12 and 2.62 mmol/L (12). There is evidence, however, that the newborn gut is not fully responsive to calcitriol until 4 weeks of age (4).
Fetal Phosphorus Homeostasis
Fetal serum phosphate is maintained ~0.5 mmol/L higher in the fetus compared to the mother, though the mechanisms for placental transport are unclear (4). The majority of phosphate, ~60–70 mg/kg/day, is also accumulated during the third trimester of gestation and is stored primarily within bone as hydroxyapatite (5, 6). Phosphate is integral to endochondral bone formation by mediating hypertrophic chondrocyte apoptosis, and in the mineralisation of fetal bone as it is incorporated into the osteoid allowing calcium to bind to it (13).
The homeostatic mechanisms by which phosphate concentrations are “sensed” in humans are not fully understood, though it is possibly via a plasma membrane complex and that intestinal lumen levels are involved in feedback-regulation on renal phosphate reabsorption (14, 15). Elevation of extracellular phosphate activates FGF23, the primary endocrine regulator of phosphate that is produced by osteocytes and osteoblasts in bone. A complex signalling cascade is then activated when FGF23 binds with co-receptor Klotho to the FGF-receptor (FGFR1) in the kidney (16). The three primary actions of FGF23 are: promoting phosphaturia by phosphorylation of the sodium/hydrogen exchange regulatory factor (NHERF1 coded by SLC9A3R1) and down-regulation of NaPi type 2a/2c co-transporters in the renal proximal tubule (coded by SLC34A1 and SLC34A3, respectively); reducing calcitriol metabolism by downregulation of 1-alpha-hydroxylase activity and upregulating catabolic enzyme 24-hydroxylase activity; and have a direct effect on parathyroid glands to reduce PTH secretion (14–17). Mutations in any of these genes can cause variable degrees of nephrocalcinosis, hypophosphataemia, hypercalcaemia, and rickets (18–20). There is emerging evidence that phosphate also has a direct effect on the parathyroid glands and CaSR, in that hyperphosphataemia directly inhibits CaSR activity which, in turn, stimulates PTH secretion and thus promotes renal phosphate wasting from the proximal renal tubule (21).
Immediately after birth phosphate concentrations are low, ~2.6 mmol/L, and rise during the first 48 h of life (6). This rise is likely to be due to immature renal excretion mechanisms (4). The main source of phosphate is dietary, so the method of infant feeding will determine phosphate loading. After birth, normal concentrations of phosphate are dependent on growth and must be interpreted within the context of age-related laboratory reference ranges.
Fetal Magnesium Homeostasis
Like calcium and phosphate, fetal magnesium concentrations in utero are also maintained independently of maternal concentrations, only 0.05 mmol/L higher though, and accrual of ~3–5 mg/kg/day primarily occurs in the third trimester gestation (4, 6, 22). Magnesium is absorbed via TRPM6 and TRPM7 transcellular transporters in the gut and renal tubules, however, and the precise mechanisms controlling placental magnesium transfer and fetal homeostasis remain unknown (4). Magnesium is an important cation that binds to the CaSR, causing modest influence on PTH secretion, and hypomagnesemia can blunt effective PTH secretion (23, 24).
Fetal Vitamin D Homeostasis
Vitamin D plays a much more important role in postnatal life rather than assisting transplacental mineral homeostasis or fetal mineral accretion. Whilst maternal 25-hydroxyvitamin D readily crosses the placenta to achieve fetal concentrations that are 75–100% of maternal concentrations (1), maternal calcitriol does not cross the placenta but is synthesised primarily in fetal kidneys to achieve fetal concentrations 50% that of maternal (4). These low concentrations are likely suppressed by the elevated fetal serum calcium and phosphate, and low concentrations of PTH. However, it has been demonstrated in animal models that the 1,25(OH)2D vitamin D receptor (VDR), and thus by proxy calcitriol, is not actually required in utero for the fetus to achieve normal calcium, phosphorus, or PTH homeostasis, or for normal skeletal mineralisation as the placenta is providing the required mineral transfer (25, 26). It is after birth that the role of calcitriol becomes vital.
After birth the level of vitamin D (cholecalciferol) intake depends on the mode of feeding as the gut becomes the main source of absorption. Breast milk primarily contains vitamin D in the form of cholecalciferol, as very little 25(OH) vitamin D passes from maternal serum into breast milk. Whilst there is good correlation between maternal cholecalciferol intake and infant serum 25(OH) vitamin D concentrations (27), breast milk contains only a small amount of cholecalciferol – no more than 25 IU/L – which is insufficient to meet newborn daily requirements (28, 29). Though infant formulae are fortified with vitamin D, it is the international consensus guidance that all infants should be supplemented with cholecalciferol 400 IU/day for every infant until 12 months of age regardless of feeding mode (30).
