

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 11 March 2021
Sec. Neonatology
Volume 9 - 2021 | https://doi.org/10.3389/fped.2021.592571
This article is part of the Research TopicResearch Model Innovations in Advancing Neonatal CareView all 21 articles
 Poh Kuan Wong1
Poh Kuan Wong1 Fook Choe Cheah2
Fook Choe Cheah2 Saiful Effendi Syafruddin3
Saiful Effendi Syafruddin3 M. Aiman Mohtar3
M. Aiman Mohtar3 Norazrina Azmi1
Norazrina Azmi1 Pei Yuen Ng1
Pei Yuen Ng1 Eng Wee Chua1*
Eng Wee Chua1*Hereditary or developmental neurological disorders (HNDs or DNDs) affect the quality of life and contribute to the high mortality rates among neonates. Most HNDs are incurable, and the search for new and effective treatments is hampered by challenges peculiar to the human brain, which is guarded by the near-impervious blood-brain barrier. Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR), a gene-editing tool repurposed from bacterial defense systems against viruses, has been touted by some as a panacea for genetic diseases. CRISPR has expedited the research into HNDs, enabling the generation of in vitro and in vivo models to simulate the changes in human physiology caused by genetic variation. In this review, we describe the basic principles and workings of CRISPR and the modifications that have been made to broaden its applications. Then, we review important CRISPR-based studies that have opened new doors to the treatment of HNDs such as fragile X syndrome and Down syndrome. We also discuss how CRISPR can be used to generate research models to examine the effects of genetic variation and caffeine therapy on the developing brain. Several drawbacks of CRISPR may preclude its use at the clinics, particularly the vulnerability of neuronal cells to the adverse effect of gene editing, and the inefficiency of CRISPR delivery into the brain. In concluding the review, we offer some suggestions for enhancing the gene-editing efficacy of CRISPR and how it may be morphed into safe and effective therapy for HNDs and other brain disorders.
The human brain is the most complex organ in our body, consisting of a multitude of neurons communicating with each other. It is the command center that governs our bodily functions, including senses, movements, emotions, language, communication, thoughts, and memory. The intricate neural circuits of the brain are built in utero and continue to grow till adulthood. The process is orchestrated by a collection of genes that encode signals for triggering neural cell differentiation and migration; but many of the genes are still unknown (1, 2). Defects in these genes impair prenatal brain development and cause hereditary neurological disorders (HNDs) (3). Currently, there is no cure for most of the HNDs, as the underlying pathogenesis is often obscure and poorly understood; and effective treatments of HNDs are impeded by the blood-brain barrier (BBB) that prevents drugs from being delivered to their target sites. For many of the known HNDs, symptomatic treatments are the only feasible avenues to clinical care (4, 5). However, a major caveat is that some HNDs may not become manifest until after the neonatal period, and critical treatment opportunities may be missed (6).
Gene-editing systems, such as Zinc Finger Nucleases (ZFN) and Transcription Activator-Like Effector Nuclease (TALEN), are potentially powerful approaches for the disease-modifying treatment of HNDs (7). However, they are complex, time-consuming, and have low gene-editing efficiency. To date, Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) is the most efficient and simplest genome editing system and has been widely used in different cell types and organisms to edit single or multiple target genes (8). CRISPR can be directed to different genetic loci simply by redesigning the sgRNAs (Table 1), unlike ZFN and TALEN which would require time-consuming synthesis of new guiding proteins (9). The ease of reconstructing sgRNAs enables CRISPR to target multiple loci simultaneously, needing only an assortment of gene-specific sgRNAs. Moreover, wild-type Cas9 can be reprogrammed into catalytically inactive Cas9 (dead Cas9 or dCas9) that can modulate target gene expression when it is fused to transcriptional modifiers (10).
The number of clinical trials looking into CRISPR-based therapy, especially that of cancer, is growing, (11). Recently, a patient with Leber congenital amaurosis, a hereditary disorder that causes blindness, became the first patient to undergo in vivo gene editing using CRISPR (ClinicalTrials.gov identifier: NCT03872479). Although CRISPR has not yet reached the clinical stages of testing in humans with HNDs, pre-clinical results have demonstrated the efficacy of CRISPR in correcting faulty genes associated with HNDs (12). Therefore, CRISPR-based treatments could help to reduce the mortality and morbidity in neonates who suffer HNDs. A viable treatment strategy would be pre-emptive CRISPR gene editing that could prevent the causal genetic defects from developing into full-blown HNDs (6).
Besides, CRISPR can be utilized to generate models to study the effects of genetic variation on drug response (13, 14). Caffeine has a beneficial effect on the developing brain, improving cognitive outcomes in infants treated with it (15). However, caffeine sensitivity varies between neonates, possibly owing to genetic variation. The outcome of caffeine therapy was shown to be adversely affected by rs16851030, a DNA variant located in the 3'-untranslated region of the ADORA1 gene—the target of caffeine. Individuals who are homozygous for the rs16851030 C-allele may respond better to caffeine than those who harbor the T-allele (16).
In this review, we discuss how CRISPR may progress from laboratory benches into clinics to improve neonatal care. We cover a particularly interesting but challenging subject—the management of HNDs, which affect the vulnerable, growing brains of newborns. We include only relevant HNDs that are manifest during the neonatal period and those that occur later. We also discuss how CRISPR may help us to understand the genetic basis of the variable caffeine sensitivity among neonates with apnea of prematurity, given the large amount of evidence pointing to a beneficial effect of caffeine on brain development.
Gene therapy has the potential to treat a wide range of inherited diseases, such as cystic fibrosis and muscular dystrophy (17, 18). Traditional gene therapy replaces faulty genes with the correct versions or, with the help of a vector, introduces new genes into cells to produce functional proteins (19) (Figure 1). However, not all gene constructs can fit into a vector. The gene expression system has a size limit, and large genes are difficult to package and deliver into cells (20). Traditional gene therapy works well for autosomal recessive disorders as the mechanism is straightforward. Most autosomal recessive disorders are caused by loss-of-function exonic variants, and inserting normal copies of the target gene into cells is sufficient to restore protein function (21). In contrast, autosomal dominant disorders are caused by gain-of-function exonic variants and require more elaborate gene editing. This includes an exogenous supply of functional gene constructs to restore protein function, and the use of antisense oligonucleotides and small interfering RNAs to silence the transcription of disease-causing genes (22, 23). The additional requirement for gene silencing means that repeat doses would be needed to maintain therapeutic efficacy (24). This would not be necessary with gene edits created by CRISPR, as the outcome is long-lasting.
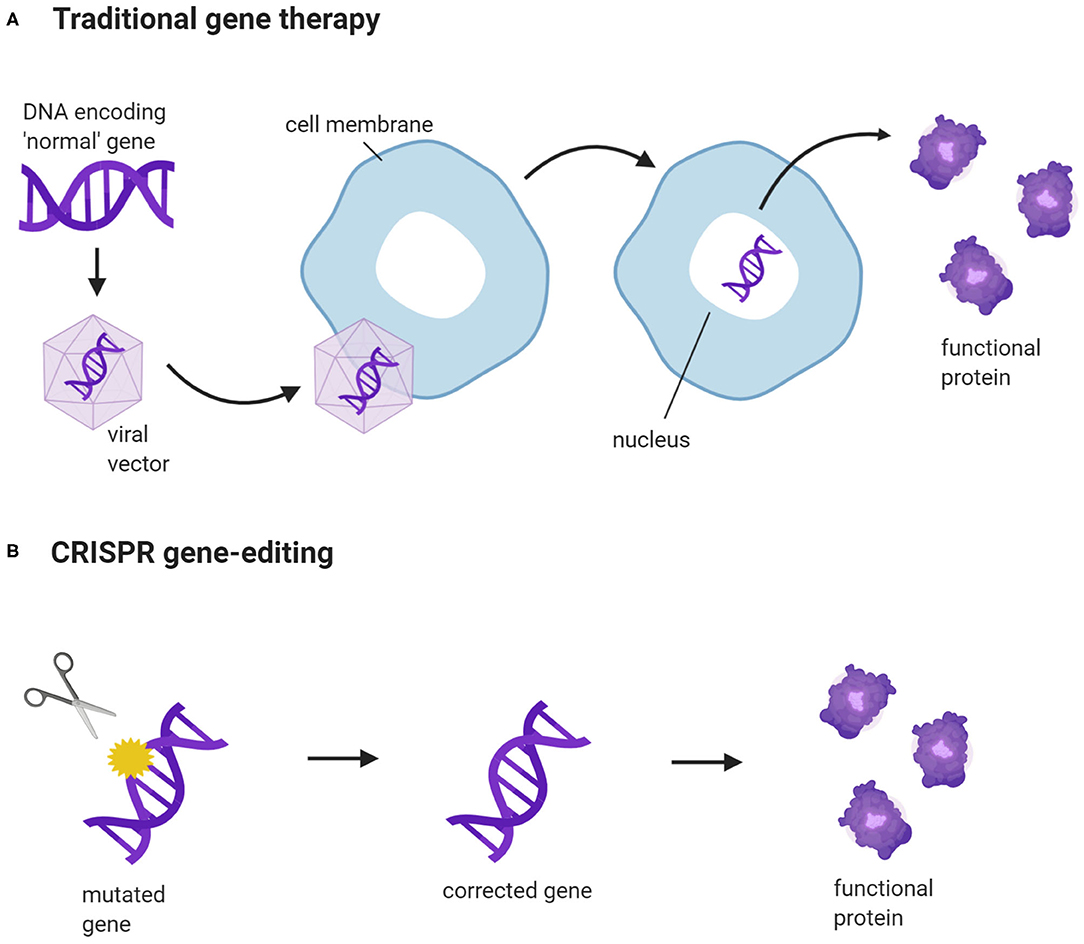
Figure 1. CRISPR gene-editing vs. traditional gene therapy. (A) A new gene construct is packaged into a viral vector. The vector binds to the cell membrane and releases the gene construct into the cell nucleus. This enables the cells to produce normal, functional proteins, encoded by the construct. (B) CRISPR targets a specific gene and corrects a disease-causing DNA variant. The corrected gene can now be transcribed and translated into a functional protein. (Created with BioRender.com).
