
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 05 February 2021
Sec. Pediatric Hematology and Hematological Malignancies
Volume 8 - 2020 | https://doi.org/10.3389/fped.2020.614521
 Nerea Vega-García1,2*
Nerea Vega-García1,2* Sara Perez-Jaume3
Sara Perez-Jaume3 Elena Esperanza-Cebollada1,2
Elena Esperanza-Cebollada1,2 Clara Vicente-Garcés1,2
Clara Vicente-Garcés1,2 Montserrat Torrebadell1,2,4
Montserrat Torrebadell1,2,4 Antonio Jiménez-Velasco5
Antonio Jiménez-Velasco5 Margarita Ortega6Marta Llop7,8Lorea Abad9
Margarita Ortega6Marta Llop7,8Lorea Abad9 José Manuel Vagace10
José Manuel Vagace10 Alfredo Minguela11
Alfredo Minguela11 Marta Pratcorona12
Marta Pratcorona12 Joaquín Sánchez-Garcia13Clara B. García-Calderón14María Teresa Gómez-Casares15
Joaquín Sánchez-Garcia13Clara B. García-Calderón14María Teresa Gómez-Casares15 Estela Martín-Clavero16
Estela Martín-Clavero16 Adela Escudero17
Adela Escudero17 Marta Riñón Martinez-Gallo18
Marta Riñón Martinez-Gallo18 Luz Muñoz19María Rosario Velasco20
Luz Muñoz19María Rosario Velasco20 Marina García-Morin21Albert Català2,4,22
Marina García-Morin21Albert Català2,4,22 Antonia Pascual23
Antonia Pascual23 Pablo Velasco24José Mª. Fernández25
Pablo Velasco24José Mª. Fernández25 Alvaro Lassaletta26
Alvaro Lassaletta26 José Luis Fuster27Isabel Badell28Águeda Molinos-Quintana14
José Luis Fuster27Isabel Badell28Águeda Molinos-Quintana14 Antonio Molinés29
Antonio Molinés29 Pilar Guerra-García30,31
Pilar Guerra-García30,31 Antonio Pérez-Martínez17,31Miriam García-Abós32Reyes Robles Ortiz33Sandra Pisa34
Antonio Pérez-Martínez17,31Miriam García-Abós32Reyes Robles Ortiz33Sandra Pisa34 Rosa Adán35
Rosa Adán35 Cristina Díaz de Heredia24José Luis Dapena2,22
Cristina Díaz de Heredia24José Luis Dapena2,22 Susana Rives2,4,22
Susana Rives2,4,22 Manuel Ramírez-Orellana9
Manuel Ramírez-Orellana9 Mireia Camós1,2,4* on behalf of the Biological Committee of the Group of Leukaemia of the Spanish Society of Paediatric Haematology Oncology (SEHOP Group)
Mireia Camós1,2,4* on behalf of the Biological Committee of the Group of Leukaemia of the Spanish Society of Paediatric Haematology Oncology (SEHOP Group)Robust and applicable risk-stratifying genetic factors at diagnosis in pediatric T-cell acute lymphoblastic leukemia (T-ALL) are still lacking, and most protocols rely on measurable residual disease (MRD) assessment. In our study, we aimed to analyze the impact of NOTCH1, FBXW7, PTEN, and RAS mutations, the measurable residual disease (MRD) levels assessed by flow cytometry (FCM-MRD) and other reported risk factors in a Spanish cohort of pediatric T-ALL patients. We included 199 patients treated with SEHOP and PETHEMA consecutive protocols from 1998 to 2019. We observed a better outcome of patients included in the newest SEHOP-PETHEMA-2013 protocol compared to the previous SHOP-2005 cohort. FCM-MRD significantly predicted outcome in both protocols, but the impact at early and late time points differed between protocols. The impact of FCM-MRD at late time points was more evident in SEHOP-PETHEMA 2013, whereas in SHOP-2005 FCM-MRD was predictive of outcome at early time points. Genetics impact was different in SHOP-2005 and SEHOP-PETHEMA-2013 cohorts: NOTCH1 mutations impacted on overall survival only in the SEHOP-PETHEMA-2013 cohort, whereas homozygous deletions of CDKN2A/B had a significantly higher CIR in SHOP-2005 patients. We applied the clinical classification combining oncogenetics, WBC count and MRD levels at the end of induction as previously reported by the FRALLE group. Using this score, we identified different subgroups of patients with statistically different outcome in both Spanish cohorts. In SHOP-2005, the FRALLE classifier identified a subgroup of high-risk patients with poorer survival. In the newest protocol SEHOP-PETHEMA-2013, a very low-risk group of patients with excellent outcome and no relapses was detected, with borderline significance. Overall, FCM-MRD, WBC count and oncogenetics may refine the risk-stratification, helping to design tailored approaches for pediatric T-ALL patients.
Compared to the particularly good outcome of pediatric patients with B-cell precursor acute lymphoblastic leukemia (BCP-ALL), T-ALL patients still do worse, and 15–20% of pediatric and 40% of adult T-ALL patients relapse. Importantly, relapsed T-ALL is often highly resistant, and such patients present a dismal prognosis (1, 2). Hence, prevention of relapse is imperative, and patients are allocated to intensive and often very toxic therapeutic regimes with short- and long-term side effects. A precise risk stratification of T-ALL patients would allow to dynamically adjust the intensity of treatment, helping to balance the risks of relapse and treatment-related toxicity. Current therapeutic protocols aim to tailor treatment by applying less intensive therapy to low-risk patients, thus reducing the risk of treatment-related toxicity, while reserving intensive therapy to high-risk patients. In contrast to BCP-ALL, the new biological insights in T-ALL have scarcely been incorporated to current protocols, and risk stratification relies mainly on response to treatment. Thus, although white blood cell (WBC) count and early response to prednisone at day 8 are used, measurable residual disease (MRD) quantification post-remission remains the most important risk factor (3–5). T-ALL patients show a slower blast clearance compared to BCP-ALL, and the MRD detection at a late time point (day 78) may define better the risk of relapse in T-ALL patients than earlier evaluation at end of induction (day 33) in some therapeutic protocols (6). The MRD gold standard method is real-time quantitative polymerase chain reaction for immunoglobulin and T-cell receptor clonality (IG-TR PCR-MRD) (6–8). Flow-cytometry MRD measurement (FCM-MRD), based on the leukemia-associated immunophenotype, is also used for risk stratification in some collaborative groups and provides reliable identification of patients eligible for reduced-intensity therapy in T-ALL and BCP-ALL (9–12). However, even in the setting of MRD-oriented protocols, some T-ALL patients relapse, and it is important to identify additional prognostic factors.
Multiple genetic abnormalities have been identified in T-ALL cases (13, 14), including proto-oncogene activation, tumor-suppressor gene deletions and constitutive activation of the NOTCH1 pathway, present in 40–60% of patients (15–17). The prognostic impact of NOTCH1 and FBXW7 mutations is controversial; it has been usually associated with favorable prognosis (18–20), but other studies have found no significant effect (21, 22), and some authors have reported that mutant FBXW7 independently predicts an inferior survival (23). Recent studies indicate that the impact of NOTCH1/FBXW7 mutations could be modulated by concomitant mutations in PTEN and RAS genes (3). PTEN loss through mutation or genomic deletion occurs in up to 35% of T-ALL pediatric patients, and ~10% harbor N- or K-RAS mutations. Although these abnormalities are usually associated with unfavorable prognosis, their impact in pediatric T-ALL patients is still unclear (24–27). The Group for Research in Adult Acute Lymphoblastic Leukemia (GRAALL) validated in adult T-ALL patients the clinical utility of a new oncogenetic classifier based on the mutational status of NOTCH1/FBXW7/PTEN/RAS (NFPR) genes (28). In this model, the presence of NOTCH1 and FBXW7 mutations without lesions involving PTEN/RAS defined the genetic low-risk subgroup (gLoR), and other genotypes were allocated to the genetic high-risk group (gHiR). In contrast, in a study in 145 pediatric T-ALL patients treated with the MRC UKALL2003 trial, neither PTEN nor RAS significantly impacted on outcome, and none of these mutations changed the highly favorable outcome of patients with double NOTCH1/FBXW7 mutations (29). Recently, the FRALLE group (French Acute Lymphoblastic Leukemia Study Group) suggested a new classifier based on WBC count, MRD by IG-TR PCR-MRD and the oncogenetic mutational status, which improved prediction of the relapse risk for their pediatric T-ALL patients (3).