Neonatal Hypocalcaemia
Neonatal hypocalcaemia is defined in two ways: one, in term and pre-term infants with birth weight >1,500 g as total serum calcium <2.0 mmol/L or ionised calcium <1.1 mmol/L; and two, in pre-term infants with low birth weight (LBW) <1,500 g as a total serum calcium <1.75 mmol/L or ionised calcium <1 mmol/L (31). Clinical signs of hypocalcaemia are difficult to elicit in newborns and hypocalcaemia is often asymptomatic within 72 h of birth. Acute hypocalcaemia can present as apnoea, irritability, jitteriness, muscle cramps, tetany (including laryngospasm), seizures, cardiac arrhythmias, and QT-segment prolongation. Chronic hypocalcaemia can be more subtle, presenting with dental enamel hypoplasia, subcapsular cataracts, cardiomyopathy, congestive cardiac failure, and basal ganglia calcifications.
The causes of neonatal hypocalcaemia are summarised in Table 1. Parathyroid glands can take >48 h to become responsive to the fetal-to-neonatal transition and important causes of hypocalcaemia can be helpfully thought of as early onset (<72 postnatal hours), or late onset (>72 postnatal hours) (31–33). Several genetic mutations have been found to cause Primary Hypoparathyroidism and should be considered when hypocalcaemia lasts >72 h. Isolated causes of hypoparathyroidism include GMC2 or PTH-gene mutations, autosomal dominant activating CASR or GNA11 mutations, and X-linked SOX3 mutations (8). Mutations in CASR result in distinct phenotypes causing either hyper- or hypocalcaemia. Activating (gain-of-function) CASR mutations decrease the set-point of CaSR and PTH is not secreted when low calcium levels would normally trigger PTH release, resulting in Autosomal Dominant Hypocalcaemia (ADH). Clinically this presents as hypocalcaemia associated with an inappropriately normal-high urinary calcium excretion, presumably due to increased activity of the CaSR in the kidney.

Table 1. Summary of the various causes of neonatal hypocalcaemia.
Hypoparathyroidism can also be associated with more complex syndromes including: 22q Deletion Syndrome; CHARGE association (CHD7); Autoimmune polyglandular syndrome type 1 (AIRE); Hypoparathyroidism, sensorineural deafness, and renal dysplasia (HDR) syndrome (GATA3); mitochondrial disorders; Sanjad-Sakati and Kenney-Caffey syndromes (TBCE or FAM111A) (34). The most prevalent of these complex syndromes is DiGeorge syndrome (DGS) due to deletions in the chromosome region 22q11 involving the candidate gene TBX1, however microdeletions on chromosome 10 (GATA3 or NEBL) may cause similar phenotypes also associated with cardiac abnormalities (35). Hypocalcaemia in DGS is usually transient and due to underdeveloped parathyroid glands, occurring in 60% of patients mostly during the neonatal period (36). Even normocalcaemic DGS infants are likely to have serum calcium concentrations in the lower half of the normal range, so all should be routinely screened for hypocalcaemia (37).
Osteopetrosis is a very rare cause of neonatal hypocalcaemia, where defective osteoclasts are unable to remodel bone. Hypocalcaemic tetany and seizures can be a presenting feature due to the inability to mobilise calcium stores from bone (38). Osteopetrosis is also associated with increased bone mass, fragility fractures, and bone marrow failure (39). It is usually identifiable with plain radiograph with osteosclerosis and “bone-within-bone” appearance on skeletal survey.
Neonatal Hypercalcaemia
Whilst there is no consensus on definition, neonatal hypercalcaemia may be considered when calcium is greater than two standard deviations above the normal mean (ionised calcium above 1.32 mmol/L or adjusted serum calcium >2.6 mmol/L) (40), or a total serum calcium >2.9 mmol/L (41). Clinical features in the newborn can be difficult to identify and may include polyuria, polydipsia, lethargy, vomiting, abdominal pain, failure to thrive, irritability, and seizures. The causes of hypercalcaemia associated with appropriately suppressed PTH secretion are extensive. Neonatal sepsis is the most common cause that lasts longer than two consecutive days, possibly due to extra-renal macrophage production of calcitriol and/or increased cytokine activity (41). Other important common causes include: subcutaneous fat necrosis where granulomatous inflammatory cells express increased calcitriol; increased calcium and phosphate intake in infants receiving parenteral nutrition; Vitamin D intoxication; and Williams-Beuren Syndrome due to gene deletion on chromosome 7q11.23, which classically present with mild hypercalcaemia (2.9 mmol/L) associated with supravalvar aortic stenosis and distinctive facial features (42).