Deaths reported following gene therapy have cast serious concerns on its safety. For example, two patients suffered liver dysfunction and died after they received high doses of adeno-associated viruses (AAV) that delivered a gene therapy for X-linked myotubular myopathy (25). An 18-year-old patient passed away 4 days after he was given an intra-artery dose of a gene therapy that was designed to remedy ornithine transcarbamylase deficiency. The cause of death was determined to be a severe immune reaction to the AAV vector that carried the corrective gene (26).
CRISPR is an adaptive immune defense used by archaea and bacteria against viruses (27). Upon infection by a virus, the host will integrate fragments of the viral genetic material into its genome, which will serve as memory for recognizing and destroying the virus in subsequent infections. The virus will be targeted and destroyed by the CRISPR-associated protein (Cas), an endonuclease that cleaves DNA strands. Between 2011 and 2013, substantial efforts by many researchers led to the successful repurposing of this CRISPR-Cas system to enable gene editing in eukaryotes. The mechanics of CRISPR-Cas are simple and readily reproducible outside the microbes (28, 29).
CRISPR has been widely used to edit single or multiple target genes in a variety of cells and organisms (8). For a detailed discussion of the mechanisms of CRISPR and the requirements for successful gene edits, the readers are referred to another review (30). A functional CRISPR toolkit needs only: (I) the Cas nuclease, commonly the Cas9, and (II) a guide RNA complementary to the target sequence. The basic scheme can be altered to suit a variety of gene-editing needs (28). The guide RNA directs the Cas9 nuclease to the targeted DNA region, which must contain a protospacer adjacent motif (PAM) at the 3′ end. The binding of Cas9 nuclease to the target region induces DNA double-strand breaks (DSBs), which subsequently trigger endogenous mechanisms to repair the DSBs. DSBs can be repaired either by non-homologous end joining (NHEJ) or homology-directed repair (HDR). In actively dividing human cells, NHEJ is the prevailing DNA repair mechanism, remedying 75% of naturally occurring DSBs, while HDR is responsible for the remaining 25% (31). NHEJ is an error-prone process and causes random DNA insertions or deletions (indels), which could generate frameshift mutations (32). Thus, NHEJ is useful when the resultant edits are intended to abolish gene expression. For precise gene edits such as single-base substitutions, a donor repair template is needed to shift the DNA repair pathways from NHEJ to HDR (33) (Figure 2A). HDR enhances the precision of CRISPR, but the issue remains that unlike NHEJ which is active throughout the cell cycle, HDR is only active at the G2 and S phases. This decreases the efficiency of HDR; and the problem is exacerbated in post-mitotic cells, which are not actively dividing (34).
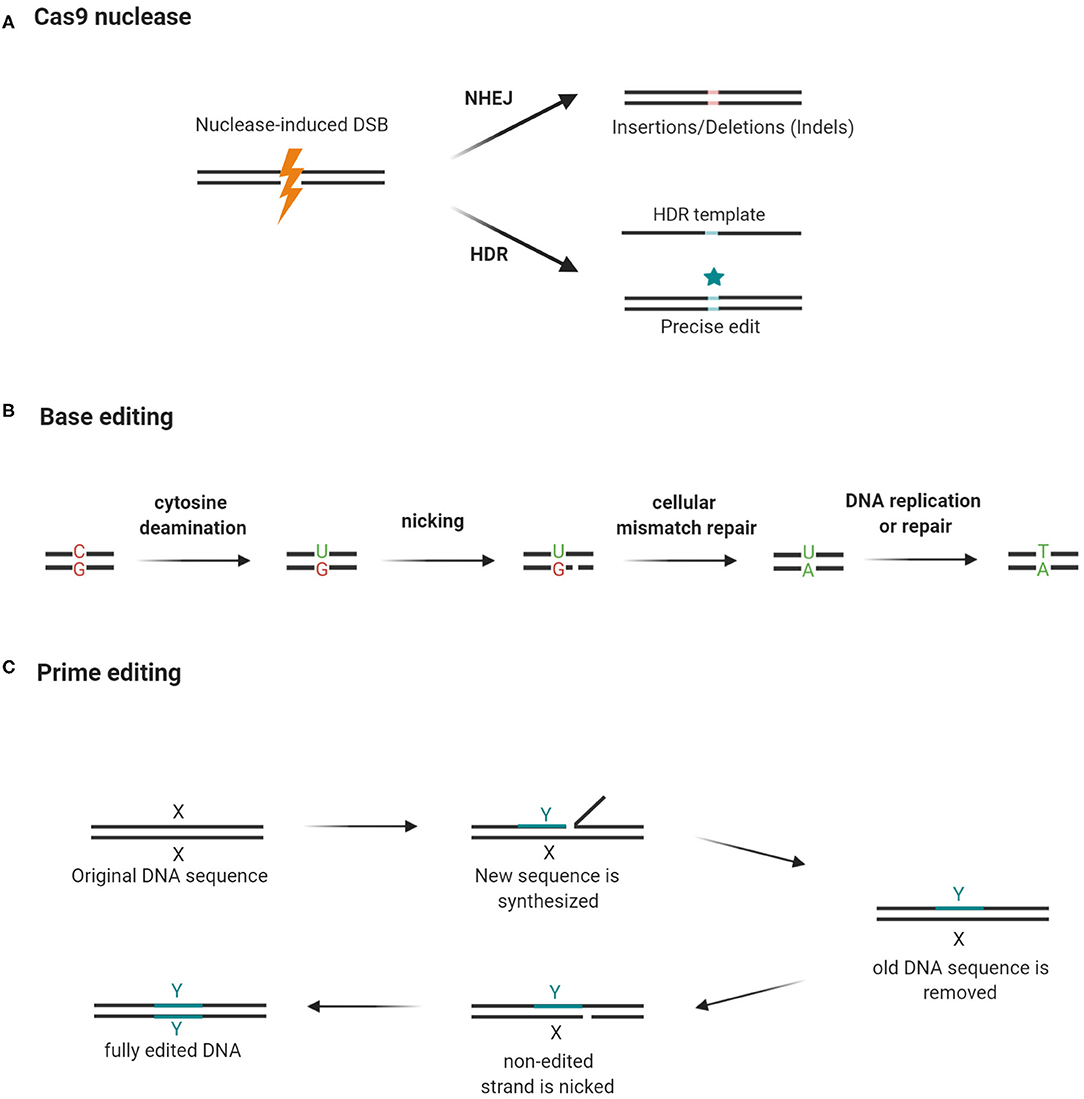
Figure 2. Cas9 nuclease, base editing, and prime editing. (A) Guide RNA guides Cas9 nuclease to cut a DNA segment that is 3 bases upstream of the PAM. The resultant double-strand break (DSB) triggers NHEJ, which may cause frame-shifting indels and abolish gene expression. By using a repair template, the repair machinery can be shifted to HDR and introduce precise edits while rejoining the broken DNA strands. (B) Cytidine deaminase converts cytosine to uracil while deoxyadenosine deaminase (not shown in the figure) converts adenosine to guanine. Cas9 nickase cuts the opposite strand and triggers a mismatch repair mechanism. As a result, in repairing the nick previously created by the Cas9 nickase, the cell uses the edited DNA strand as a template and copies the “mutation” into the complementary strand. (C) Prime editing requires a prime editor and a prime editing guide RNA (pegRNA) to modify gene sequences. The prime editor is a chimera of a Cas9 nickase and a reverse transcriptase (RT). The pegRNA guides the prime editor to the target site where editing should occur. It also carries a primer-binding site (PBS) and a short stretch of a template sequence containing the desired edit. The reverse transcriptase converts the template sequence into complementary DNA, which is then incorporated into the target site after the original DNA sequence is excised by an endogenous endonuclease. Then, the edited strand serves as a template for the repair of the unedited strand after it is nicked by Cas9 nickase. Hence, both DNA strands have the desired edit. X: original DNA sequence; Y: edited DNA sequence (Created with BioRender.com).
The discovery of DNA base editors in 2016 offered an HDR-independent solution to the problem (35). These base editors utilize Cas9-nickase or dCas9 conjugated with deaminase to induce single-base transitions from C to T or A to G (36, 37) (Figure 2B). Prime editing expands the types of base substitutions that can be created by the base editors, using its dual-functioning guide RNA to prime the synthesis of DNA edits by a reverse transcriptase (Figure 2C). Instructed by a template embedded in the guide RNA, the reverse transcriptase can create precise indels or any of the 12 possible point mutations (C → T, G → A, A → G, T → C, C → A, C → G, G → C, G → T, A → C, A → T, T → A, and T → G) without the need for DSBs or an HDR template (37).
Precise single-base editing would be an important, clinically relevant modality of gene editing, as ~50% of disease-causing mutations are single-nucleotide substitutions rather than small indels. The Cas9-deaminase base editors may find use in correcting those mutations and treating the associated disorders (37, 38). For instance, a base editor has been shown to correct a mutation that caused Niemann-Pick disease type C and accumulation of lipids in mouse brain tissue (39). Prime editing is expected to surpass the base editors in therapeutic utility, as it could edit up to 89% of known genetic variants associated with human diseases (37).
Genes hold the blueprint for how the brain matures and functions. However, the roles of many genes in the developing human brain are still poorly understood, making the search for new HND treatments a difficult undertaking (40). In vitro and in vivo disease models are useful for understanding the molecular mechanisms and the pathogeneses of HNDs and exploring novel therapies. However, the generation of disease models using the conventional transgenesis technique, which introduces an altered version of a gene (harbored in a vector) into a host organism, is time-consuming and inefficient. Cells naturally reject foreign substances, so the expression of the mutant gene is usually lost after several rounds of cell division (41). CRISPR obviates this limitation, creating inheritable and permanent changes in nuclear (native) DNA (42).
CRISPR has been used to rapidly create in vivo and in vitro models to elucidate the pathogenetic mechanisms of genetic diseases and to identify potential treatments (Table 2). For instance, to facilitate the study on how the loss of UBE3A, which regulates synaptic development, in neurons leads to Angelman syndrome, in vitro and in vivo (rat) models were generated by knocking out UBE3A using CRISPR. UBE3A-deficient rats showed signs similar to what have been observed in patients with Angelman syndrome, namely cognitive and motor impairment. Neurons lacking functional UBE3A lose the ability to fire mature action potentials—the electrical signals that connect neurons and underpin the workings of the brain (43, 44). By silencing UBE3A, CRISPR has helped to pinpoint the genetic switch of neural circuits and the causal gene for Angelman syndrome.