Copy number alterations (CNA) have been reported as prognostic markers, both in BCP-ALL and in T-ALL (30, 31). CDKN2A/B loci deletion is present in up to 70% of T-ALL patients. In adult T-ALL, CDKN2A/B homozygous deletion (CDKN2A/Bhomo) was associated with a favorable prognosis only within the mature/cortical group (32), while other studies showed a correlation with poor survival or no association with outcome (31, 33). Overall, the role of oncogenetics as risk factors in T-ALL is controversial and seems to differ according to the treatment protocol.
We present a retrospective collaborative study of the Biological Committee of the Leukemia Group of the SEHOP (Spanish Society of Pediatric Hematology and Oncology) on a series of 199 pediatric T-ALL patients treated according to SEHOP and PETHEMA (Programa para el Estudio de la Terapéutica en Hemopatías Malignas) Cooperative Groups. In both protocols, we aimed to: (1) confirm the clinical impact of FCM-MRD measurement, (2) assess the prognostic significance of MRD at late time points, and (3) test whether the previously reported oncogenetic classifier by the FRALLE group could refine the risk stratification in our patients receiving BFM-based protocols.
The study design is shown in Supplementary Figure 1. This is a retrospective study, designed to include pediatric patients diagnosed with T-ALL in Spain with available biological material. The participation in the study was offered to all the Spanish centers belonging to the SEHOP Group of Leukemias. DNA samples from 26 different Spanish centers were sent to Hospital Sant Joan de Déu to perform the oncogenetic studies. The referring centers provided data about clinical and basic biological characterization of their patients at diagnosis and the levels of MRD during follow-up. The clinic-biological data of the patients included in our study are detailed in Table 1 and Supplementary Table 1. A total of 199 pediatric T-ALL patients treated according to SEHOP and PETHEMA Cooperative Groups from 1998 to 2019 were collected. We analyzed the molecular features of 189 patients, after excluding cases with T-cell lymphoblastic lymphoma, and patients with insufficient sample or no clinical data. For the survival analysis, we excluded those patients treated as per PETHEMA protocols, as they had been treated heterogeneously according to different therapeutic regimes. The 142 patients selected for the survival analyses were treated according to the consecutive protocols SHOP-2005 (n = 51) and SEHOP-PETHEMA-2013 (n = 91). These patients were representative of the whole SHOP-2005 and SEHOP-PETHEMA-2013 population in terms of clinical characteristics at diagnosis and outcome and only differed in a longer follow-up for the patients included in the study (Supplementary Table 2).
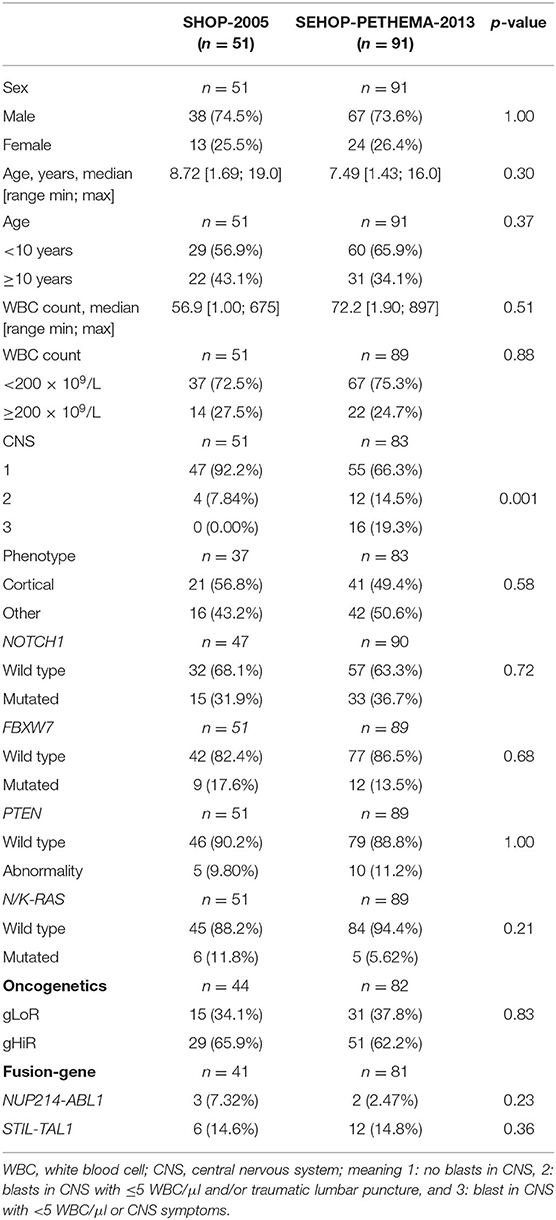
Table 1. Clinical and molecular features of patients included in SEHOP-PETHEMA-2013 and SHOP-2005 protocols for the survival analyses.
SHOP-2005 (2005–2013) and SEHOP-PETHEMA-2013 (2013-present) are two consecutive therapeutic protocols BFM (Berlin-Frankfurt-Münster)-inspired. The former has been previously described (5), and the full details of SEHOP-PETHEMA-2013 protocol are provided in the Supplementary Material. Patients with T-ALL were not eligible for standard risk group stratification in either protocol. To summarize the main differences between both protocols, in the newer SEHOP-PETHEMA-2013: (1) prednisone response was incorporated as initial stratification criteria; (2) an Induction IB course was added; (3) craniospinal irradiation was omitted and increased number of doses of triple intrathecal therapy were used instead; (4) there was a more intensive use of asparaginase, with more doses of asparaginase, use of PEG-asparaginase and longer periods of asparagine depletion; (5) there were more restrictive indications of allogeneic stem cell transplantation (allo-SCT), mainly based on FCM-MRD criteria, and (6) eight-colors FCM-MRD was performed in reference laboratories, in contrast with decentralized 4-color FCM-MRD in SHOP-2005 (see below).
The immunophenotyping by FCM was performed in each laboratory using the combination of antibodies and panels detailed for each protocol in the Supplementary Material. Briefly, in SHOP-2005 protocol, 4-color panels were used in each center for identification and monitoring of leukemia-associated immunophenotypes (LAIPs), according to the guidelines of group EGIL (34). For residual disease analysis, either patient-tailored antibody combinations or those employed at diagnosis were used. In this protocol, MRD levels >0.1% were considered positive and used for clinical decisions. However, lower values were reviewed and recorded for the analysis in the present study. In SEHOP-PETHEMA-2013 protocol, an 8-colors panel according to EuroFlow consortium was recommended at diagnosis (35), and FCM-MRD was centralized in 10 reference laboratories. For MRD detection down to the 0.01% level (with a required resolution of at least 20 events to refer a sample as positive), an optimum of 500,000 nucleated cells needed to be acquired. MRD was assessed at day 15, end of induction (time point 1, TP1) and after the second course of chemotherapy, around day 78 (timepoint 2, TP2). In this protocol, MRD levels >0.01% were considered positive, and the thresholds considered for clinical decisions varied according to the timepoint. Thus, patients were stratified to high-risk arm if FCM-MRD levels were >10% at day 15, >1% at the end of induction (TP1) and >0.1% at the end of consolidation (TP2). Patients were allocated to receive an allo-SCT in case of: (1) not complete remission (CR) after TP1; (2) day 33 FCM-MRD level >1%, and FCM-MRD level >0.1% at TP2; or (3) persistence of positive FCM-MRD (>0.01%) after high-risk blocks of chemotherapy.
Globally, irrespectively, of the protocol treatment, we used a sensitivity threshold of 0.01% in this study to try to reproduce the previously reported results in the FRALLE and GRAALL protocols (3, 28).