There has been a number of new genetic discoveries for various causes of neonatal hypercalcaemia. Identification of the active calcium placental transport mechanism, transmembrane calcium-selective channel TRPV6, provides genetic explanation for a condition previously labelled “Transient Neonatal Hyperparathyroidism,” which was thought to be have been caused by congenital vitamin D deficiency. Newly identified compound heterozygous missense mutations in TRPV6 were found to prevent adequate transplacental calcium transport and cause potentially lethal skeletal abnormalities (undermineralised bone, fractures, periosteal, and metaphyseal changes), elevated PTH, hypomagnesaemia and hypovitaminosis D (33, 43–45). New genetic mutations in vitamin D metabolism and urinary phosphate excretion have also been identified as causes of previously labelled “Idiopathic Infantile Hypercalcaemia.” Loss-of-function mutation in CYP24A1 [encoding vitamin D breakdown enzyme 25(OH) vitamin D3 24-hydroxylase] and in SLC34A1 (encoding renal proximal tubular NaPi co-transporter) and NHERF1 (a modifier of SLC34A1) cause accumulation of calcitriol, hypercalcaemia, hypercalciuria, and nephrocalcinosis (19, 46). An awareness of these last two conditions is important in reducing long-term kidney disease in adulthood. Other causes of neonatal hypercalcaemia currently do not have an identifiable cause and are truly idiopathic.
When hypercalcaemia is associated with inappropriately detectable PTH (within laboratory “normal” range or elevated), then causes to consider include rare inactivating (loss-of-function) CASR gene mutations which result in CaSR insensitivity and thus PTH secretion is not switched-off until higher-than-normal calcium concentrations, causing Familial Hypocalciuric Hypercalcaemia (FHH). Clinically this presents as generally asymptomatic hypercalcaemia, inappropriately detectable concentrations of PTH, associated with reduced renal calcium excretion. The mode of inheritance appears to cause a “dosage effect” with regard to the severity of hypercalcaemia (47). In contrast, homozygous or compound heterozygous loss-of-function CASR mutations, or heterozygous mutations where the mother is not affected, cause Neonatal Severe Hyperparathyroidism (NSHPT), which is a severe phenotype that is associated with life-threatening hypercalcaemia, hyperparathyroid bone disease and multiple fractures. Early diagnosis is critical to prevent death or neuromotor delay (48).
Rickets and Rickets-Like Disorders
Rickets is a disorder of the growth plate resulting from defective chondrocyte apoptosis and osteoid mineralisation. Rickets can be sub-classified as: calciopenic (due to dietary deficiency of vitamin D or calcium, or due to defects of vitamin D metabolism or action); or phosphopenic (due to renal phosphate wasting or deficiency of phosphate intake). A concurrent serum PTH measurement can be useful when distinguishing between calciopenic (PTH should be elevated) and phosphopenic rickets (PTH likely within normal laboratory reference range or only modest elevation). The radiological and clinical features depend on the child's age at presentation and underlying cause, but include metaphyseal widening, under-mineralised bone matrix (osteomalacia), delayed closure of fontanelles, softening of the skull bones (craniotabes), parietal and frontal bone bossing, craniosynostosis, bowing of long bones, pseudofracture (Looser zones) and fractures, sequelae of hypocalcaemia (seizures, tetany, dilated cardiomyopathy), failure to thrive, decreased muscle tone, and delayed motor milestones (30, 48, 49). It is important to note that craniotabes and ulnar cupping can be normal variants seen in healthy neonates without correlation to maternal or neonatal vitamin D concentrations (4).
Nutritional rickets are the most common form of rickets in the newborn period, and consensus guidelines recommend vitamin D sufficiency of 25(OH) vitamin D >50 nmol/L (30). Breastfed infants of vitamin D deficient mothers, especially with darker skin pigmentation, are high risk and should be routinely screened. The resulting hypocalcaemia is exacerbated by an immature newborn PTH-response. There is significant controversy about the appropriateness of the term “congenital rickets,” as mothers were found to have compounding conditions (such as malnutrition or malabsorption) that interfered with vitamin D metabolism. Therefore, “neonatal rickets” is the preferred terminology (4). Genetic mutations in vitamin D synthesis (25-hydroxylase and 1-alpha-hydroxylase deficiencies) or action (VDR mutations) are rarer causes of vitamin D associated rickets. The demand for skeletal mineral delivery is high, especially in preterm babies, and rachitic skeletal changes that are absent at birth can develop rapidly within 16 days after delivery (50).
Hypophosphataemic rickets uncommonly presents in the newborn. Special consideration is needed, however, to ensure adequate enteral or parenteral supplementation meets the increased skeletal mineralisation demands. Feeding with amino-acid elemental formulas, such as Neocate®, and high-dose antacids, has been associated with reduced bioavailability of phosphate resulting in hypophosphataemic rickets and fractures (51, 52). Hemizygous mutations in phosphate-regulating endopeptidase (PHEX) gene lead to overexpression of FGF23 and cause X-Linked hypophosphataemic rickets (XLH) (53). While the renal phosphate wasting is present from birth, XLH does not tend to become clinically apparent until child begins to weight-bear.