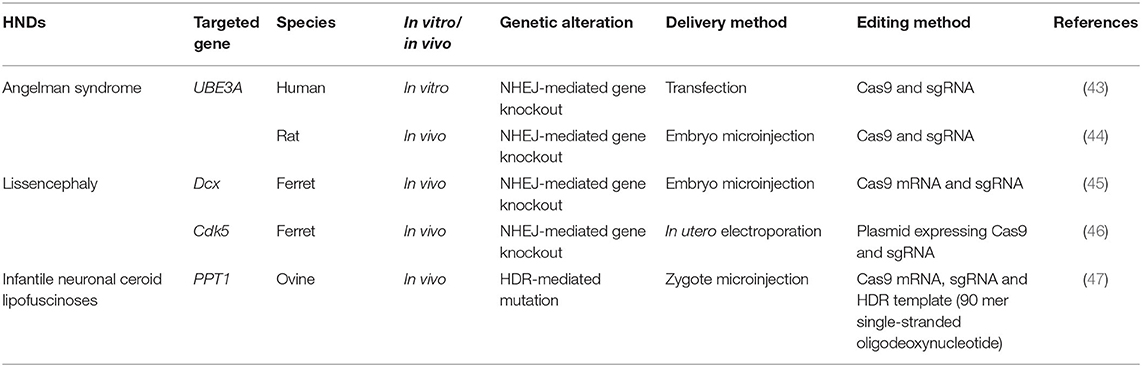
Table 2. HNDs models generated by CRISPR.
Mice and rats are popular choices of model organisms for studies of human diseases. However, they are not always compatible with human HNDs. Though the brains of humans, mice, and rats share the same general layout, they differ in some key aspects, making it impossible to create valid mouse or rat models for certain HNDs. The cortex of the human brain is heavily folded to house dense networks of neurons that perform high-level cognitive functions—an anatomical feature that is absent from the brains of mice and rats (48, 49). Beneath the cortical sheath, neurons are grouped by the genes they actively express into a variety of functional classes. Many of the neuron classes are conserved between humans and mice, but some putative counterparts were found to have notably varied patterns of gene expression (50). This means the molecular workings of some human brain diseases are species-specific and may only be accurately replicated in animals that are closely related to humans.
Medium-sized animals, such as sheep, monkeys, pigs, and ferrets resemble humans more closely than mice or rats and are therefore better model organisms for use in CRISPR-based studies of HNDs (51). For instance, CRISPR-mediated genome editing was applied to develop a ferret model to study lissencephaly, which causes loss of cortical folding in the human brain (45). In this study, a CRISPR system targeting Dcx was injected into single-cell ferret embryos, which were then implanted into surrogate females. This abolished the function of Dcx and resulted in the birth of ferrets who had smooth brains, confirming the importance of Dcx in enabling neuronal migration during cortical folding (45). Infants born with lissencephaly have small brains and severe intellectual disability (40). In another study, by delivering a CRISPR system using in utero electroporation, researchers proved that Cdk5 can be another gene required for cortical folding, as knocking out this gene resulted in smooth-surfaced brains (46). Both studies used ferrets, as cortical folds are present in ferrets but not in rodents (45, 46).
It is evident that the choice of an animal model depends on the characteristics of the disease in question. For instance, a CRISPR-ovine model is the logical choice for infantile neuronal ceroid lipofuscinoses, where deleterious mutations in the palmitoyl-protein thioesterase 1 (PPT1) gene cause progressive death of nerve cells. The incurable disease affects children and severely reduces their life expectancy to ~10% of the average lifespan of humans, as death typically occurs at ~9 years of age. Sheep are more effective disease models than ferrets in this case, though both have brain structures similar to humans. The longer lifespans of sheep would allow us to thoroughly map out the development of the disease (47, 52, 53).
Overall, the studies curated in Table 1 have clearly shown the utility of CRISPR, when coupled with medium-sized animal models, in helping us to understand the pathogeneses of HNDs. However, the high costs, long breeding periods, ethical concerns, and strict regulations may still limit the use of those animal models in the future (51, 54).
Besides its potential in the generation of effective models of human diseases, CRISPR can also be used to treat HNDs (Table 3). Here we discuss in detail the pre-clinical findings reported by studies of a variety of HNDs, namely fragile X syndrome, Down syndrome, and sphingolipidoses. CRISPR-based therapy could be achieved using different approaches in the clinical settings, namely in vitro germline editing, in utero gene editing, and in vivo and ex vivo gene editing (Figure 3). We detail the rationales and challenges of different strategies for CRISPR editing of HND-causing DNA variants in Table 4.
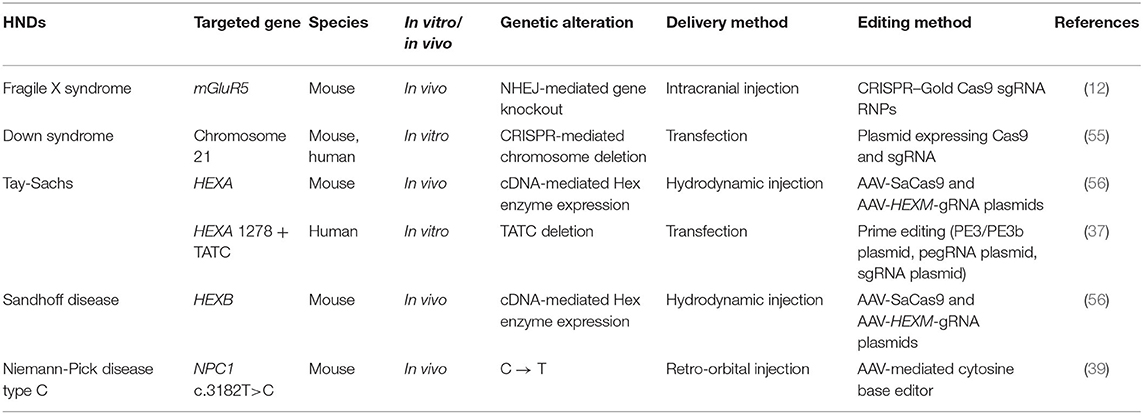
Table 3. CRISPR-mediated treatment of HNDs.
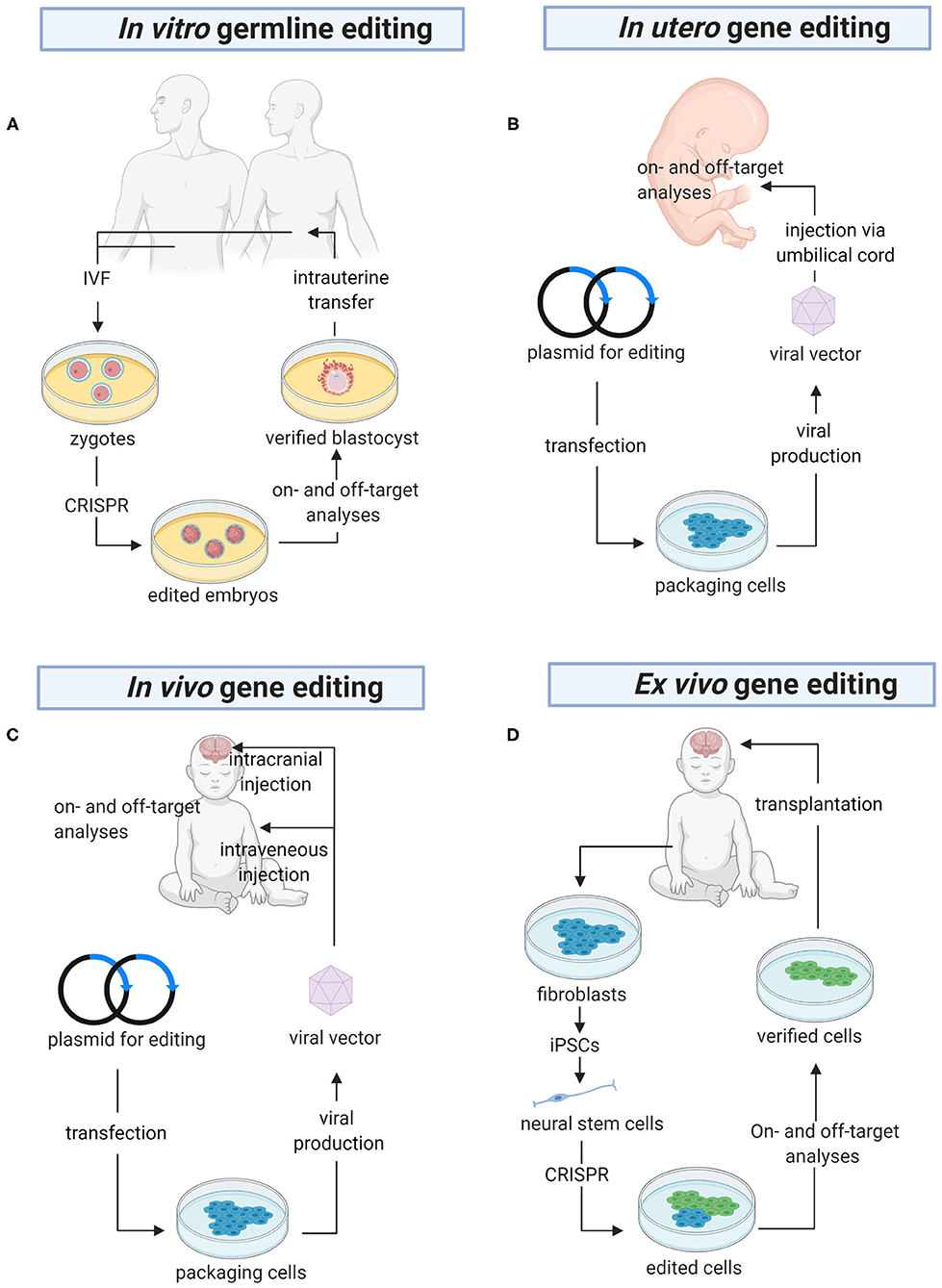
Figure 3. The future of gene editing in HNDs. (A) In vitro germline editing is initiated with the creation of zygotes. CRISPR constructs are microinjected into the zygotes, which are allowed to grow into embryos harboring the desired DNA edits. PGD is carried out to ensure there are no off-target mutations before the embryos are transferred into the uterus. (B) A viral vector harboring a genome editor is injected into the umbilical cord for direct delivery into the fetus. Alternatively, the editor can be delivered using a non-viral vector (not shown in the figure). Before the baby is born, a variety of tests will be performed to confirm on-target gene edits and detect off-target mutations. (C) CRISPR is packaged in a viral or non-viral vector for systemic delivery or direct injection into the brain. (D) Ex vivo gene editing begins with isolation of fibroblasts from the patients. The cells are reprogrammed into iPSCs, differentiated into neural stem cells, and CRISPR-edited. Then, the edited cells are analyzed for on- and off-target gene edits before they are transplanted into the brain. PGD, pre-implantation genetic diagnosis; IVF, in vitro fertilization; iPSCs, induced pluripotent stem cells (Created with BioRender.com).