We analyzed the hotspot regions reported for NOTCH1 (exons 26, 27, 28, 34), FBXW7 (exons 9 and 10), PTEN (exon 7), NRAS (exons 1 and 2), and KRAS (exons 1 and 2) by Sanger sequencing as previously described (25, 36). CNAs were screened by MLPA using the SALSA MLPA P383 T-ALL kit (MRC Holland, Amsterdam, The Netherlands), according to the manufacturer's instructions. We used the Coffalyser software v.140721.1958 for the analysis (MRC-Holland, Amsterdam, The Netherlands). The P383 T-ALL kit assesses alterations (deletions or amplifications) in transcription factors (LEF1 and MYB), genes involved in signal transduction (PTEN, NF1, and PTPN2), in cell cycle (CDKN2A, CDKN2B, and CASP8AP2), in epigenetic regulation (EZH2, SUZ12, and PHF6), and identifies the STIL-TAL1 and NUP214-ABL1 fusion genes. Full experimental details are given in the Supplementary Methods.
To assess the predictive value of the conventional and new oncogenetic risk factors, we analyzed the prognostic impact of the NFPR mutational status, first addressing each gene individually, and then grouped as previously reported (20). We applied the GRAALL oncogenetic classifier (28), and grouped our patients into gLoR and gHiR to compare their outcome. We also analyzed our patients with the FRALLE score, combining the oncogenetic classifier by GRAALL group with WBC count ≥200 × 109/L and the MRD levels at TP1 (3), plus an additional time point at TP2.
Statistical analyses were carried out using R software (37), considering all p-values lower than 0.05 to be statistically significant. Details are provided in Supporting Information.
The ethical issues are detailed in Supplementary Material.
Molecular findings are shown in Table 1 and Supplementary Figure 2. Sixty-four patients (35%) harbored NOTCH1 mutations (NOTCH1mut), 45% at HD-N domain, 5% at HD-C domain and 9% at PEST domain. No mutations were found at TAD domain (Supplementary Figure 3). Out of NOTCH1mut patients, eleven cases with HD-C mutations also had concomitant mutations at the PEST domain (NOTCH1double). FBXW7 was mutated in 25 cases (13%), alone in 16 patients and combined with NOTCH1mut in 9 cases. Overall, we observed NOTCH1/FBXW7 mutations in 80/182 cases (44%), in line with previous reports, albeit in the lower range. PTEN abnormalities (mutation or major deletion, PTENabn) were found in 30 patients (20%), including 9 with NOTCH1/FBXW7 mutations. K/N-RAS mutations were identified in 14 patients (8%). Overall, 108 patients (57%) harbored at least one mutation/deletion in NOTCH1, FBXW7, PTEN, or K/N-RAS. The mutations included point mutations, insertions, deletions and indels (Supplementary Table 3 and Supplementary Figure 3). CDKN2A and CDKN2B were deleted in 109 out of the 160 tested cases (68%) and 95 patients (60%), respectively, including 89 homozygous deletions for both genes.
The association of NOTCH1, FBXW7, PTENabn, and K/N-RAS status with clinical variables is shown in Supplementary Table 4. Globally, and in each protocol, NOTCH1 mutations significantly associated with females, while K-RAS mutations associated with WBC count ≥200 × 109/L.
We could classify 168 patients according to the previously reported oncogenetic stratifier (Table 1). Fifty-eight patients (34.5%) presented NOTCH1/FBXW7 mutations without a PTEN/RAS mutation (NFmutPRwt) and were considered as gLoR; the remaining 110 cases (65.5%) were classified as gHiR. The gHiR group included 15 positive patients for both NOTCH1/FBXW7 and PTEN/RAS mutations, 26 patients harboring PTEN/RAS mutations only and 69 patients with all genes in a wild-type status. The distribution of oncogenetic risk-groups between SHOP-2005 and SEHOP-PETHEMA-2013 cohorts was similar (Table 1).
The outcome of the 142 assessable patients according to the therapeutic protocol is shown in Supplementary Figures 4, 5. The median follow-up of the patients was 7.7 years for SHOP-2005 and 2.8 years for SEHOP-PETHEMA-2013 cohort. Overall, we observed a non-significantly better outcome in the newest protocol: the 5-year overall survival (OS) was 85.9 ± 4.0 vs. 76.5 ± 5.9% (p = 0.22), and the disease-free survival (DFS) was 85.3 ± 4.1 vs. 70.5 ± 6.4% (p = 0.080) for SEHOP-PETHEMA-2013 and SHOP-2005 patients, respectively. Noticeably, the cumulative incidence of relapse (CIR) was not statistically different in both protocols (12.1 ± 3.8% vs. 18.1 ± 5.5%, p = 0.49).
FCM-MRD data in the SHOP-2005 cohort was available for 46 and 35 out of 51 patients at TP1 and TP2, respectively. At TP1, patients with MRD <0.01% (n = 33) showed a significantly better outcome than patients with MRD ≥ 0.01%, with a 5-year OS of 84.8 vs. 53.8%, a 5-year DFS of 78.7 vs. 46.2%, and a CIR of 12.6 vs. 38.5% (p = 0.019, p = 0.009, and p = 0.039, respectively). At TP2, with less data available, we observed only a borderline significant difference with worse OS in patients with FCM-MRD ≥ 0.01% (p = 0.050; Supplementary Figure 6).
In SEHOP-PETHEMA-2013 patients, FCM-MRD data was available in 84 and 62 out of 91 cases at TP1 and TP2, respectively. In contrast with the former protocol, FCM-MRD significantly impacted on outcome at TP2 (5-year DFS for MRD <0.01% 92.6 vs. 68.6% for MRD ≥ 0.01%, p = 0.026; Supplementary Figure 7), whereas at TP1 only impacted on OS with a borderline significance (p = 0.049).
In SHOP-2005 cohort, NOTCH1 mutations did not significantly impact on outcome. Of note, all patients with NOTCH1double mutations (n = 3) were alive in continuous CR (data not shown). Using the combination of the NFPR mutational status defined in the GRAAL oncogenetic stratifier (gLoR vs. gHiR), we observed no differences in patients'outcome (Figure 1A). In contrast, patients with CDKN2A/Bhomo had a trend toward a worse DFS (p = 0.079) and a significantly higher CIR than patients with normal or heterozygous deletions of CDKN2A/B (p = 0.009, Supplementary Figure 8a).
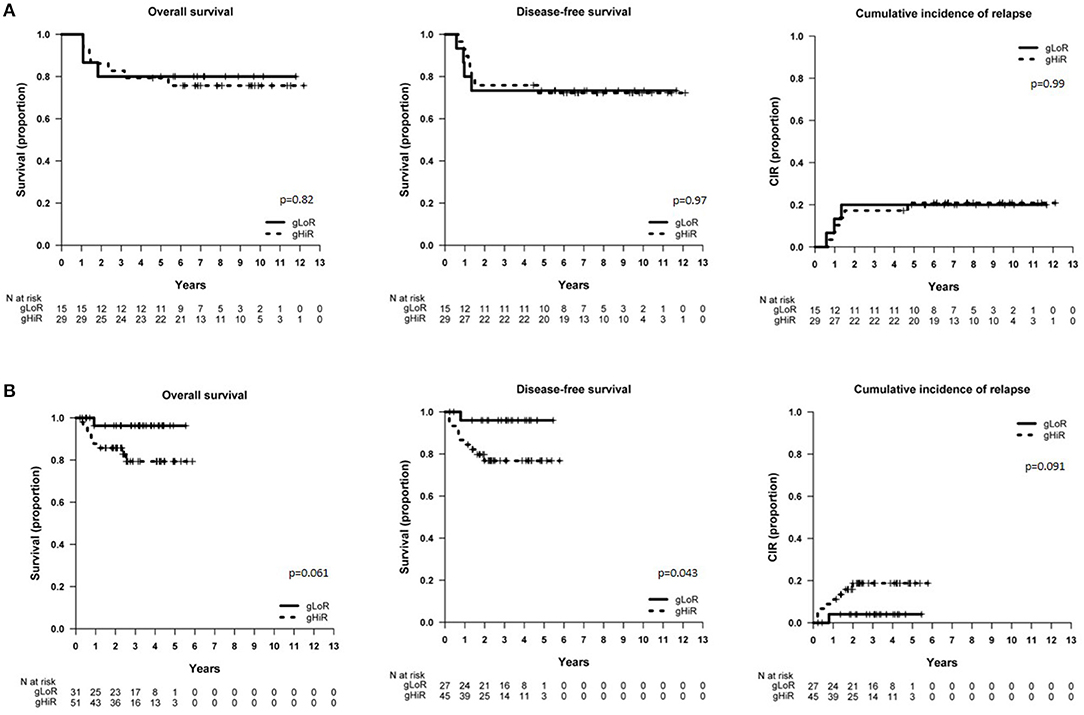
Figure 1. Survival of patients included in SHOP-2005 and SEHOP-PETHEMA-2013 protocol according to oncogenetics. (A) OS, DFS, and CIR in patients included in the SHOP-2005 protocol with gLoR vs. gHiR; (B) OS, DFS, and CIR in patients included in the SEHOP-PETHEMA-2013 protocol with gLoR vs. gHiR. gLoR: presence of NOTCH1 and FBXW7 mutations without lesions involving PTEN/RAS; gHiR: all the other genotypes.