The differential diagnosis for rachitic-appearing skeletal changes in newborns is large and includes neonatal hyperparathyroidism, skeletal dysplasias, hypophosphatasia, metaphyseal chondrodysplasia, osteogenesis imperfecta (OI), and vitamin C deficiency (Scurvy) (49). Of these, hypophosphatasia caused by loss-of-function TNSALP mutations, in its severest forms (perinatal and infantile) can present with profound skeletal hypomineralisation and bone deformity, hypercalcaemia with downregulation of PTH, and hypercalciuria (54).
Bone Fragility
When considering conditions that present in utero and the neonatal period with bone fragility, OI and OI-like disorders may jump to mind. However, they do not tend to present with biochemical mineral disturbance. The most likely cause of fragile or poorly mineralised bones associated with mineral disturbance in the newborn is Metabolic Bone Disease of Prematurity (MBDP), which has multiple contributors to its characteristic biochemical and radiological findings, see Table 2. Clinical features can develop between 3 and 12 weeks of age, so it is important to screen routinely for biochemical evidence of MBDP with serum alkaline phosphatase (ALP), albumin-adjusted calcium, phosphate, and PTH concentrations from 4 weeks of age in at-risk groups (5, 55). In the premature infant gut phosphate is more readily absorbed than calcium so it is important to ascertain whether MBDP is due to hypophosphatemia or hypocalcaemia. A PTH level paired with serum calcium and phosphate, and urine renal tubular resorption of phosphate (TRP) measurement will help differentiate between hypophosphatemia or hypocalcaemia. Secondary elevation of PTH will occur to maintain normocalcaemia, whereas this compensation does not tend to occur with hypophosphatemia and is associated with decreased phosphaturia (56). Initiating the correct supplementation depending on deficiency is important, as phosphate supplementation in the hypocalcaemic state will bind ionised calcium, exacerbating hypocalcaemia, driving PTH higher, exacerbating renal phosphate loss, and worsening MBPD. While there is no biochemical cut-off consensus to diagnose MBDP, in preterm infants <33 weeks of gestation the combination of bone turnover marker ALP >900 IU/L and phosphate <1.8 mmol/L is associated with sensitivity of 70% and specificity of 100% of having low bone mineral density at 3 months corrected age (57). In practice, lower threshold ALP 500–800 IU/L in infants <34 weeks of gestation is used to implement supplementation (58, 59).
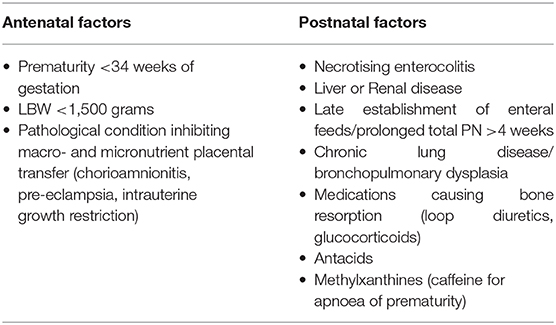
Table 2. Factors contributing to Metabolic Bone Disease of Prematurity.
Radiological changes occur late as bones will only appear generally osteopenic on X-ray when >20% of bone mineral is lost (60). Fragility fractures can occur with incidence reported between 17 and 34%, usually after 10 weeks of age in long bones or ribs up until 6 months of uncorrected gestational age (5). Although all fractures are painful, rib fractures often remain undetected by parents and clinical staff until found incidentally on routine chest X-ray (61). Routine screening with X-ray for MBDP is not indicated in the absence of biochemical disease. Techniques such as dual energy X-ray absorptiometry, peripheral quantitative computed tomography, and quantitative ultrasound have been utilised in research settings (60). There are, however, no normative data <5 years of age, which limits their widespread clinical use.
Prevention of MBDP is limited by multiple factors including the inability of premature infants to tolerate full enteral feeding volumes, the inability to deliver high-dose calcium and phosphate via parenteral nutrition (PN) due to solubility and precipitation limits, and reduced gut absorption of calcium and phosphate. The recommended range delivered via PN route is calcium 40–120 mg/kg/day (1.3–3 mmol/kg/day) and phosphate 31–71 mg/kg/day (1–2.3 mmol/kg/day), and via enteral route is calcium 120–200 mg/kg/day and phosphate 60–140 mg/kg/day (62, 63). Enteral calcium and phosphate supplementation should not be given simultaneously or with milk-meals, to avoid precipitation (56). Effective treatment of MBDP should include routine supplementation with cholecalciferol once on full enteral feeding, aiming for serum 25(OH) vitamin D >50 nmol/L (56, 58). Once treatment of MBDP has been initiated ongoing monitoring of ALP, albumin-adjusted calcium, phosphate, serum creatinine levels, and urine creatinine and TRP is important to avoid hypercalcaemia, hyperphosphataemia, and nephrocalcinosis.