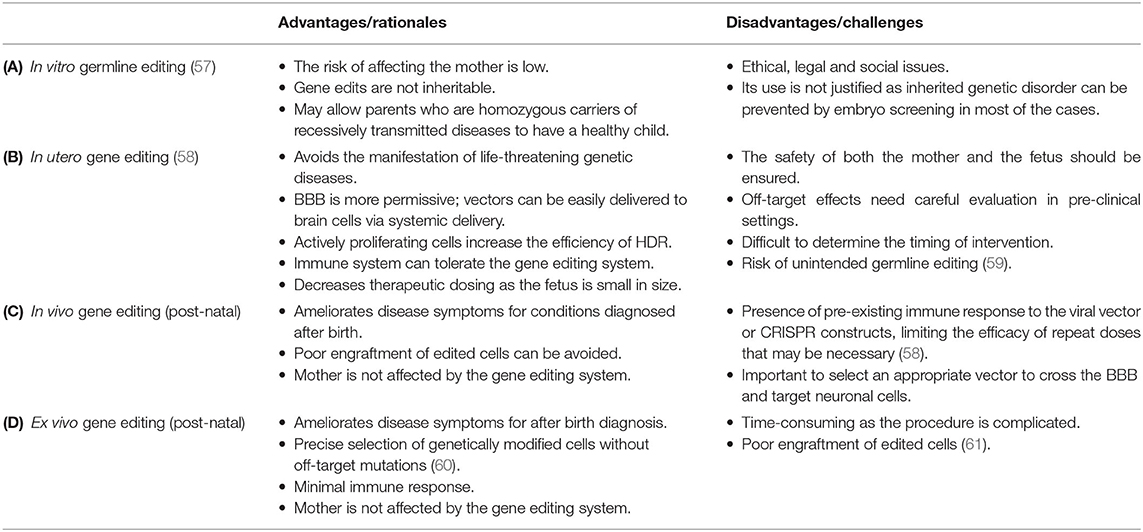
Table 4. Advantages and disadvantages of different strategies of CRISPR-based gene editing in HNDs.
CRISPR-Gold, a non-viral delivery system, was found to effectively edit mGluR5, an autism-associated gene, in a mouse model of fragile X syndrome. mGluR5 editing reduces the signaling between brain cells and thus decreases the repetitive behaviors caused by this disorder (Figure 4). In the study that examined the therapeutic potential of CRISPR-Gold, the CRISPR system was injected directly into mouse brains to limit gene editing to the striatum, which mediates repetitive behaviors. As a result, mGluR5 mRNA and protein levels were reduced by 40–50%, and this was sufficient to rescue the treated mice from repetitive behaviors (12). However, it could be a challenge for researchers to determine the extent of mGluR5 reduction that would cause a similar effect in humans. Fragile X syndrome is associated with an imbalance of glutamatergic and GABAergic signaling (64). mGluR5 serves a role in excitatory glutamatergic neurotransmission and completely knocking out this gene can further disrupt GABAergic signaling and impair human brain function (65, 66). Hence, the nuanced balance between the glutamatergic and GABAergic signals in the brain dictates the level of mGluR5 inhibition that would be therapeutic in humans (67).
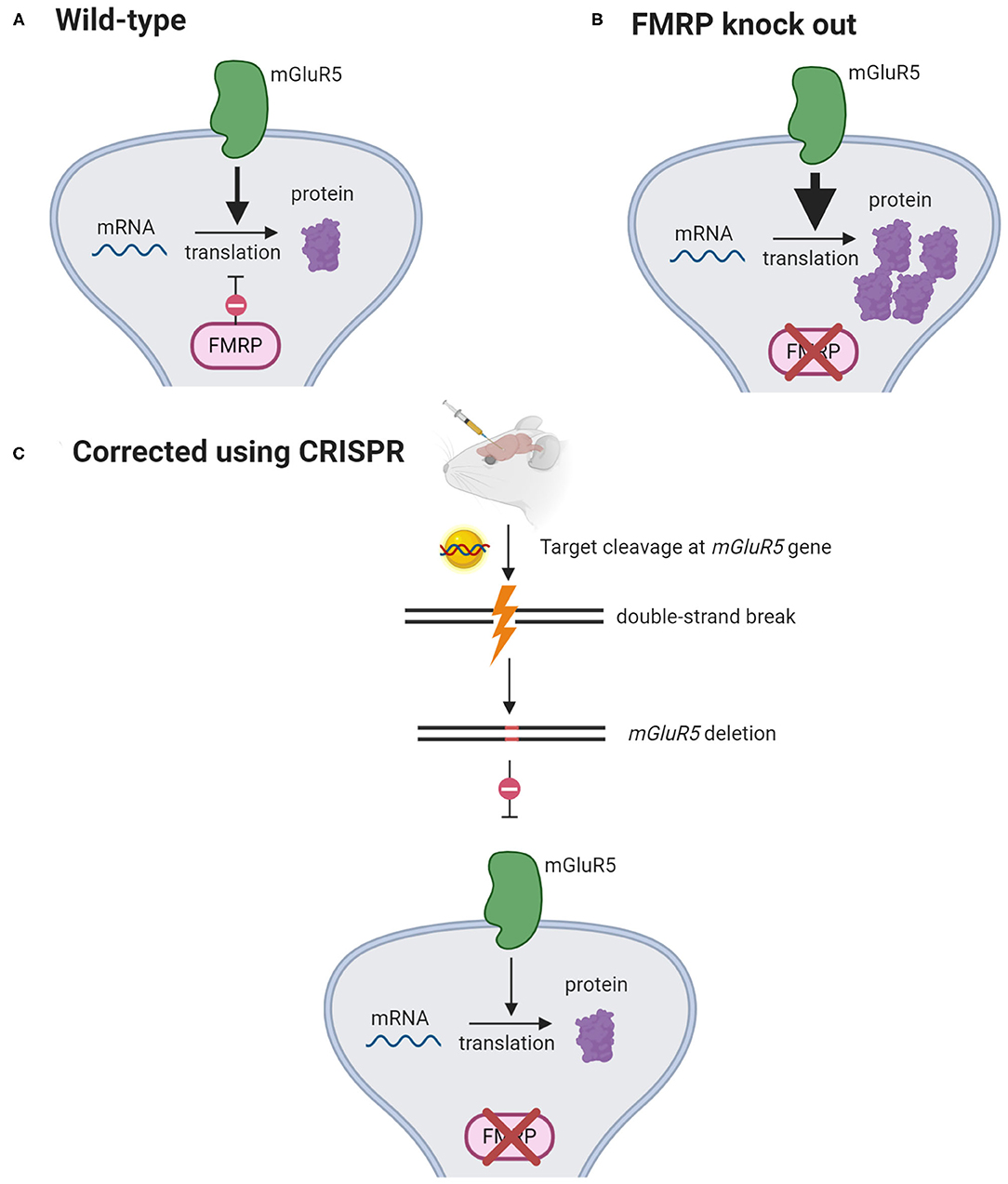
Figure 4. mGluR5 signaling reduction rescues fragile X syndrome in mice. (A) mGluR5 signaling activates protein synthesis. FMRP opposes mGluR5 and suppresses the translation of mRNAs into proteins. (B) In FMRP-knockout mice, mGluR5 signaling is exaggerated, causing excessive synthesis of proteins. (C) CRISPR-Gold is injected intracranially to knock out mGlur5 gene. This has been shown to decrease mGlurR5 mRNAs by 40–50% and restore protein synthesis to its normal levels (12). (Created with BioRender.com).
Down syndrome, also known as trisomy 21, is caused by an error in cell division that leads to an extra chromosome 21 (68). It is a well-known genetic disorder that impairs neurodevelopment in newborns. The extra chromosome 21 causes overexpression of >100 genes that drive brain development or function (69). Several gene editing strategies, including CRISPR, have been applied to eliminate the surplus chromosome (70). For instance, two gRNAs were designed to target repetitive sequences at the long arm of chromosome 21, induce cleavage at multiple sites, and eliminate the whole chromosome. The deletion of an entire chromosome is challenging as it is difficult to efficiently induce multiple DNA cleavages. Although the initial trial of the chromosome-removing strategy was successful in stem cells derived from patients with Down syndrome, the same outcome was not replicable in embryos, probably because chromosomal deletion was lethal to embryonic cells (55). Recently, two alternative strategies were proposed. The suggestions were aimed at inactivating instead of deleting the extra chromosome 21 (71). Guided by sgRNAs, the Cas9 nuclease could home in on and cut off the Down syndrome critical regions in chromosome 21, which harbor the culprit genes that cause Down syndrome and inhibit neuronal development. Alternatively, the enzyme could edit out a non-functional segment within chromosome 21 in exchange for a regulatory DNA construct which contains XIST that inactivates the chromosome (71, 72). With this proposed approach, chromosomal inactivation by XIST which normally occurs at the pluripotent stage could be induced in non-pluripotent neural stem cells and differentiated neurons (73). Both the proposed methods could rescue neurogenesis and improve cognitive performance in Down syndrome patients.