In patients treated with SEHOP-PETHEMA-2013 protocol, those cases with NOTCH1mut showed a better OS (p = 0.045), and all patients with NOTCH1double mutations (n = 7) were alive in continuous CR (data not shown). In contrast to the former protocol, those SEHOP-PETHEMA-2013 patients classified as gLoR presented a significantly better DFS (p = 0.043), and a trend toward a better OS and a lower CIR (Figure 1B). Patients with CDKN2A/Bhomo had a worse outcome, but the difference was not statistically significant (Supplementary Figure 8b).
Overall, we observed a different impact of oncogenetics on the SHOP-2005 and SEHOP-PETHEMA-2013 series of patients.
The univariate and multivariate analyses of OS and DFS for patients in SHOP-2005 and SEHOP-PETHEMA-2013 protocols is shown in Table 2. FCM-MRD was significantly predictive of outcome in both SHOP-2005 and SEHOP-PETHEMA-2013 cohorts (Supplementary Figures 6, 7), being statistically significant at early TP1 only for SHOP-2005 (p = 0.035) and at late TP2 only for SEHOP-PETHEMA 2013 protocol (p = 0.048). However, other classical risk factors impacted differently depending on the protocol: patients with hyperleukocytosis ≥200 × 109/L showed a significantly worse DFS only in SHOP-2005 series. Sex, age, and CNS infiltration did not impact on outcome in either protocol.
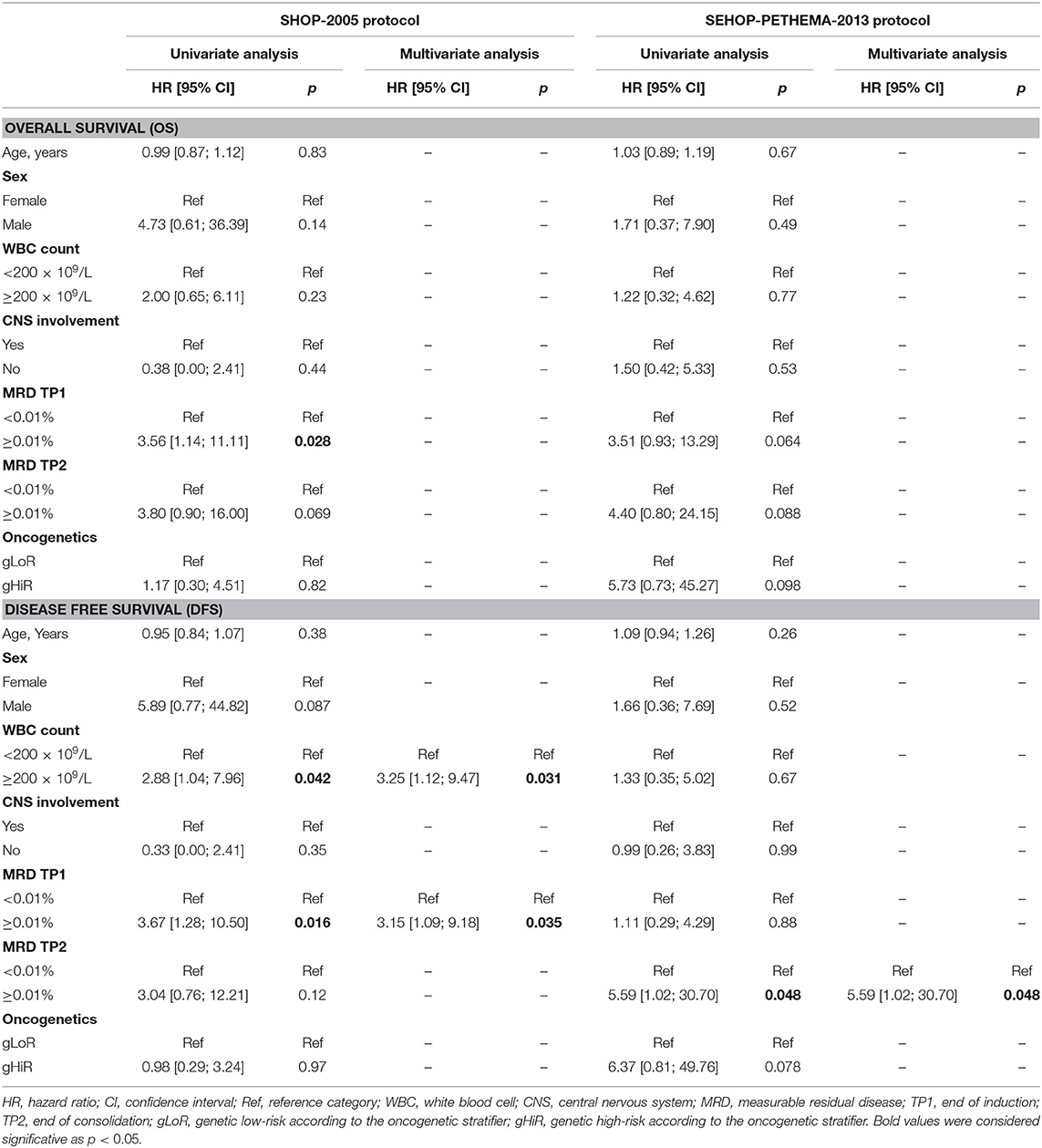
Table 2. Univariate and multivariate analysis of OS and DFS for patients in SHOP-2005 and SEHOP-PETHEMA-2013 protocols.
We used different combinations of classical and new risk factors to gain additional information on the relapse risk. First, we wanted to test in our series of patients the clinical classification reported by Petit et al. (3) integrating oncogenetics, MRD at TP1 (threshold 0.01%) and WBC count. By doing that, we observed a different distribution of subgroups in the analysis by protocols: in SHOP-2005, the outcome analysis following the clinical classification defined by the FRALLE group showed a poorer survival of high-risk patients as compared to the similar outcome of the low- and intermediate-risk patients (OS p = 0.25, DFS p = 0.024, and CIR p = 0.044; data not shown). In contrast, in SEHOP-PETHEMA-2013 patients, the low-risk patients had a better not significant outcome as compared to the survival of intermediate- and high-risk groups (p = 0.15, data not shown). We grouped the latter groups with similar outcome, and found that the clinical classifier was able to identify with borderline significance low-risk patients with 100% OS and no relapses (p = 0.059, p = 0.05, and p = 0.06 for OS, DFS, and CIR, respectively) (Figure 2).