Investigations to Arrange
Essential investigations to assess calcium and phosphate homeostasis include concurrent serum calcium, phosphate, magnesium, PTH, albumin, ALP, electrolytes and renal function, and 25-hydroxyvitamin D levels. Most laboratories provide a calcium corrected for albumin concentration, if not then the following formula will give you the albumin-adjusted serum calcium mmol/L: measured total serum calcium mmol/L + 0.02 × (40 gm/L – measured serum albumin gm/L). High risk infants such as LBW, prematurity <34 weeks, infant of diabetic mother, and prenatal asphyxia should be routinely screened for hypocalcaemia within the first 48 h of age.
If neonatal blood quantities are particularly scarce, then a capillary or venous blood gas will give an ionised calcium value, and serum phosphate can be used as an indirect marker of PTH activity (i.e., low phosphate concentrations reflect high PTH activity and vice versa). Additional investigations such as 1,25(OH)2D and serum DNA for genetic analysis may be required but this should follow discussion with local Paediatric Endocrinology service.
Urinary electrolytes and glucose should be part of routine analysis, to calculate renal calcium: creatinine ratio, and to assess renal tubular function.
Radiology should be considered in specific circumstances, namely when bone mineralisation or skeletal dysplasia is a concern. Usually, diagnosis can be made on a limited series to reduce the neonate's radiation exposure including plain films of anterior-posterior chest and metaphysis of a long bone (e.g., unilateral distal femur or wrist). Specific skull films (looking for Wormian bones) or a full skeletal survey are required if bone fragility or skeletal dysplasia are a concern. The need for these more extensive investigations should be discussed with a local specialist paediatric Radiologist.
Treatment
Appropriate treatment depends on the cause. Where the primary cause is mineral deficiency, additional supplements of calcium, phosphate and, if necessary, magnesium should be given. This can usually be achieved orally but, if demineralisation is severe then intravenous infusion may be required. Vitamin D deficiency should always be corrected.
Hypoparathyroidism, particularly if symptomatic, may require treatment with vitamin D analogues calcitriol or its prohormone alfacalcidol, but caution must be taken to ensure that hypercalciuria and nephrocalcinosis do not result from this treatment. New treatment options in the form of subcutaneous injections of synthetic human PTH teriparatide (hPTH 1-34) and recombinant human PTH (rhPTH 1-84) have been used, particularly where activating mutations of CaSR are the cause of the hypoparathyroidism (23, 64, 65). Calcilytic agents, which reduce the sensitivity of CaSR, are also being investigated as novel therapies for activating CaSR mutations (66).
Hyperparathyroidism can sometimes be corrected with the use of bisphosphonate therapy (although this can lead to an increase in PTH secretion) and/or calcimimetic agents such as cinacalcet (which activate the CaSR, thus increasing the receptor's sensitivity and reducing PTH secretion). Total parathyroidectomy may occasionally be required for intractable NSHPT. Burosumab, a monoclonal antibody to FGF23, is not yet licensed for infants with XLH under 1 year of age but is available in the United Kingdom under an Early Access to Medicines scheme.
Role of Genomics
Genetics plays an important part in diagnosis as an increasing proportion of cases have been found to have a genetic basis. Liaison with a clinical geneticist can be invaluable.
Conclusion
The physiology of mineral metabolism differs considerably between fetal and post-natal life. The neonatal period is one of transition from one to the other and a thorough understanding of these processes is required to be able to diagnose and treat the various conditions when they arise.
Author Contributions
TT-M: writing—original draft, review, and editing. JA: conceptualisation, supervision, writing—review, and editing. All authors have approved the final version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Allgrove J, Adami S, Manning RM, O'Riordan JL. Cytochemical bioassay of parathyroid hormone in maternal and cord blood. Arch Dis Child. (1985) 60:110. doi: 10.1136/adc.60.2.110
2. Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Bioph Res Co. (2000) 277:494–8. doi: 10.1006/bbrc.2000.3696
3. White KE, Evans WE, O'Riordan JLH, Speer MC, Econs MJ, Lorenz-Depiereux B, et al. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. (2000) 26:345–8. doi: 10.1038/81664
4. Kovacs CS. Bone development and mineral homeostasis in the fetus and neonate: roles of the calciotropic and phosphotropic hormones. Physiol Rev. (2014) 94:1143–218. doi: 10.1152/physrev.00014.2014
5. Chinoy A, Mughal MZ, Padidela R. Metabolic bone disease of prematurity: causes, recognition, prevention, treatment and long-term consequences. Archives Dis Child - Fetal Neonatal Ed. (2019) 104:F560. doi: 10.1136/archdischild-2018-316330
6. Karpen HE. Mineral homeostasis and effects on bone mineralization in the preterm neonate. Clin Perinatol. (2018) 45:129–41. doi: 10.1016/j.clp.2017.11.005
7. Simmonds CS, Karsenty G, Karaplis AC, Kovacs CS. Parathyroid hormone regulates fetal-placental mineral homeostasis. J Bone Miner Res. (2010) 25:594–605. doi: 10.1359/jbmr.090825
8. Peissig K, Condie BG, Manley NR. Embryology of the parathyroid glands. Endocrin Metab Clin. (2018) 47:733–42. doi: 10.1016/j.ecl.2018.07.002
9. Grigorieva IV, Thakker RV. Transcription factors in parathyroid development: lessons from hypoparathyroid disorders. Ann Ny Acad Sci. (2011) 1237:24–38. doi: 10.1111/j.1749-6632.2011.06221.x
10. Miao D, He B, Karaplis AC, Goltzman D. Parathyroid hormone is essential for normal fetal bone formation. J Clin Invest. (2002) 109:1173–82. doi: 10.1172/jci14817
11. Kovacs CS, Chafe LL, Fudge NJ, Friel JK, Manley NR. PTH regulates fetal blood calcium and skeletal mineralization independently of PTHrP. Endocrinology. (2001) 142:4983–93. doi: 10.1210/endo.142.11.8509
12. Medicine I of. Dietary Reference Intakes for Calcium and Vitamin D. (2011). Available online at: http://nap.edu/13050 (accessed January 23, 2021).