CRISPR has also demonstrated its genome editing efficacy in mouse models of Tay-Sachs and Sandhoff diseases. Mutations in HEXA, which encodes the Hex α subunit, lead to Tay-Sachs disease while mutations in HEXB, which encodes the Hex β subunit, cause Sandhoff disease. Mutations in the HEXA and HEXB genes reduce the activity of beta-hexosaminidase, which breaks down GM2 ganglioside, a normal component of the neuronal membrane. As a result, GM2 ganglioside accumulates to a level which is toxic to neurons in the brain and the spinal cord, causing intellectual disability and seizures (74). Instead of targeting the brain, a CRISPR system delivered by AAV was used to turn hepatocytes into machinery that produces a modified human Hex μ subunit, by integrating cDNA encoding the protein into the albumin gene. The enzymes expressed and secreted from the edited hepatocytes were then carried by the bloodstream to the brain to break down GM2 ganglioside (56).
With new techniques being rapidly developed, several alternative strategies—one of which being prime editing—have become available for editing HEXA mutations in Tay-Sachs disease (37). The most common mutation found in patients with Tay-Sachs disease is a 4-bp insertion, i.e., TATC in exon 11 of the HEXA gene (75). In an in vitro model, prime editing was shown to correct the mutation by removing the 4-bp insertion without DSB (37). To determine which CRISPR system is optimal, criteria such as safety, costs, delivery vectors, and how well the system works in cells should be considered. Most importantly, more supporting evidence should be garnered from pre-clinical studies before moving into clinical trials.
Caffeine, an antagonist of adenosine A1 (ADORA1) and A2A receptors (ADORA2A), is a key modality of the management of apnoea of prematurity. Administration of caffeine in pre-mature, apneic infants was found to improve symptoms and significantly reduce death rates and the severity of neurocognitive impairment (76). Caffeine has also been shown to have a variety of neuroprotective effects in vitro and in vivo. It protects against cell death and preserves background electrical activity in the hypoxic-ischemic brains (77–80) and enhances the connection between neurons by activating genes that control neuron projection. This is especially important to the developing brains of infants. Together, the protective mechanisms of caffeine act to improve neurodevelopment in preterm infants (81).
Individual genetic differences can affect the pharmacology of some drugs and cause inter-individual variability in drug response. This would affect drug therapy outcomes. Because of genetic variation, not all infants given caffeine will respond optimally to the drug. Rs16851030, a DNA variant located in the 3′-untranslated region of the ADORA1 gene, was shown to adversely affect the outcome of caffeine therapy. A prospective case-control study was conducted to assess the variability of caffeine sensitivity in relation to single-nucleotide variants in the ADORA1 gene. All infants who were >28 weeks old and homozygous for the rs16851030 reference C-allele were found to have responded favorably to caffeine therapy, in comparison with a 57% response rate among those harboring the alternate T-allele. The discrepancies in the treatment outcomes may be due to the influence of rs16851030 on the expression of the adenosine A1 receptor (16); but this remains an unconfirmed theory. Furthermore, we do not know whether the genetic variation in caffeine response could be overcome by dosage adjustment (82). In caffeine-sensitive individuals, the augmented effects of caffeine could be offset by lowering caffeine doses to avoid toxicity, such as tachycardia and seizure (83). Conversely, individuals who are less caffeine-responsive may benefit from higher caffeine doses to reduce the risk of apnea and to prevent complications such as hypoxia-induced brain damage. CRISPR has been used to create a mutant breast cancer cell line with a single base edit to elucidate the mechanism of drug resistance (14). Therefore, by creating a cell-based model using CRISPR and treating the cells carrying the C- or T-allele with different concentrations of caffeine, we could then gauge whether the underlying genetic influence could be overcome by adjustments to the dosage of caffeine. The resultant findings would be valuable for optimizing the management of apnea of prematurity and improving neurodevelopmental outcomes in preterm infants.
Using a base editor to investigate how rs16851030 affects the outcome of caffeine therapy could be challenging as there are multiple Cs around the target C (in brackets) within the editing window, as shown in the flanking sequences of rs16851030, TCTTAGATGTTGGTGGTGCAGC[C/T]CCAGGACCAAGCTTAAGGAGAG. The editing of additional Cs can cause harmful effects. CRISPR base editors with narrow editing windows were reported recently but they still would not be able to precisely edit the target C, owing to bystander effects (84, 85); and the editing may result in a non-T (86, 87). Prime editing would be a fitting alternative to the base editors (Figure 2C), as it creates precise point mutations by directly copying the desired gene edits into the target DNA segments (37). Another advantage of prime editing is that unlike the other base editors, it can perform gene editing in post-mitotic cells, including neurons in the brain (37).
In this section, we detail the roadblocks to CRISPR attaining its maximal utility in neuroscience research: off-target effects, potential difficulties in crossing the blood-brain barrier, and immunogenicity of Cas9 and vulnerability of neuronal cells to the adverse effect of CRISPR editing (or the system that delivers it).
The specificity of CRISPR is ensured by its companion gRNA, which consists of sequences complementary to the target DNA region. However, the specificity is not absolute, and unintended binding between gRNA and non-target DNA sequences is possible. Off-target activity must be avoided because it can lead to adverse side effects. Current tactics for curbing off-target editing have focused on two key aspects of how CRISPR operates, i.e., the need for specific gRNAs, and the fundamental gene editing mechanics.
DISCOVER-Seq (discovery of in situ Cas off-targets and verification by sequencing) is a tool for detecting possible in vitro or in vivo off-targets of CRISPR, helping researchers to validate the guide RNAs they have designed in silico. By checking the interaction between a DSB repair protein, MRE11, with Cas9 cut sites, DISCOVER-Seq can identify the exact locations in the genome where a cut has been made by CRISPR. The MRE11-bound DNA segments are captured by chromatin immunoprecipitation and sequenced on a high-throughput platform. DISCOVER-Seq is superior to other tools as it can be used to detect off-target events in vivo (88). This may in then inform corresponding strategies to eradicate the off-target editing.
Prime editing overhauls the mechanism of base editing and could be an option that is relatively free of off-target editing. It was reported to have increased target specificity (37). However, the safety and efficacy of prime editors in neuronal cells are still unclear. Further studies are needed to explore the utility of this newly developed gene editor in the neuroscience space.
For gene expression studies and treatment of HNDs, the main challenge is to deliver a CRISPR system across the BBB. The BBB prevents the entry of foreign substances into the brain, including toxins and pathogens (89). The protective mechanism is a two-edged sword, as it also cuts off the access of CRISPR systems to the brain. There are several strategies to tackle the BBB, such as viral delivery and nanoparticles. AAV is a popular method for shipping CRISPR expression constructs to the target brain cells. To make room for CRISPR, the virus is emptied of its protein-coding genes, leaving only the capsids and the sequences that regulate DNA replication. The passage of AAV-CRISPR across the BBB is made possible by the inherent ability of viruses to bind to and invade host cells (90).
However, wild-type AAVs are inefficient in crossing the BBB and need direct injection into the brain (91). Besides, they have low transduction efficiency in vitro and in vivo (92, 93). To counter the drawbacks, a 7-mer peptide, PHP.B, was inserted into the capsids of wild-type AAVs to facilitate the penetration of BBB and to increase transduction efficiency in neuronal cells (93, 94). However, a high viral load of AAV-PHP.B would be required (≥1 ×1012 vg per adult mouse) for genetic modification in the brain, and this translates into a high risk of immune reactions. With the development of AAV-PHP.eB, which varies from AAV-PHP.B at only two amino acids adjacent to the initial 7-mer peptide insertion, neuronal cells can be edited using a lower viral load (95). Some studies showed that PHP.B and PHP.eB require the LY6A receptor (lymphocyte antigen 6 complex) to reach the mouse brain. Ly6a disruption decreases, while Ly6a overexpression enhances, transduction efficiency (96, 97). Nonetheless, this mechanism utilized by AAVs to cross the BBB is mouse-specific and there is no direct homolog to Ly6a in humans. Further experiments should focus on pinpointing a gene which can be targeted to increase AAV transduction in the human brain (98).
Another way to deliver the CRISPR system across the BBB has been developed recently using in vitro BBB models and holds promises in the eradication of neuroHIV. It was achieved by packaging the CRISPR system bound with magneto-electric nanoparticles (MENPs) in a nanoformulation. A magnetic field was then applied on the nanoformulation to release CRISPR from the surfaces of MENPs and to facilitate cell uptake. This would then result in intracellular release of CRISPR and inhibition of HIV (99). Neonates acquire neuroHIV when the virus enters their brains, and this could delay their brain development (100, 101). Although HIV is not an inherited disease, the same approach to delivery across the BBB could be applied in the treatment of HNDs. However, it is unclear whether the BBB has fully formed during the neonatal period (89). This would affect the concentration of a CRISPR system to be safely delivered into the brain. Future efforts should focus on determining the optimum concentration of CRISPR before this technique can be adopted clinically.
The most common Cas9 orthologs are derived from Staphylococcus aureus (SaCas9) and Streptococcus pyogenes (SpCas9) (102). Because of their bacterial origins, Cas9 proteins face pre-existing adaptive immune responses in humans. Antibodies against SaCas9 and SpCas9 have been detected in 86 and 73%, respectively, of the serum samples obtained from cord blood donors (103). The potential immunogenicity of Cas9 proteins warrants caution in future clinical trials examining the use of CRISPR in neonates.
Besides, the delivery methods of CRISPR systems may also induce immune responses and impact neuronal cells. Viral vectors are commonly used to deliver CRISPR constructs across the BBB (12, 104). However, viral delivery causes protracted CRISPR expression, which is toxic to neuronal cells and alters neuronal phenotypes (12). Moreover, it has been demonstrated that persistent Cas9 expression elicits cytotoxic immune response, which removes genetically edited cells. The removal of modified cells in the brain could lead to adverse consequences, as brain cells have limited capacity to regenerate (105). The finding also means that CRISPR edits are not necessarily permanent, so repeat administration of CRISPR therapy would be required (106). Hence, non-viral delivery methods have received great interest recently; for instance, gold nanoparticles have been used to deliver Cas9 ribonucleoproteins targeting mGluR5 in a mouse model of fragile X syndrome (12). Gold nanoparticles are safe, as they were not found to cause cytotoxicity or changes to neuronal functions at low doses. Also, they did not induce immune responses—a common problem arising from the delivery of CRISPR systems via viral methods (12).