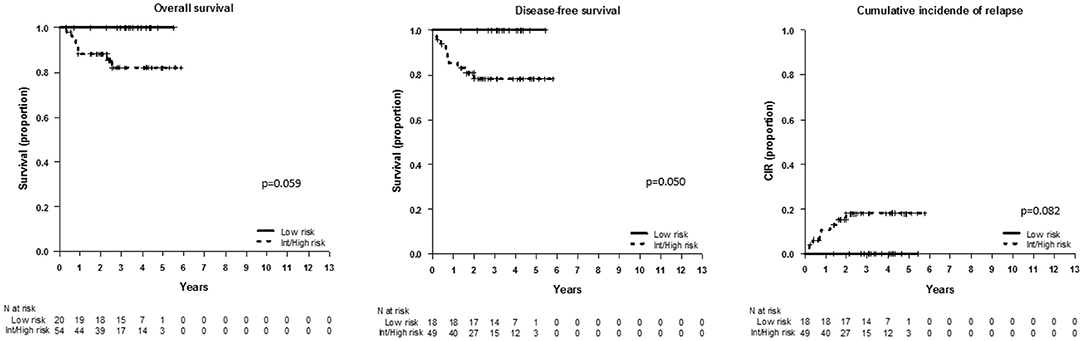
Figure 2. Outcome of patients included in SEHOP-PETHEMA-2013 according to the clinical classifier defined by Petit et al. (3). OS, DFS, and CIR in SEHOP-PETHEMA-2013 patients according to the FRALLE group combination, comparing the outcome of low-risk patients with the intermediate-risk and high-risk pooled together. The clinical classifier identified a subgroup of low-risk patients with excellent outcome, 100% OS at 5 years and no relapses. We observed a trend to the statistical significance in the outcome of the low-risk group vs. the remaining patients.
Taken together, the oncogenetics, FCM-MRD at TP1 and WBC count, combined as defined by Petit et al. (3) in the FRALLE group, impacted significantly on outcome, and identified different risk populations in the analyzed cohorts.
Most pediatric T-ALL protocols only rely on MRD assessment to guide stratification in risk-groups, and robust and applicable risk-stratifying genetic factors at diagnosis are still lacking. The FRALLE group built a classifier based on WBC count, MRD at TP1 and an oncogenetic stratifier according to NFPR mutational status. However, some questions remain open regarding the FRALLE classifier: (1) would it be predictive of outcome in patients treated with BFM-based pediatric protocols?; (2) could MRD at TP2 be applied?, and (3) would this classifier be similarly predictive of patients' outcome when assessing MRD by FCM? To answer these questions, we analyzed a large cohort of pediatric T-ALL patients treated with consecutive Spanish protocols. We confirmed MRD as a prognostic risk-factor throughout different protocols, also when measured by flow-cytometry. However, oncogenetics impacted differently according to treatment, confirming that therapeutic modifications can lead to changes in the prognostic impact of biological variables like NOTCH1 mutations.
We first evaluated the outcome of Spanish pediatric T-ALL patients in consecutive protocols. We observed a not statistically significant improvement in the 5-year DFS with SEHOP-PETHEMA-2013 protocol, as compared with the older SHOP-2005 protocol. The differences between both protocols could partly explain the different outcome. Briefly, both are BFM-inspired protocols, but in SEHOP-PETHEMA-2013 an induction IB course with cyclophosphamide, cytarabine, and mercaptopurine was incorporated. In this regard, an intensive induction and multiagent consolidation including cyclophosphamide is strongly recommended in T-ALL (38), and omitting the induction IB resulted in inferior outcome in large reported cohorts (39, 40). Also, a more intense asparaginase scheme including PEG-asparaginase was given in SEHOP-PETHEMA-2013. Thus, as compared to SHOP-2005 protocol, patients received increased doses of asparaginase and longer periods of asparagine depletion. The response to treatment was also differently assessed: in SEHOP-PETHEMA-2013 the prednisone response was added as stratifying criteria, and the non-centralized 4-colors FCM-MRD was replaced by 8-colors FCM-MRD with agreed panels, centralized in reference laboratories. Moreover, a lower number of patients underwent an allo-SCT in SEHOP-PETHEMA-2013 (Supplementary Figure 4), and had a lower non-leukemic mortality (data not shown). Overall, the intensification of the chemotherapy, a better stratification of patients and lower non-leukemic mortality in the latter protocol, could have contributed to the improvement of the survival in SEHOP-PETHEMA-2013 cohort. Similarly, a survival benefit dependent on mainly reducing toxicity alongside improving efficacy was recently described in adult T-ALL patients (41).
We then assessed the prognostic impact of conventional and new risk factors within each protocol. Hyperleukocytosis ≥200 × 109/L was only predictive of a significantly worse DFS in SHOP-2005 cohort. Next, we analyzed the prognostic value of FCM-MRD in our cohorts. FCM-MRD levels impacted significantly on patients' outcome in both SHOP-2005 and SEHOP-PETHEMA-2013 protocols, proving the usefulness of FCM-MRD with a threshold of 0.01% to discriminate prognosis. This is especially relevant in the difficult setting of flow-cytometry assessment of MRD in T-ALL patients. However, FCM-MRD impacted differently according to the protocol and time point. The impact of MRD at late time points in T-ALL patients may vary between protocols, as reported by AIEOP-BFM and NOPHO Groups (6, 11). The discrepancies could be explained by differences in the treatment protocols, stratification criteria, cutoff levels and methods (11). In our study, FCM-MRD discriminated outcome both at early and late time points in SHOP-2005, whereas significant differences were observed in SEHOP-PETHEMA-2013 cohort at TP2. This could be driven by differences between both protocols in terms of intensity of treatment, with earlier stratification criteria using prednisone response, and early intensification therapy being added in SEHOP-PETHEMA-2013 patients, mimicking the backbone of current BFM protocols. Importantly, methodological differences in the FCM-MRD assessment between SHOP-2005 and SEHOP-PETHEMA-2013 can play a role in the results. Thus, in the latter protocol, 8-color FCM-MRD centralized in reference laboratories, replaced the non-centralized 4-color FCM-MRD performed in SHOP-2005. The increased number of markers in SEHOP-PETHEMA-2013 should, theoretically, improve the accuracy of the analysis, but we did not investigate the concordance between FCM and other methods like IG-TR MRD. On the other hand, the sensitivity in the FCM studies performed in SEHOP-PETHEMA-2013 was expected to be higher than in SHOP-2005 due to a higher number of acquired cells, but this point has not been confirmed nor a straightforward comparison between methodologies has been performed. Globally, in the SEHOP-PETHEMA-2013 cohort, FCM-MRD reproduced previous remarks about the relevance of late time points in BFM-based protocols. Overall, although the number of patients in each protocol is low, and even considering the less controlled setting in the older protocols, FCM-MRD appeared as a robust risk-factor in our Spanish series of patients.
As reported, we confirmed in our cohorts recurrent mutations in pediatric T-ALL patients. We found alterations in NOTCH1/FBXW7 genes in 44% of our patients. The incidence of NOTCH1 mutated cases in our series is low. However, similar incidences have been published in other series, ranging from 23 to 70.8% (42, 43). Specifically, 23 and 40% of NOTCH1 mutations were reported in Turkish (44) and Japanese cohorts of patients (45), respectively. FBXW7 mutations also range from 8.6 to 30.8% in the literature. Overall, the frequency of NFPR mutations, and mutation rates of CDKN2A/B deletions, were in the range of previously reported incidences (42, 46–48). When we classified patients into gLoR and gHiR, patients were grouped in the same way in both protocols. However, we found a lower proportion of gLoR patients compared to the FRALLE group, in parallel to the lower incidence of NOTCH1/FBXW7 mutations in our cohort. The highly variable frequency in NOTCH1/FBXW7 mutations could be explained by different factors including bias in sample storage, ethnicity, methodological aspects, and the total number of patients.
Regarding oncogenetic impact, we first studied the impact of individual genetic alterations. NOTCH1 mutations had a favorable clinical impact in SEHOP-PETHEMA-2013 cohort, and all patients with NOTCH1double mutations were alive in continuous complete remission. Neither PTEN nor K/N-RAS mutations had a significant impact on outcome. As reported, the homozygous deletion of CDKN2A/B associated with higher CIR in SHOP-2005 patients. In this line, recent studies have shown that the lack of deletions involving CDKN2A/ARF/CDKN2B locus, combined with undetectable MRD (≤ 0.01%) values, allowed the identification of a subset of adult T-ALL patients with better OS in the absence of allo-SCT (31).