13. Sabbagh Y, Carpenter TO, Demay MB. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci USA. (2005) 102:9637–42. doi: 10.1073/pnas.0502249102
14. Chande S, Bergwitz C. Role of phosphate sensing in bone and mineral metabolism. Nat Rev Endocrinol. (2018) 14:637–55. doi: 10.1038/s41574-018-0076-3
15. Takashi Y, Fukumoto S. Phosphate-sensing and regulatory mechanism of FGF23 production. J Endocrinol Invest. (2020) 43:877–83. doi: 10.1007/s40618-020-01205-9
16. Razzaque MS. The FGF23–Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. (2009) 5:611–9. doi: 10.1038/nrendo.2009.196
17. Andrukhova O, Zeitz U, Goetz R, Mohammadi M, Lanske B, Erben RG. FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2–SGK1 signaling pathway. Bone. (2012) 51:621–8. doi: 10.1016/j.bone.2012.05.015
18. Karim Z, Gérard B, Bakouh N, Alili R, Leroy C, Beck L, et al. NHERF1 mutations and responsiveness of renal parathyroid hormone. New Engl J Med. (2008) 359:1128–35. doi: 10.1056/nejmoa0802836
19. Schlingmann KP, Ruminska J, Kaufmann M, Dursun I, Patti M, Kranz B, et al. Autosomal-recessive mutations in SLC34A1 encoding sodium-phosphate cotransporter 2A cause idiopathic infantile hypercalcemia. J Am Soc Nephrol. (2016) 27:604–14. doi: 10.1681/asn.2014101025
20. Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genetics. (2006) 78:179–92. doi: 10.1086/499409
21. Centeno PP, Herberger A, Mun H-C, Tu C, Nemeth EF, Chang W, et al. Phosphate acts directly on the calcium-sensing receptor to stimulate parathyroid hormone secretion. Nat Commun. (2019) 10:4693. doi: 10.1038/s41467-019-12399-9
22. Mihatsch W, Fewtrell M, Goulet O, Molgaard C, Picaud J-C, Senterre T, et al. ESPGHAN/ESPEN/ESPR/CSPEN guidelines on pediatric parenteral nutrition: calcium, phosphorus and magnesium. Clin Nutr. (2018) 37:2360–5. doi: 10.1016/j.clnu.2018.06.950
23. Mannstadt M, Bilezikian JP, Thakker RV, Hannan FM, Clarke BL, Rejnmark L, et al. Hypoparathyroidism. Nat Rev Dis Primers. (2017) 3:17055. doi: 10.1038/nrdp.2017.55
24. Allgrove J, Shaw NJ. Calcium and Bone Disorders in Children and Adolescents. 2nd revised edition. Basel: Kager (2015). doi: 10.1159/isbn.978-3-318-05467-5
25. Kovacs CS, Woodland ML, Fudge NJ, Friel JK. The vitamin D receptor is not required for fetal mineral homeostasis or for the regulation of placental calcium transfer in mice. Am J Physiol-Endoc M. (2005) 289:E133–44. doi: 10.1152/ajpendo.00354.2004
26. Ryan BA, Kovacs CS. Calciotropic and phosphotropic hormones in fetal and neonatal bone development. Seminars Fetal Neonatal Med. (2020) 25:101062. doi: 10.1016/j.siny.2019.101062
27. Thiele DK, Senti JL, Anderson CM. Maternal vitamin D supplementation to meet the needs of the breastfed infant. J Hum Lact. (2013) 29:163–70. doi: 10.1177/0890334413477916
28. Holick MF. Resurrection of vitamin D deficiency and rickets. J Clin Invest. (2006) 116:2062–72. doi: 10.1172/jci29449
29. Bae YJ, Kratzsch J. Vitamin D and calcium in the human breast milk. Best Pract Res Cl En. (2018) 32:39–45. doi: 10.1016/j.beem.2018.01.007
30. Munns CF, Shaw N, Kiely M, Specker BL, Thacher TD, Ozono K, et al. Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab. (2016) 101:394–415. doi: 10.1210/jc.2015-2175
31. Vuralli D. Clinical approach to hypocalcemia in newborn period and infancy: who should be treated? Int J Pediatrics. (2019) 2019:1–7. doi: 10.1155/2019/4318075
32. Nadar R, Shaw N. Investigation and management of hypocalcaemia. Arch Dis Childhood. (2020) 105:399–405. doi: 10.1136/archdischild-2019-317482