Overall, nanoparticles seem an ideal carrier for delivering CRISPR systems into brain cells. Nanoparticles are versatile as their surfaces can be engineered to target specific cells. For instance, gold nanoparticles coated with exosomes have been shown to be able to cross the BBB via endocytosis (107, 108). To selectively target brain cells, the exosomes can be modified with a neuron-specific peptide derived from the rabies virus glycoprotein. This peptide specifically binds to the acetylcholine receptors expressed by neuronal cells (109, 110). Therefore, by modifying the surfaces of nanoparticles, we can ensure a CRISPR system is able to pass through the BBB and reach its target brain cells.
In the last few years, CRISPR-driven research is rapidly increasing, and new cell and animal models have been created to elucidate the pathogenesis of HNDs. This is important groundwork for future research into new therapies. The family of CRISPR-Cas9 gene editors has been growing steadily. A variety of base editors and prime editors are continually being discovered that may improve the precision and efficacy of gene editing. Studies trialing the gene editors have resulted in various success rates. The simple mechanics of CRISPR make it a robust gene-editing tool; however, off-target editing is still a major concern and could have severe consequences (111). The safety of CRISPR editing should be guaranteed in two aspects. First, enhancing the precision of gene editing should remain at the core of future CRISPR-centric research. It may be helpful to pinpoint “hotspots” in the genome where off-target edits are most likely to arise. This may then lead to strategies that can effectively curb off-target editing in those DNA segments. Second, the chosen mode of CRISPR delivery should be non-toxic to neuronal cells and non-immunogenic. The body's immune response may suppress CRISPR gene therapy, and pose a health risk to the person receiving the treatment. Screening for potential immunological or allergic reactions to CRISPR should be performed before commencing therapy.
An appealing use of the CRISPR technique would be pre-emptive in utero editing of pathogenic gene mutations coupled with prenatal genetic testing (112–114). This would be better than delaying gene therapy until after birth, when the disease would have become manifest and the damage established. Some of the HNDs develop before birth; for instance, lissencephaly impairs cortical folding and is irreversible once the prenatal brain development is completed (115).
However, a long road lies ahead for the adoption of CRISPR-based gene editing in the clinics. What is therapeutic and what is not; or defining which genetic diseases should take priority for CRISPR therapy, are some difficult choices to make even in settings with relatively abundant health care resources. CRISPR therapy is likely to be costly—some estimates have priced it at USD $0.5 to $2 million, so funding it would be a challenge for most countries or insurance companies. Owing to the exorbitant costs of emerging gene therapies, health insurers may become increasingly selective in choosing their clients, excluding those diagnosed with “pre-existing conditions” (116, 117).
Ethical concerns are also important considerations before CRISPR can be used in humans. Genome editing in clinical settings is currently limited to somatic cells, as this is less likely than germline editing to be misused for non-ethical purposes. Potential problems may arise if “designer babies” are created using CRISPR. For instance, undetected off-target effects can be passed down to future generations and the undesirable negative consequences may be grave. Other ethical considerations include using CRISPR to achieve better phenotypic characteristics, such as height, intelligence, and athletic performance. This highlights the need for strict regulations and judicial frameworks on human germline editing. The National Institute of Health supports the call for an international moratorium on human germline editing in the clinical settings until certain conditions are met (118). Global discussions involving scientists and ethicists are needed to address how germline editing should be performed and ethically acceptable before the moratorium can be lifted.
In summary, CRISPR is an effective research tool for studying HNDs. If important safety and ethical concerns can be addressed, it has immense potential as a new treatment modality for HNDs. We expect more established CRISPR-based treatment strategies that bring new hopes for HNDs in the future.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by a grant from the Ministry of Higher Education, Malaysia, Fundamental Research Grant Scheme (FRGS/1/2019/SKK08/UKM/03/4).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Cardoso AR, Lopes-Marques M, Silva RM, Serrano C, Amorim A, Prata MJ, et al. Essential genetic findings in neurodevelopmental disorders. Hum Genom. (2019) 13:31. doi: 10.1186/s40246-019-0216-4
2. Stiles J, Brown TT, Haist F, Jernigan TL. Brain and cognitive development. Handb Child Psychol Dev Sci. (2015) 2:1–54. doi: 10.1002/9781118963418.childpsy202
3. Qu H, Lei H, Fang X. Big data and the brain: peeking at the future. Genom Proteomics Bioinform. (2019) 17:333–6. doi: 10.1016/j.gpb.2019.11.003
4. Ingusci S, Verlengia G, Soukupova M, Zucchini S, Simonato M. Gene therapy tools for brain diseases. Front Pharmacol. (2019) 10:724. doi: 10.3389/fphar.2019.00724
5. Simonato M, Bennett J, Boulis NM, Castro MG, Fink DJ, Goins WF, et al. Progress in gene therapy for neurological disorders. Nat Rev Neurol. (2013) 9:277–91. doi: 10.1038/nrneurol.2013.56
6. Sondhi D, Peterson DA, Edelstein AM, del Fierro K, Hackett NR, Crystal RG. Survival advantage of neonatal CNS gene transfer for late infantile neuronal ceroid lipofuscinosis. Exp Neurol. (2008) 213:18–27. doi: 10.1016/j.expneurol.2008.04.022
7. Laoharawee K, DeKelver RC, Podetz-Pedersen KM, Rohde M, Sproul S, Nguyen H-O, et al. Dose-dependent prevention of metabolic and neurologic disease in murine MPS II by ZFN-mediated in vivo genome editing. Mol Ther. (2018) 26:1127–36. doi: 10.1016/j.ymthe.2018.03.002
8. Gupta RM, Musunuru K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J Clin Invest. (2014) 124:4154–61. doi: 10.1172/JCI72992
9. Li H, Yang Y, Hong W, Huang M, Wu M, Zhao X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct Targeted Ther. (2020) 5:1. doi: 10.1038/s41392-019-0089-y
10. McCarty NS, Graham AE, Studená L, Ledesma-Amaro R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat Commun. (2020) 11:1281. doi: 10.1038/s41467-020-15053-x
11. Stadtmauer EA, Fraietta JA. CRISPR-engineered T cells in patients with refractory cancer. Science. (2020) 367:eab7365. doi: 10.1126/science.aba7365
12. Lee B, Lee K, Panda S, Gonzales-Rojas R, Chong A, Bugay V, et al. Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile X syndrome from exaggerated repetitive behaviours. Nat Biomed Eng. (2018) 2:497–507. doi: 10.1038/s41551-018-0252-8
13. Abe S, Kobayashi K, Oji A, Sakuma T, Kazuki K, Takehara S, et al. Modification of single-nucleotide polymorphism in a fully humanized CYP3A mouse by genome editing technology. Sci Rep. (2017) 7:15189. doi: 10.1038/s41598-017-15033-0
14. Harrod A, Fulton J, Nguyen VTM, Periyasamy M, Ramos-Garcia L, Lai CF, et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene. (2017) 36:2286–96. doi: 10.1038/onc.2016.382
15. Moschino L, Zivanovic S, Hartley C, Trevisanuto D, Baraldi E, Roehr CC. Caffeine in preterm infants: where are we in 2020? ERJ Open Res. (2020) 6:00330-2019. doi: 10.1183/23120541.00330-2019
16. Kumral A, Tuzun F, Yesilirmak DC, Duman N, Ozkan H. Genetic basis of apnoea of prematurity and caffeine treatment response: role of adenosine receptor polymorphisms: genetic basis of apnoea of prematurity. Acta paediatrica (Oslo, Norway: 1992). (2012) 101:e299–303. doi: 10.1111/j.1651-2227.2012.02664.x
17. Burney TJ, Davies JC. Gene therapy for the treatment of cystic fibrosis. Appl Clin Genet. (2012) 5:29–36. doi: 10.2147/TACG.S8873
18. Asher DR, Thapa K, Dharia SD, Khan N, Potter RA, Rodino-Klapac LR, et al. Clinical development on the frontier: gene therapy for duchenne muscular dystrophy. Expert Opin Biol Ther. (2020) 20:263–74. doi: 10.1080/14712598.2020.1725469
19. Goswami R, Subramanian G, Silayeva L, Newkirk I, Doctor D, Chawla K, et al. Gene therapy leaves a vicious cycle. Front Oncol. (2019) 9:297. doi: 10.3389/fonc.2019.00297
20. Niibori Y, Lee SJ, Minassian BA, Hampson DR. Sexually divergent mortality and partial phenotypic rescue after gene therapy in a mouse model of dravet syndrome. Hum Gene Ther. (2019) 31:339–51. doi: 10.1089/hum.2019.225
21. Turner TN, Douville C, Kim D, Stenson PD, Cooper DN, Chakravarti A, et al. Proteins linked to autosomal dominant and autosomal recessive disorders harbor characteristic rare missense mutation distribution patterns. Hum Mol Genet. (2015) 24:5995–6002. doi: 10.1093/hmg/ddv309
22. Yu-Wai-Man P. Genetic manipulation for inherited neurodegenerative diseases: myth or reality? Br J Ophthalmol. (2016) 100:1322. doi: 10.1136/bjophthalmol-2015-308329
23. Watts JK, Corey DR. Silencing disease genes in the laboratory and the clinic. J Pathol. (2012) 226:365–79. doi: 10.1002/path.2993
24. Aguiar S, van der Gaag B, Cortese FAB. RNAi mechanisms in Huntington's disease therapy: siRNA versus shRNA. Transl Neurodegen. (2017) 6:30. doi: 10.1186/s40035-017-0101-9
25. High-dose AAV gene therapy deaths. Nat Biotechnol. (2020) 38:910. doi: 10.1038/s41587-020-0642-9
27. Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, Semenova E. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun. (2012) 3:945. doi: 10.1038/ncomms1937
28. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (New York, NY). (2012) 337:816–21. doi: 10.1126/science.1225829
29. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science (New York, NY). (2013) 339:819–23. doi: 10.1126/science.1231143
30. Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. (2020) 38:824–44. doi: 10.1038/s41587-020-0561-9
31. Mao Z, Bozzella M, Seluanov A, Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair. (2008) 7:1765–71. doi: 10.1016/j.dnarep.2008.06.018
32. Ryu SM, Hur JW, Kim K. Evolution of CRISPR towards accurate and efficient mammal genome engineering. BMB Rep. (2019) 52:475–81. doi: 10.5483/BMBRep.2019.52.8.149
33. Okamoto S, Amaishi Y, Maki I, Enoki T, Mineno J. Highly efficient genome editing for single-base substitutions using optimized ssODNs with Cas9-RNPs. Sci Rep. (2019) 9:4811. doi: 10.1038/s41598-019-41121-4
34. Devkota S. The road less traveled: strategies to enhance the frequency of homology-directed repair (HDR) for increased efficiency of CRISPR/Cas-mediated transgenesis. BMB Rep. (2018) 51:437–43. doi: 10.5483/BMBRep.2018.51.9.187
35. Zeng Y, Li J, Li G, Huang S, Yu W, Zhang Y, et al. Correction of the marfan syndrome pathogenic FBN1 mutation by base editing in human cells and heterozygous embryos. Mol Ther. (2018) 26:2631–7. doi: 10.1016/j.ymthe.2018.08.007
36. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. (2016) 533:420–4. doi: 10.1038/nature17946
37. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. (2019) 576:149–57. doi: 10.1038/s41586-019-1711-4
38. Zhou C, Zhang M, Wei Y, Sun Y, Sun Y, Pan H, et al. Highly efficient base editing in human tripronuclear zygotes. Protein Cell. (2017) 8:772–5. doi: 10.1007/s13238-017-0459-6
39. Levy JM, Yeh WH. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat Biomed Eng. (2020) 4:97–110. doi: 10.1038/s41551-019-0501-5
40. Dixon-Salazar TJ, Gleeson JG. Genetic regulation of human brain development: lessons from Mendelian diseases. Ann NY Acad Sci. (2010) 1214:156–67. doi: 10.1111/j.1749-6632.2010.05819.x
41. Burgio G. Redefining mouse transgenesis with CRISPR/Cas9 genome editing technology. Genome Biol. (2018) 19:27. doi: 10.1186/s13059-018-1424-2
42. Foulkes AL, Soda T, Farrell M, Giusti-Rodríguez P, Lázaro-Muñoz G. Legal and ethical implications of crispr applications in psychiatry. N C Law Rev. (2019) 97:1359–98.