The analysis of the combination of oncogenetic abnormalities yielded different results according to the protocols. Hence, the application of the oncogenetic classifier (NFPR) to our cohorts showed no impact in SHOP-2005 patients and a trend to a better OS and CIR, and a significantly better DFS in SEHOP-PETHEMA-2013 patients. We next tested the clinical classification reported by the FRALLE group, integrating oncogenetics, MRD at TP1 (threshold 0.01%) and WBC count. The FRALLE classifier allowed to identify different subgroups of patients with statistically different outcome in the whole cohort of Spanish patients. In the newest protocol SEHOP-PETHEMA-2013, a very low-risk group could be segregated with this score. Thereby, we reproduced the results of the FRALLE group in different, BFM-inspired therapeutic protocols. However, the results in SHOP-2005 and SEHOP-PETHEMA-2013 analyzed individually varied, probably due to differences between protocols.
The FRALLE-2000 protocol differed in several aspects from SHOP-2005 and SEHOP-PETHEMA-2013. Briefly, FRALLE-2000 and SHOP-2005 had similar indications of cranial irradiation, given both prophylactically in a subgroup of patients and therapeutically in all T-ALL patients, whereas cranial irradiation was omitted in SEHOP-PETHEMA-2013. The FRALLE-2000 protocol had more cyclophosphamide and anthracycline cumulative doses than SHOP-2005. On the other hand, SEHOP-PETHEMA-2013 had a prolonged intensification with PEG-asparaginase compared with FRALLE-2000 and SHOP-2005 protocols. Taken together, we observed that, even in the setting of different protocols, the oncogenetics, FCM-MRD at TP1, and WBC count, may be useful to refine the risk-stratification in pediatric T-ALL patients. In BFM-based protocols, the FRALLE classifier allowed to identify a subgroup of patients with very low-risk, who could be considered as potential candidates to de-intensify treatment. Our current protocol, SEHOP-PETHEMA-2013, aims to intensify treatment, and no de-intensification is considered. Interestingly, following the FRALLE risk stratification, a quarter of our intermediate-risk patients (20/76, 26%) could be considered as candidates to receive less intense treatment, and be moved to a lower-risk group. Our results, if confirmed in larger series, could help to consider the option of selected de-intensification therapy in future clinical trials.
Some factors may limit our study. Centralization in reference laboratories and biobanking was not mandatory in former protocols. Also, to assess the possible impact of differences in the therapeutic regimes, we analyzed the outcome of each protocol separately. Hence, the low number of patients in each group may have limited the power of statistics and precluded the finding of significant results. Finally, the median follow-up of SEHOP-PETHEMA-2013 cohort is short (2.8 years), but it covers most of the expected T-ALL relapses.
In summary, we present a national collaborative study of clinical and genetic prognostic factors in a large series of T-ALL pediatric patients. Notably, FCM-MRD predicted outcome in both Spanish protocols, being more important in TP2 in SEHOP-PETHEMA-2013. We could reproduce the FRALLE group's results in our BFM-based SEHOP protocols, and observed the predictive value of the combination of FCM-MRD, WBC count and oncogenetics to predict outcome at TP1. This classifier allowed, in our current protocol SEHOP-PETHEMA-2013, to identify a low-risk subgroup of patients with excellent outcome. Further studies in the context of controlled clinical trials would be necessary to confirm if the de-intensification therapy of a selected low-risk group of patients would lead to the same good outcome. Our results provide data that could be clinically relevant, as may help to apply tailored risk-directed treatments to reduce both toxicity and relapse.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by Comitè Ètic d'Investigació amb medicaments (CEIm) de la Fundació Sant Joan de Déu. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
NV-G and MC designed the study and MC supervised the project. NV-G, EE-C, and CV-G performed the molecular analyses of the study. NV-G and SP-J analyzed the data and performed the statistical analyses. NV-G and MC wrote the paper with the contribution of SP-J, MT, JD, and SR. MT, AJ-V, MO, ML, LA, JV, AM, MP, JS, CG-C, MG-C, EM-C, AE, MM-G, LM, MV, MG-M and MC diagnosed patients and performed the main biological characterization of samples. AC, AP, AJ-V, JMF, AL, JLF, IB, AMo, AM-Q, PG-G, APM, MG, RR, SP, RA, JD, SR and MR-O recruited patients. All authors read and approved the final version of the manuscript. Other centers and investigators belonging to the Biological Committee and other integrands of the Group of Leukemia of the Spanish Society of Pediatric Hematology and Oncology (SEHOP) contributing to this study are listed in the participant investigators.
The authors thank the contribution of other clinicians and researchers from different centers integrated in the Group of Leukemia of the Spanish Society of Pediatric Hematology and Oncology (SEHOP), for collecting data, providing and caring for study patients: Hospital Sant Joan de Déu (Nuria Conde, Sandra Pont, Sara Montesdeoca, Ignacio Isola); Hospital Niño Jesús (Ana Castillo), Hospital Universitari i Politècnic La Fe (Inés Gomez-Seguí), Hospital Carlos Haya (Antonia Pascual); Hospital General de Alicante (Fabián Tarín, María Tasso); Hospital Virgen de la Arrixaca (María Victoria Martinez-Sánchez, José Antonio Campillo, Eduardo Ramos-Eibal, Esther Llinares, Ana María Galera, María del Mar Bermúdez); Hospital Vall d'Hebrón (Anna Varela-Magallón); Hospital Virgen de las Nieves (Francisco Ruiz-Cabello, Pilar Jiménez, Mª José Ortega); Hospital Reina Sofía (Carmen Martínez); Hospital Virgen del Rocío (José Antonio Pérez-Simón); Hospital Gregorio Marañón (Carolina Martínez-Laperche); Hospital Santa Creu i Sant Pau (Josep Nomdedéu); Hospital Virgen de la Salud (Nerea Domínguez); Hospital Maternoinfantil de Badajoz (María Dolores de la Maya, María Belén Moreno); Hospital Marqués de Valdecillas (Mónica López-Duarte), Complejo Hospitalario de Navarra (María Sagaseta de Ilurdoz); Hospital Virgen de la Macarena (David García, Helga Benítez, Ana Fernández-Teijeiro); Hospital Son Espases (Marta Bernués, Laia Ferrés, José Antonio Salinas).
This study has been supported by a grant from Instituto de Salud Carlos III (PI12/02417; PI16/00246), integrated into Plan Nacional de I+D+I and co-funded by ISCIII—Subdirección General de Evaluación y Fomento de la Investigación Sanitaria—and Fondo Europeo de Desarrollo Regional (FEDER), CIBERER.