33. Thomas TC, Smith JM, White PC, Adhikari S. Transient neonatal hypocalcemia: presentation and outcomes. Pediatrics. (2012) 129:e1461–7. doi: 10.1542/peds.2011-2659
34. Abate EG, Clarke BL. Review of hypoparathyroidism. Front Endocrinol. (2017) 7:172. doi: 10.3389/fendo.2016.00172
35. Gordon RJ, Levine MA. Genetic disorders of parathyroid development and function. Endocrin Metab Clin. (2018) 47:809–23. doi: 10.1016/j.ecl.2018.07.007
36. Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. (1997) 34:798. doi: 10.1136/jmg.34.10.798
37. Taylor SC, Morris G, Wilson D, Davies SJ, Gregory JW. Hypoparathyroidism and 22q11 deletion syndrome. Arch Dis Child. (2003) 88:520. doi: 10.1136/adc.88.6.520
38. Wu CC, Econs MJ, DiMeglio LA, Insogna KL, Levine MA, Orchard PJ, et al. Diagnosis and management of osteopetrosis: consensus guidelines from the osteopetrosis working group. J Clin Endocrinol Metab. (2017) 102:3111–23. doi: 10.1210/jc.2017-01127
39. Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis. (2009) 4:5. doi: 10.1186/1750-1172-4-5
40. Stokes VJ, Nielsen MF, Hannan FM, Thakker RV. Hypercalcemic disorders in children. J Bone Miner Res. (2017) 32:2157–70. doi: 10.1002/jbmr.3296
41. McNeilly JD, Boal R, Shaikh MG, Ahmed SF. Frequency and aetiology of hypercalcaemia. Arch Dis Child. (2016) 101:344. doi: 10.1136/archdischild-2015-309029
42. Pober BR. Williams–beuren syndrome. New Engl J Med. (2010) 362:239–52. doi: 10.1056/nejmra0903074
43. Suzuki Y, Kovacs CS, Takanaga H, Peng J, Landowski CP, Hediger MA. Calcium channel TRPV6 is involved in murine maternal–fetal calcium transport. J Bone Miner Res. (2008) 23:1249–56. doi: 10.1359/jbmr.080314
44. Burren CP, Caswell R, Castle B, Welch CR, Hilliard TN, Smithson SF, et al. TRPV6 compound heterozygous variants result in impaired placental calcium transport and severe undermineralization and dysplasia of the fetal skeleton. Am J Med Genet A. (2018) 176:1950–5. doi: 10.1002/ajmg.a.40484
45. Suzuki Y, Chitayat D, Sawada H, Deardorff MA, McLaughlin HM, Begtrup A, et al. TRPV6 variants interfere with maternal-fetal calcium transport through the placenta and cause transient neonatal hyperparathyroidism. Am J Hum Genetics. (2018) 102:1104–14. doi: 10.1016/j.ajhg.2018.04.006
46. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, et al. Mutations in CYP24A1 and Idiopathic Infantile hypercalcemia. New Engl J Med. (2011) 365:410–21. doi: 10.1056/nejmoa1103864
47. Marx SJ, Sinaii N. Neonatal severe hyperparathyroidism: novel insights from calcium, PTH, and the CASR gene. J Clin Endocrinol Metab. (2020) 105:1061–78. doi: 10.1210/clinem/dgz233
48. Egbuna OI, Brown EM. Hypercalcaemic and hypocalcaemic conditions due to calcium-sensing receptor mutations. Best Pract Res Clin Rheumatol. (2008) 22:129–48. doi: 10.1016/j.berh.2007.11.006
49. Carpenter TO, Shaw NJ, Portale AA, Ward LM, Abrams SA, Pettifor JM. Rickets. Nat Rev Dis Primers. (2017) 3:17101. doi: 10.1038/nrdp.2017.101
50. Sann L, David L, Frederich A, Bovier-Lapierre M, Bourgeois J, Romand-Monier M, et al. Congenital rickets study of the evolution of secondary hyperparathyroidism. Acta Paediatr. (1977) 66:323–7. doi: 10.1111/j.1651-2227.1977.tb07901.x
51. Insogna KL, Bordley DR, Caro JF, Lockwood DH. Osteomalacia and weakness from excessive antacid ingestion. JAMA. (1980) 244:2544–6. doi: 10.1001/jama.1980.03310220042025
52. Ballesteros LFG, Ma NS, Gordon RJ, Ward L, Backeljauw P, Wasserman H, et al. Unexpected widespread hypophosphatemia and bone disease associated with elemental formula use in infants and children. Bone. (2017) 97:287–92. doi: 10.1016/j.bone.2017.02.003