43. Fink JJ, Robinson TM, Germain ND, Sirois CL, Bolduc KA, Ward AJ, et al. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat Commun. (2017) 8:15038. doi: 10.1038/ncomms15038
44. Dodge A, Peters MM, Greene HE, Dietrick C, Botelho R, Chung D, et al. Generation of a novel rat model of angelman syndrome with a complete Ube3a gene deletion. Autism Res. (2020) 13:397–409. doi: 10.1002/aur.2267
45. Kou Z, Wu Q, Kou X, Yin C, Wang H, Zuo Z, et al. CRISPR/Cas9-mediated genome engineering of the ferret. Cell Res. (2015) 25:1372–5. doi: 10.1038/cr.2015.130
46. Shinmyo Y, Terashita Y, Dinh Duong TA, Horiike T, Kawasumi M, Hosomichi K, et al. Folding of the cerebral cortex requires Cdk5 in upper-layer neurons in gyrencephalic mammals. Cell Rep. (2017) 20:2131–43. doi: 10.1016/j.celrep.2017.08.024
47. Eaton SL, Proudfoot C, Lillico SG, Skehel P, Kline RA, Hamer K, et al. CRISPR/Cas9 mediated generation of an ovine model for infantile neuronal ceroid lipofuscinosis (CLN1 disease). Sci Rep. (2019) 9:9891. doi: 10.1038/s41598-019-45859-9
48. Sun T, Hevner RF. Growth and folding of the mammalian cerebral cortex: from molecules to malformations. Nat Rev Neurosci. (2014) 15:217–32. doi: 10.1038/nrn3707
49. Ellenbroek B, Youn J. Rodent models in neuroscience research: is it a rat race? Dis Model Mech. (2016) 9:1079. doi: 10.1242/dmm.026120
50. Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, et al. Conserved cell types with divergent features in human versus mouse cortex. Nature. (2019) 573:61–8.
51. Yang W, Li S, Li X-J. A CRISPR monkey model unravels a unique function of PINK1 in primate brains. Mol Neurodegen. (2019) 14:17. doi: 10.1186/s13024-019-0321-9
52. Marini RP, Otto G, Erdman S, Palley L, Fox JG. Biology and diseases of ferrets. Lab Anim Med. (2002):483–517. doi: 10.1016/B978-012263951-7/50016-8
53. Nibe K, Miwa Y, Matsunaga S, Chambers JK, Uetsuka K, Nakayama H, et al. Clinical and pathologic features of neuronal ceroid-lipofuscinosis in a ferret (Mustela putorius furo). Vet Pathol. (2011) 48:1185–9. doi: 10.1177/0300985811400441
54. Yang W, Liu Y, Tu Z, Xiao C, Yan S, Ma X, et al. CRISPR/Cas9-mediated PINK1 deletion leads to neurodegeneration in rhesus monkeys. Cell Res. (2019) 29:334–6. doi: 10.1038/s41422-019-0142-y
55. Zuo E, Huo X, Yao X, Hu X, Sun Y, Yin J, et al. CRISPR/Cas9-mediated targeted chromosome elimination. Genome Biol. (2017) 18:224. doi: 10.1186/s13059-017-1354-4
56. Ou L, Przybilla MJ, Tăbăran A-F, Overn P, O'Sullivan MG, Jiang X, et al. A novel gene editing system to treat both Tay-Sachs and Sandhoff diseases. Gene Ther. (2020) 27:226–36. doi: 10.1038/s41434-019-0120-5
57. von Hammerstein AL, Eggel M, Biller-Andorno N. Is selecting better than modifying? An investigation of arguments against germline gene editing as compared to preimplantation genetic diagnosis. BMC Med Ethics. (2019) 20:83. doi: 10.1186/s12910-019-0411-9
58. Peranteau WH, Flake AW. The future of in utero gene therapy. Mol Diagn Ther. (2020) 24:135–42. doi: 10.1007/s40291-020-00445-y
59. Coller BS. Ethics of human genome editing. Ann Rev Med. (2019) 70:289–305. doi: 10.1146/annurev-med-112717-094629
60. Jacinto FV, Link W, Ferreira BI. CRISPR/Cas9-mediated genome editing: from basic research to translational medicine. J Cell Mol Med. (2020) 24:3766–78. doi: 10.1111/jcmm.14916
61. Ho BX, Loh SJH, Chan WK, Soh BS. In vivo genome editing as a therapeutic approach. Int J Mol Sci. (2018) 19:2721. doi: 10.3390/ijms19092721
62. Rui Y, Wilson DR, Green JJ. Non-viral delivery to enable genome editing. Trends Biotechnol. (2019) 37:281–93. doi: 10.1016/j.tibtech.2018.08.010
63. Jackson M, Marks L, May GHW. The genetic basis of disease. Essays Biochem. (2018) 62:643–723. doi: 10.1042/EBC20170053
64. Paluszkiewicz SM, Martin BS, Huntsman MM. Fragile X syndrome: the GABAergic system and circuit dysfunction. Dev Neurosci. (2011) 33:349–64. doi: 10.1159/000329420
65. Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, et al. Correction of fragile X syndrome in mice. Neuron. (2007) 56:955–62. doi: 10.1016/j.neuron.2007.12.001
66. Barnes SA, Pinto-Duarte A, Kappe A, Zembrzycki A, Metzler A, Mukamel EA, et al. Disruption of mGluR5 in parvalbumin-positive interneurons induces core features of neurodevelopmental disorders. Mol Psychiatry. (2015) 20:1161–72. doi: 10.1038/mp.2015.113
67. Erickson CA, Davenport MH, Schaefer TL, Wink LK, Pedapati EV, Sweeney JA, et al. Fragile X targeted pharmacotherapy: lessons learned and future directions. J Neurodev Disord. (2017) 9:7. doi: 10.1186/s11689-017-9186-9
68. Kazemi M, Salehi M, Kheirollahi M. Down syndrome: current status, challenges and future perspectives. Int J Mol Cell Med. (2016) 5:125–33. doi: 10.1038/s41586-019-1506-7
69. Haydar TF, Reeves RH. Trisomy 21 and early brain development. Trends Neurosci. (2012) 35:81–91. doi: 10.1016/j.tins.2011.11.001
70. Akutsu SN, Fujita K, Tomioka K, Miyamoto T. Applications of genome editing technology in research on chromosome aneuploidy disorders. Cells. (2020) 9:239. doi: 10.3390/cells9010239
71. Tafazoli A, Behjati F, Farhud DD, Abbaszadegan MR. Combination of genetics and nanotechnology for down syndrome modification: a potential hypothesis and review of the literature. Iran J Public Health. (2019) 48:371–8. doi: 10.18502/ijph.v48i3.878
72. Czermiński JT, Lawrence JB. Silencing trisomy 21 with XIST in neural stem cells promotes neuronal differentiation. Dev Cell. (2020) 52:294–308.e3. doi: 10.1016/j.devcel.2019.12.015
73. Panning B. X-chromosome inactivation: the molecular basis of silencing. J Biol. (2008) 7:30. doi: 10.1186/jbiol95
74. Karimzadeh P, Jafari N, Nejad Biglari H, Jabbeh Dari S, Ahmad Abadi F, Alaee MR, et al. GM2-gangliosidosis (Sandhoff and Tay Sachs disease): diagnosis and neuroimaging findings (An Iranian pediatric case series). Iran J Child Neurol. (2014) 8:55–60.
75. Boles DJ, Proia RL. The molecular basis of HEXA mRNA deficiency caused by the most common Tay-Sachs disease mutation. Am J Hum Genet. (1995) 56:716–24.