MT reports travel and accommodation support from Novartis (outside the submitted work); travel and accommodation support from Jazz Pharma and Shire/Servier (outside the submitted work); and travel and accommodation support from Amgen (outside the submitted work). JLF is a consultant/advisory member for Amgen, Jazz Pharmaceuticals, and Novartis (outside the submitted work), receives honoraria for speaking at symposia from Amgen, Servier, Jazz Pharmaceuticals, and Pfizer (outside the submitted work) and support for attending symposia from Servier and Jazz Pharmaceuticals (outside the submitted work). AM discloses talk fees from Jazz Pharma and Shire/Servier outside the presented work and reports travel and accommodation support from Jazz Pharma, Shire/Servier, and Novartis (outside the submitted work). JD reports advisory board honorarium, speaker fees, and travel and accommodation support from Jazz Pharma and Shire/Servier; personal fees, advisory board honorarium, speaker fees, and travel and accommodation support from Novartis (outside the submitted work); advisory board honorarium, and travel and accommodation support from Amgen (outside the submitted work); advisory board honorarium, speaker fees, and travel and accommodation support from Celgene (outside the submitted work); advisory board honorarium, speaker fees, and travel and accommodation support from Sobi (outside the submitted work). SR reports advisory board honorarium, speaker fees, and travel and accommodation support from Jazz Pharma and Shire/Servier; personal fees, advisory board honorarium, speaker fees, and travel and accommodation support from Novartis (outside the submitted work); advisory board honorarium, and travel and accommodation support from Amgen (outside the submitted work); advisory board honorarium, speaker fees, and travel and accommodation support from Celgene (outside the submitted work). MC discloses talk fees from Shire/Servier outside the presented work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are indebted to patients and families and Obra Social from Hospital Sant Joan de Déu, Força Miquel, Candela polsera solidària, Pirata Barballum, Magic, and Mua projects for their support. We are also indebted with Fundación Cris contra el cáncer, Fundación Unoentrecienmil and Asociación Pablo Ugarte. We also want to thank the excellent technical support from Ms Camino Estella and Ms Lidia Ruiz. We are indebted to the Biobanc de l'Hospital Infantil Sant Joan de Déu per a la Investigació, integrated into the Spanish Biobank Network of ISCIII for the sample and data procurement.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2020.614521/full#supplementary-material
1. Teachey DT, Pui CH. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. (2019) 20:e142–54. doi: 10.1016/S1470-2045(19)30031-2
2. Schrappe M, Hunger SP, Pui CH, Saha V, Gaynon PS, Baruchel A, et al. Outcomes after Induction Failure in Childhood Acute Lymphoblastic Leukemia. N Engl J Med. (2012) 366:1371–81. doi: 10.1056/NEJMoa1110169
3. Petit A, Trinquand A, Chevret S, Ballerini P, Cayuela JM, Grardel N, et al. Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood. (2018) 131:289–300. doi: 10.1182/blood-2017-04-778829
4. Hefazi M, Litzow MR. Recent advances in the biology and treatment of T cell acute lymphoblastic leukemia. Curr Hematol Malig Rep. (2018) 13:265–74. doi: 10.1007/s11899-018-0455-9
5. Rives S, Estella J, Camós M, García-Miguel P, Verdeguer A, Couselo JM, et al. [T-cell pediatric acute lymphoblastic leukemia: analysis of survival and prognostic factors in 4 consecutive protocols of the Spanish cooperative study group SHOP]. Med Clin. (2012) 139:141–9. doi: 10.1016/j.medcli.2011.12.019
6. Schrappe M, Valsecchi MG, Bartram CR, Schrauder A, Panzer-Grümayer R, Möricke A, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. (2011) 118:2077–84. doi: 10.1182/blood-2011-03-338707
7. Bruggemann M, Kotrova M. Minimal residual disease in adult ALL: technical aspects and implications for correct clinical interpretation. Blood Adv. (2017) 1:2456–66. doi: 10.1182/bloodadvances.2017009845
8. Brüggemann M, Kotrová M, Knecht H, Bartram J, Boudjogrha M, Bystry V, et al. Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia. (2019) 33:2241–53. doi: 10.1038/s41375-019-0496-7
9. Wood B, Wu D, Crossley B, Dai Y, Williamson D, Gawad C, et al. Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood. (2018) 131:1350–9. doi: 10.1182/blood-2017-09-806521
10. Attarbaschi A, Mann G, Panzer-Grümayer R, Röttgers S, Steiner M, König M, et al. Minimal residual disease values discriminate between low and high relapse risk in children with B-cell precursor acute lymphoblastic leukemia and an intrachromosomal amplification of chromosome 21: the Austrian and German acute lymphoblastic leukemia berl. J Clin Oncol. (2008) 26:3046–50. doi: 10.1200/JCO.2008.16.1117
11. Modvig S, Madsen HO, Siitonen SM, Rosthøj S, Tierens A, Juvonen V, et al. Minimal residual disease quantification by flow cytometry provides reliable risk stratification in T-cell acute lymphoblastic leukemia. Leukemia. (2019) 33:1324–36. doi: 10.1038/s41375-018-0307-6
12. Tembhare PR, Narula G, Khanka T, Ghogale S, Chatterjee G, Patkar NV, et al. Post-induction measurable residual disease using multicolor flow cytometry is strongly predictive of inferior clinical outcome in the real-life management of childhood T-cell acute lymphoblastic leukemia: a study of 256 patients. Front Oncol. (2020) 10:577. doi: 10.3389/fonc.2020.00577
13. Girardi T, Vicente C, Cools J, De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood. (2017) 129:1113–23. doi: 10.1182/blood-2016-10-706465
14. Gianni F, Belver L, Ferrando A. The genetics and mechanisms of T-cell acute lymphoblastic leukemia. Cold Spring Harb Perspect Med. (2019) 10:a035246. doi: 10.1101/cshperspect.a035246
15. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. (2017) 49:1211–18. doi: 10.1038/ng.3909
16. Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. (2016) 16:494–507. doi: 10.1038/nrc.2016.63
17. Kimura S, Seki M, Yoshida K, Shiraishi Y, Akiyama M, Koh K, et al. NOTCH1 pathway activating mutations and clonal evolution in pediatric T-cell acute lymphoblastic leukemia. Cancer Sci. (2019) 110:784–94. doi: 10.1111/cas.13859
18. Kox C, Zimmermann M, Stanulla M, Leible S, Schrappe M, Ludwig W-D, et al. The favorable effect of activating NOTCH1 receptor mutations on long-term outcome in T-ALL patients treated on the ALL-BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia. (2010) 24:2005–13. doi: 10.1038/leu.2010.203
19. Asnafi V, Buzyn A, Le Noir S, Baleydier F, Simon A, Beldjord K, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a group for research on adult acute lymphoblastic leukemia (GRAALL) study. Blood. (2009) 113:3918–24. doi: 10.1182/blood-2008-10-184069
20. Jenkinson S, Koo K, Mansour MR, Goulden N, Vora A, Mitchell C, et al. Impact of NOTCH1/FBXW7 mutations on outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on the MRC UKALL 2003 trial. Leukemia. (2013) 27:41–7. doi: 10.1038/leu.2012.176
21. Zuurbier L, Homminga I, Calvert V, te Winkel ML, Buijs-Gladdines JGC a M, Kooi C, et al. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia. (2010) 24:2014–22. doi: 10.1038/leu.2010.204
22. Clappier E, Collette S, Grardel N, Girard S, Suarez L, Brunie G, et al. NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment, but not on outcome, in children with T-cell acute lymphoblastic leukemia (T-ALL) treated on EORTC trials 58881 and 58951. Leukemia. (2010) 24:2023–31. doi: 10.1038/leu.2010.205
23. Yuan L, Lu L, Yang Y, Sun H, Chen X, Huang Y, et al. Genetic mutational profiling analysis of T cell acute lymphoblastic leukemia reveal mutant FBXW7 as a prognostic indicator for inferior survival. Ann Hematol. (2015) 94:1817–28. doi: 10.1007/s00277-015-2474-0
24. Zuurbier L, Petricoin EF, Vuerhard MJ, Calvert V, Kooi C, Buijs-Gladdines JGC, et al. The significance of PTEN and AKT aberrations in pediatric T-cell acute lymphoblastic leukemia. Haematologica. (2012) 97:1405–13. doi: 10.3324/haematol.2011.059030
25. Bandapalli OR, Zimmermann M, Kox C, Stanulla M, Schrappe M, Ludwig WD. NOTCH1 activation clinically antagonizes the unfavorable effect of PTEN inactivation in BFM-treated children with precursor T-cell acute lymphoblastic leukemia. Haematologica. (2013) 98:928–36. doi: 10.3324/haematol.2012.073585
26. Kindler T, Cornejo MG, Scholl C, Liu J, Leeman DS, Haydu JE, et al. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to gamma-secretase inhibitors. Blood. (2008) 112:3373–82. doi: 10.1182/blood-2008-03-147587
27. Perentesis JP, Bhatia S, Boyle E, Shao Y, Shu XO, Steinbuch M, et al. RAS oncogene mutations and outcome of therapy for childhood acute lymphoblastic leukemia. Leukemia. (2004) 18:685–92. doi: 10.1038/sj.leu.2403272
28. Trinquand A, Tanguy-Schmidt A, Abdelali RB, Lambert J, Beldjord K, Lengliné E, et al. Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a group for research in adult acute lymphoblastic leukemia study. J Clin Oncol. (2013) 31:4333–42. doi: 10.1200/JCO.2012.48.5292
29. Jenkinson S, Goulden N, Vora A, Linch DC, Gale RE. Impact of PTEN abnormalities on outcome in pediatric patients with T-cell acute lymphoblastic leukemia treated on the MRC UKALL2003 trial. Leukemia. (2016) 30:39–47. doi: 10.1038/leu.2015.206
30. Ribera J, Zamora L, Morgades M, Mallo M, Solanes N, Batlle M, et al. Copy number profiling of adult relapsed B-cell precursor acute lymphoblastic leukemia reveals potential leukemia progression mechanisms. Genes Chromosom Cancer. (2017) 56:810–20. doi: 10.1002/gcc.22486
31. Genescà E, Lazarenkov A, Morgades M, Berbis G, Ruíz-Xivillé N, Gómez-Marzo P, et al. Frequency and clinical impact of CDKN2A/ARF/CDKN2B gene deletions as assessed by in-depth genetic analyses in adult T cell acute lymphoblastic leukemia. J Hematol Oncol. (2018) 11:9–12. doi: 10.1186/s13045-018-0639-8
32. Van Vlierberghe P, Ambesi-Impiombato A, De Keersmaecker K, Hadler M, Paietta E, Tallman MS, et al. Prognostic relevance of integrated genetic profiling in adult T-cell acute lymphoblastic leukemia. Blood. (2013) 122:74–82. doi: 10.1182/blood-2013-03-491092
33. Marks DI, Paietta EM, Moorman AV, Richards SM, Buck G, DeWald G, et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood. (2009) 114:5136–45. doi: 10.1182/blood-2009-08-231217
34. Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A, et al. Proposals for the immunological classification of acute leukemias. European group for the immunological characterization of leukemias (EGIL). Leukemia. (1995) 9:1783–6.