53. Bacchetta J, Bardet C, Prié D. FGF23, hypophosphatemia and genetic disorders. Metabolis. (2019) 103:153865. doi: 10.1016/j.metabol.2019.01.006
54. Whyte MP. Hypophosphatasia — aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. (2016) 12:233–46. doi: 10.1038/nrendo.2016.14
55. Abrams SA. Calcium and vitamin D requirements of enterally fed preterm infants. Pediatrics. (2013) 131:e1676–83. doi: 10.1542/peds.2013-0420
56. Chinoy A, Mughal MZ, Padidela R. Current status in therapeutic interventions of neonatal bone mineral metabolic disorders. Seminars Fetal Neonatal Med. (2020) 25:101075. doi: 10.1016/j.siny.2019.101075
57. Backström M, Kouri T, Kuusela A-L, Sievänen H, Koivisto A-M, Ikonen R, et al. Bone isoenzyme of serum alkaline phosphatase and serum inorganic phosphate in metabolic bone disease of prematurity. Acta Paediatr. (2000) 89:867–73. doi: 10.1111/j.1651-2227.2000.tb00395.x
58. Chin LK, Doan J, Teoh YS, Stewart A, Forrest P, Simm PJ. Outcomes of standardised approach to metabolic bone disease of prematurity. J Paediatr Child H. (2018) 54:665–70. doi: 10.1111/jpc.13813
59. Rayannavar A, Calabria AC. Screening for metabolic bone disease of prematurity. Seminars Fetal Neonatal Med. (2020) 25:101086. doi: 10.1016/j.siny.2020.101086
60. Faienza MF, D'Amato E, Natale MP, Grano M, Chiarito M, Brunetti G, et al. Metabolic bone disease of prematurity: diagnosis and management. Front Pediatr. (2019) 7:143. doi: 10.3389/fped.2019.00143
61. Bishop N, Sprigg A, Dalton A. Unexplained fractures in infancy: looking for fragile bones. Arch Dis Child. (2007) 92:251. doi: 10.1136/adc.2006.106120
62. Nehra D, Carlson SJ, Fallon EM, Kalish B, Potemkin AK, Gura KM, et al. Clinical guidelines. Jpen-Parenter Enter. (2013) 37:570–98. doi: 10.1177/0148607113487216
63. Agostoni C, Buonocore G, Carnielli V, Curtis MD, Darmaun D, Decsi T, et al. Enteral nutrient supply for preterm infants: commentary from the European society of paediatric gastroenterology, hepatology and nutrition committee on nutrition. J Pediatr Gastr Nutr. (2010) 50:85–91. doi: 10.1097/mpg.0b013e3181adaee0
64. Winer KK. Advances in the treatment of hypoparathyroidism with PTH 1–34. Bone. (2018) 120:535–41. doi: 10.1016/j.bone.2018.09.018
65. Gafni RI, Collins MT. Hypoparathyroidism. New Engl J Med. (2019) 380:1738–47. doi: 10.1056/nejmcp1800213
66. Letz S, Haag C, Schulze E, Frank-Raue K, Raue F, Hofner B, et al. Amino alcohol- (NPS-2143) and Quinazolinone-derived calcilytics (ATF936 and AXT914) differentially mitigate excessive signalling of calcium-sensing receptor mutants causing bartter syndrome type 5 and autosomal dominant hypocalcemia. PLoS ONE. (2014) 9:e115178. doi: 10.1371/journal.pone.0115178
Keywords: calcium, phosphate, magnesium, vitamin D, PTH, genetics, fibroblast growth factor 23
Citation: Taylor-Miller T and Allgrove J (2021) Endocrine Diseases of Newborn: Epidemiology, Pathogenesis, Therapeutic Options, and Outcome “Current Insights Into Disorders of Calcium and Phosphate in the Newborn”. Front. Pediatr. 9:600490. doi: 10.3389/fped.2021.600490
Received: 30 August 2020; Accepted: 13 January 2021;
Published: 05 February 2021.
Edited by:
Amanda Lesley Ogilvy-Stuart, Cambridge University Hospitals NHS Foundation Trust, United KingdomReviewed by:
Tim Cheetham, Newcastle University, United KingdomBenjamin Udoka Nwosu, University of Massachusetts Medical School, United States
Copyright © 2021 Taylor-Miller and Allgrove. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeremy Allgrove, amVyZW15LmFsbGdyb3ZlQG5ocy5uZXQ=