76. Schmidt B, Roberts RS, Davis P, Doyle LW, Barrington KJ, Ohlsson A, et al. Long-term effects of caffeine therapy for apnea of prematurity. N Engl J Med. (2007) 357:1893–902. doi: 10.1056/NEJMoa073679
77. Winerdal M, Urmaliya V, Winerdal ME, Fredholm BB, Winqvist O, Ådén U. Single Dose Caffeine Protects the Neonatal Mouse Brain against Hypoxia Ischemia. PLoS ONE. (2017) 12:e0170545. doi: 10.1371/journal.pone.0170545
78. Tanaka K, Ishitsuka Y, Kurauchi Y, Yamaguchi K, Kadowaki D, Irikura M, et al. Comparative effects of respiratory stimulants on hypoxic neuronal cell injury in SH-SY5Y cells and in hippocampal slice cultures from rat pups. Pediatr Int. (2013) 55:320–7. doi: 10.1111/ped.12079
79. Di Martino E, Bocchetta E, Tsuji S, Mukai T, Harris RA, Blomgren K, et al. Defining a time window for neuroprotection and glia modulation by caffeine after neonatal hypoxia-ischaemia. Mol Neurobiol. (2020) 57:2194–205. doi: 10.1007/s12035-020-01867-9
80. Sun H, Gonzalez F, McQuillen PS. Caffeine restores background EEG activity independent of infarct reduction after neonatal hypoxic ischemic brain injury. Dev Neurosci. (2020) 42:72–82. doi: 10.1159/000509365
81. Yu NY, Bieder A, Raman A, Mileti E, Katayama S, Einarsdottir E, et al. Acute doses of caffeine shift nervous system cell expression profiles toward promotion of neuronal projection growth. Sci Rep. (2017) 7:11458. doi: 10.1038/s41598-017-11574-6
82. Mokhtar WA, Fawzy A, Allam RM, Zidan N, Hamed MS. Association between adenosine receptor gene polymorphism and response to caffeine citrate treatment in apnea of prematurity; An Egyptian single-center study. Egypt Pediatr Assoc Gazette. (2018) 66:115–20. doi: 10.1016/j.epag.2018.09.001
83. Kumar VHS, Lipshultz SE. Caffeine and clinical outcomes in premature neonates. Children (Basel, Switzerland). (2019) 6:118. doi: 10.3390/children6110118
84. Kim YB, Komor AC, Levy JM. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol. (2017) 35:371–6. doi: 10.1038/nbt.3803
85. Tan J, Zhang F, Karcher D, Bock R. Expanding the genome-targeting scope and the site selectivity of high-precision base editors. Nat Commun. (2020) 11:629. doi: 10.1038/s41467-020-14465-z
86. Lee HK, Smith HE, Liu C, Willi M, Hennighausen L. Cytosine base editor 4 but not adenine base editor generates off-target mutations in mouse embryos. Commun Biol. (2020) 3:19. doi: 10.1038/s42003-019-0745-3
87. Jin S, Zong Y, Gao Q, Zhu Z, Wang Y, Qin P, et al. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science (New York, NY). (2019) 364:292–5. doi: 10.1126/science.aaw7166
88. Wienert B, Wyman SK. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science. (2019) 364:286–9. doi: 10.1126/science.aav9023
89. Ek CJ, Dziegielewska KM, Habgood MD, Saunders NR. Barriers in the developing brain and Neurotoxicology. Neurotoxicology. (2012) 33:586–604. doi: 10.1016/j.neuro.2011.12.009
90. Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. (2019) 18:358–78. doi: 10.1038/s41573-019-0012-9
91. Dong X. Current strategies for brain drug delivery. Theranostics. (2018) 8:1481–93. doi: 10.7150/thno.21254
92. Hanlon KS, Meltzer JC, Buzhdygan T, Cheng MJ, Sena-Esteves M, Bennett RE, et al. Selection of an efficient AAV vector for robust CNS transgene expression. Mol Ther Methods Clin Dev. (2019) 15:320–32. doi: 10.1016/j.omtm.2019.10.007
93. Lau CH, Ho JW, Lo PK, Tin C. Targeted transgene activation in the brain tissue by systemic delivery of engineered AAV1 expressing CRISPRa. Mol Ther Nucleic Acids. (2019) 16:637–49. doi: 10.1016/j.omtn.2019.04.015
94. Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol. (2016) 34:204–9. doi: 10.1038/nbt.3440
95. Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. (2017) 20:1172–9. doi: 10.1038/nn.4593
96. Hordeaux J, Yuan Y, Clark PM, Wang Q, Martino RA, Sims JJ, et al. The GPI-linked protein LY6A drives AAV-PHP.B transport across the blood-brain barrier. Mol Ther. (2019) 27:912–21. doi: 10.1016/j.ymthe.2019.02.013
97. Huang Q, Chan KY, Tobey IG, Chan YA, Poterba T, Boutros CL, et al. Delivering genes across the blood-brain barrier: LY6A, a novel cellular receptor for AAV-PHP.B capsids. PLoS ONE. (2019) 14:e0225206. doi: 10.1371/journal.pone.0225206
98. Batista AR, King OD, Reardon CP, Davis C, Shankaracharya, Philip V, et al. Ly6a differential expression in blood-brain barrier is responsible for strain specific central nervous system transduction profile of AAV-PHP.B. Hum Gene Ther. (2019) 31:90–102. doi: 10.1089/hum.2019.186
99. Kaushik A, Yndart A, Atluri V, Tiwari S, Tomitaka A, Gupta P, et al. Magnetically guided non-invasive CRISPR-Cas9/gRNA delivery across blood-brain barrier to eradicate latent HIV-1 infection. Sci Rep. (2019) 9:3928. doi: 10.1038/s41598-019-40222-4
100. Van Rie A, Harrington PR, Dow A, Robertson K. Neurologic and neurodevelopmental manifestations of pediatric HIV/AIDS: a global perspective. EJPN. (2007) 11:1–9. doi: 10.1016/j.ejpn.2006.10.006
101. Wilmshurst JM, Hammond CK, Donald K, Hoare J, Cohen K, Eley B. NeuroAIDS in children. Handb Clin Neurol. (2018) 152:99–116. doi: 10.1016/B978-0-444-63849-6.00008-6
102. Yourik P, Fuchs RT, Mabuchi M, Curcuru JL, Robb GB. Staphylococcus aureus Cas9 is a multiple-turnover enzyme. RNA (New York, NY). (2019) 25:35–44. doi: 10.1261/rna.067355.118
103. Charlesworth CT, Deshpande PS, Dever DP, Camarena J, Lemgart VT, Cromer MK, et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med. (2019) 25:249–54. doi: 10.1038/s41591-018-0326-x
104. Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y, Trombetta J, et al. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol. (2015) 33:102–6. doi: 10.1038/nbt.3055
105. Li A, Tanner MR, Lee CM, Hurley AE, De Giorgi M, Jarrett KE, et al. AAV-CRISPR gene editing is negated by pre-existing immunity to Cas9. Mol Ther. (2020) 28:1432–41. doi: 10.1016/j.ymthe.2020.04.017
106. Yang S, Chang R, Yang H, Zhao T, Hong Y, Kong HE, et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington's disease. J Clin Invest. (2017) 127:2719–24. doi: 10.1172/JCI92087
107. Betzer O, Perets N, Angel A, Motiei M, Sadan T, Yadid G, et al. In vivo neuroimaging of exosomes using gold nanoparticles. ACS Nano. (2017) 11:10883–93. doi: 10.1021/acsnano.7b04495
108. Chen CC, Liu L, Ma F, Wong CW, Guo XE, Chacko JV, et al. Elucidation of exosome migration across the blood-brain barrier model in vitro. Cell Mol Bioeng. (2016) 9:509–29. doi: 10.1007/s12195-016-0458-3
109. Khongkow M, Yata T, Boonrungsiman S, Ruktanonchai UR, Graham D, Namdee K. Surface modification of gold nanoparticles with neuron-targeted exosome for enhanced blood-brain barrier penetration. Sci Rep. (2019) 9:8278. doi: 10.1038/s41598-019-44569-6
110. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJA. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. (2011) 29:341–5. doi: 10.1038/nbt.1807
111. Zhang X-H, Tee LY, Wang X-G, Huang Q-S, Yang S-H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids. (2015) 4:e264. doi: 10.1038/mtna.2015.37
112. Ma H, Marti-Gutierrez N, Park SW, Wu J, Lee Y, Suzuki K, et al. Correction of a pathogenic gene mutation in human embryos. Nature. (2017) 548:413–9. doi: 10.1038/nature23305
113. Rose BI, Brown S. Genetically modified babies and a first application of clustered regularly interspaced short palindromic repeats (CRISPR-Cas9). Obstet Gynecol. (2019) 134:157–62. doi: 10.1097/AOG.0000000000003327
114. Rossidis AC, Stratigis JD, Chadwick AC, Hartman HA, Ahn NJ, Li H, et al. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat Med. (2018) 24:1513–8. doi: 10.1038/s41591-018-0184-6
115. Alhasan M, Mathkour M, Milburn JM. Clinical images: postterm newborn with lissencephaly presented with seizure: case report and review of literature. Ochsner J. (2015) 15:127–9.
117. Wilson RC, Carroll D. The daunting economics of therapeutic genome editing. CRISPR J. (2019) 2:280–4. doi: 10.1089/crispr.2019.0052
Keywords: Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR), gene-editing, gene therapy, hereditary neurological disorders, neonates, pharmacogenomics, caffeine, drug responsiveness
Citation: Wong PK, Cheah FC, Syafruddin SE, Mohtar MA, Azmi N, Ng PY and Chua EW (2021) CRISPR Gene-Editing Models Geared Toward Therapy for Hereditary and Developmental Neurological Disorders. Front. Pediatr. 9:592571. doi: 10.3389/fped.2021.592571
Received: 07 August 2020; Accepted: 19 February 2021;
Published: 11 March 2021.
Edited by:
Evan Yale Snyder, Sanford Burnham Institute for Medical Research, United StatesReviewed by:
Surendra Sharma, Women & Infants Hospital of Rhode Island, United StatesCopyright © 2021 Wong, Cheah, Syafruddin, Mohtar, Azmi, Ng and Chua. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eng Wee Chua, Y2V3ODU5MTFAdWttLmVkdS5teQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.