35. van Dongen JJM, Orfao A. EuroFlow: resetting leukemia and lymphoma immunophenotyping. Basis for companion diagnostics and personalized medicine. Leukemia. (2012) 26:1899–907. doi: 10.1038/leu.2012.121
36. Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. (2004) 306:269–71. doi: 10.1126/science.1102160
37. R Core Team. R: A Language and Environment for Statistical Computing. (2019). Available online at: https://www.r-project.org/
38. Teachey DT, O'Connor D. How I treat newly diagnosed T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma in children. Blood. (2020) 135:159–66. doi: 10.1182/blood.2019001557
39. Möricke A, Reiter A, Zimmermann M, Gadner H, Stanulla M, Dördelmann M, et al. Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood. (2008) 111:4477–89. doi: 10.1182/blood-2007-09-112920
40. Aricò M, Valsecchi MG, Conter V, Rizzari C, Pession A, Messina C, et al. Improved outcome in high-risk childhood acute lymphoblastic leukemia defined by prednisone-poor response treated with double berlin-frankfurt-muenster protocol ii. Blood. (2002) 100:420–6. doi: 10.1182/blood.V100.2.420
41. Barba P, Morgades M, Montesinos P, Gil C, Fox M, Ciudad J, et al. Increased survival due to lower toxicity for high-risk T-cell acute lymphoblastic leukemia patients in two consecutive pediatric-inspired PETHEMA trials. Eur J Haematol. (2019) 102:79–86. doi: 10.1111/ejh.13178
42. Liu RB, Guo JG, Liu TZ, Guo CC, Fan XX, Zhang X, et al. Meta-analysis of the clinical characteristics and prognostic relevance of NOTCH1 and FBXW7 mutation in T-cell acute lymphoblastic leukemia. Oncotarget. (2017) 8:66360–70. doi: 10.18632/oncotarget.18576
43. Noronha EP, Marques LVC, Andrade FG, Thuler LCS, Terra-Granado E, Pombo-de-Oliveira MS. The profile of immunophenotype and genotype aberrations in subsets of pediatric T-cell acute lymphoblastic leukemia. Front Oncol. (2019) 9:316. doi: 10.3389/fonc.2019.00316
44. Erbilgin Y, Sayitoglu M, Hatirnaz O, Dogru O, Akcay A, Tuysuz G, et al. Prognostic significance of NOTCH1 and FBXW7 mutations in pediatric T-ALL. Dis Markers. (2010) 28:353–60. doi: 10.1155/2010/740140
45. Park MJ, Taki T, Oda M, Watanabe T, Yumura-Yagi K, Kobayashi R, et al. FBXW7 and NOTCH1 mutations in childhood T cell acute lymphoblastic leukaemia and T cell non-Hodgkin lymphoma. Br J Haematol. (2009) 145:198–206. doi: 10.1111/j.1365-2141.2009.07607.x
46. Bonn BR, Rohde M, Zimmermann M, Krieger D, Oschlies I, Niggli F, et al. Incidence and prognostic relevance of genetic variations in T-cell lymphoblastic lymphoma in childhood and adolescence. Blood. (2013) 121:3153–60. doi: 10.1182/blood-2012-12-474148
47. Neumann M, Vosberg S, Schlee C, Heesch S, Schwartz S, Gökbuget N, et al. Mutational spectrum of adult T-ALL. Oncotarget. (2015) 6:2754–66. doi: 10.18632/oncotarget.2218
Keywords: measurable (minimal) residual disease, T-cell acute lymphoblastic leukemia, oncogenetics, NOTCH1, flow cytometry, pediatrics, risk-factors
Citation: Vega-García N, Perez-Jaume S, Esperanza-Cebollada E, Vicente-Garcés C, Torrebadell M, Jiménez-Velasco A, Ortega M, Llop M, Abad L, Vagace JM, Minguela A, Pratcorona M, Sánchez-Garcia J, García-Calderón CB, Gómez-Casares MT, Martín-Clavero E, Escudero A, Riñón Martinez-Gallo M, Muñoz L, Velasco MR, García-Morin M, Català A, Pascual A, Velasco P, Fernández JMa, Lassaletta A, Fuster JL, Badell I, Molinos-Quintana Á, Molinés A, Guerra-García P, Pérez-Martínez A, García-Abós M, Robles Ortiz R, Pisa S, Adán R, Díaz de Heredia C, Dapena JL, Rives S, Ramírez-Orellana M and Camós M (2021) Measurable Residual Disease Assessed by Flow-Cytometry Is a Stable Prognostic Factor for Pediatric T-Cell Acute Lymphoblastic Leukemia in Consecutive SEHOP Protocols Whereas the Impact of Oncogenetics Depends on Treatment. Front. Pediatr. 8:614521. doi: 10.3389/fped.2020.614521
Received: 06 October 2020; Accepted: 31 December 2020;
Published: 05 February 2021.
Edited by:
Yong-mi Kim, Children's Hospital of Los Angeles, United StatesReviewed by:
Elizabeth Macintyre, Université de Paris, FranceCopyright © 2021 Vega-García, Perez-Jaume, Esperanza-Cebollada, Vicente-Garcés, Torrebadell, Jiménez-Velasco, Ortega, Llop, Abad, Vagace, Minguela, Pratcorona, Sánchez-Garcia, García-Calderón, Gómez-Casares, Martín-Clavero, Escudero, Riñón Martinez-Gallo, Muñoz, Velasco, García-Morin, Català, Pascual, Velasco, Fernández, Lassaletta, Fuster, Badell, Molinos-Quintana, Molinés, Guerra-García, Pérez-Martínez, García-Abós, Robles Ortiz, Pisa, Adán, Díaz de Heredia, Dapena, Rives, Ramírez-Orellana and Camós. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mireia Camós, bWNhbW9zQHNqZGhvc3BpdGFsYmFyY2Vsb25hLm9yZw==; Nerea Vega-García, bnZlZ2FAc2pkaG9zcGl0YWxiYXJjZWxvbmEub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.