 Laura Bosch i Ara
Laura Bosch i Ara Harshini Katugampola1
Harshini Katugampola1- 1Department of Paediatric Endocrinology, Great Ormond Street Hospital for Children, London, United Kingdom
- 2Genetics and Genomic Medicine Programme, UCL Great Ormond Street Institute of Child Health, London, United Kingdom
Introduction: Congenital hypopituitarism (CH) is characterized by a deficiency of one or more pituitary hormones. The pituitary gland is a central regulator of growth, metabolism, and reproduction. The anterior pituitary produces and secretes growth hormone (GH), adrenocorticotropic hormone, thyroid-stimulating hormone, follicle-stimulating hormone, luteinizing hormone, and prolactin. The posterior pituitary hormone secretes antidiuretic hormone and oxytocin.
Epidemiology: The incidence is 1 in 4,000–1 in 10,000. The majority of CH cases are sporadic; however, a small number of familial cases have been identified. In the latter, a molecular basis has frequently been identified. Between 80–90% of CH cases remain unsolved in terms of molecular genetics.
Pathogenesis: Several transcription factors and signaling molecules are involved in the development of the pituitary gland. Mutations in any of these genes may result in CH including HESX1, PROP1, POU1F1, LHX3, LHX4, SOX2, SOX3, OTX2, PAX6, FGFR1, GLI2, and FGF8. Over the last 5 years, several novel genes have been identified in association with CH, but it is likely that many genes remain to be identified, as the majority of patients with CH do not have an identified mutation.
Clinical manifestations: Genotype-phenotype correlations are difficult to establish. There is a high phenotypic variability associated with different genetic mutations. The clinical spectrum includes severe midline developmental disorders, hypopituitarism (in isolation or combined with other congenital abnormalities), and isolated hormone deficiencies.
Diagnosis and treatment: Key investigations include MRI and baseline and dynamic pituitary function tests. However, dynamic tests of GH secretion cannot be performed in the neonatal period, and a diagnosis of GH deficiency may be based on auxology, MRI findings, and low growth factor concentrations. Once a hormone deficit is confirmed, hormone replacement should be started. If onset is acute with hypoglycaemia, cortisol deficiency should be excluded, and if identified this should be rapidly treated, as should TSH deficiency. This review aims to give an overview of CH including management of this complex condition.
Introduction
Congenital hypopituitarism (CH) is defined as the deficiency of one or more hormones produced by the anterior pituitary (AP) or released from the posterior pituitary (PP). Its estimated incidence is between 1 in 4,000 and 1 in 10,000 live births (1).
The pituitary gland is the central regulator of growth, metabolism, reproduction and homeostasis. It is located in the midline of the brain within the sella turcica and consists of three lobes of dual embryologic origin. The adenohypophysis (anterior and intermediate lobes) originates from Rathke's pouch, an invagination of the oral ectoderm, whereas the neurohypophysis (posterior lobe) develops from the neural ectoderm of the ventral diencephalon.
The AP consists of five different cell lineages producing six hormones: somatotrophs (growth hormone, GH), gonadotrophs (follicle stimulating hormone, FSH, and luteinising hormone, LH), corticotrophs (adrenocorticotropic hormone, ACTH), thyrotrophs (thyroid stimulating hormone, TSH), and lactotrophs (prolactin, PRL). The intermediate lobe contains melanotrophs, which secrete proopiomelanocortin (POMC), a major precursor to endorphins, and melanocyte-stimulating hormone (MSH). The PP lobe releases two hormones, oxytocin and antidiuretic hormone (ADH, also known as vasopressin), which are produced in the hypothalamus (supraoptic and paraventricular nuclei) and transported axonally via the pituitary stalk to be stored and released from the PP.
The hypothalamic parvocellular neurosecretory system is responsible for the release of specific AP hormones. It consists of neurons secreting thyrotrophin-releasing hormone (TRH) stimulating secretion of TSH and PRL, corticotrophin-releasing hormone (CRH) that acts to stimulate the secretion of ACTH, gonadotrophin releasing hormone (GnRH) that stimulates release of FSH and LH, growth hormone releasing hormone (GHRH) that stimulates the secretion of GH, somatostatin (SS) that negatively regulates GH secretion, and dopamine that inhibits secretion of PRL. These hypothalamic factors are rapidly transported to the AP via the hypophyseal portal blood system (2, 3).
The aim of this review is to describe the range of mechanisms underlying CH, clinical findings during the neonatal period, diagnosis, treatment, and future therapeutic options.
Etiology
CH may occur due to developmental defects of the pituitary gland, in some cases as a result of genetic mutations. Acquired forms of hypopituitarism, secondary to perinatal or neonatal events, rarely occur in the neonatal period. CH may present as isolated or combined pituitary hormone deficiencies (CPHD), and may be part of a syndrome involving extra-pituitary abnormalities.
In the majority of cases, the etiology of CH is unknown. The overall incidence of genetic mutations in these patients is low (16% of cases can currently be explained by mutations in known genes) indicating that many genes remain to be identified (4). PROP1 mutations are the most frequent known cause of both familial and sporadic congenital CPHD. Mutations in other genes have also been described, but appear to be much rarer. However, it is likely during the next few years that novel genetic determinants of pituitary disorders will probably be identified with the availability of next generation sequencing technology (5).
Embryology and Genetics
The development of the pituitary gland is a multifactorial process that results from the temporo-spatial interactions of transcription factors and signaling molecules. These occur in distinct and sequential developmental steps. Although direct evidence in humans is lacking, the process of pituitary development is highly conserved across all vertebrate species including rodents, and development of the mouse pituitary, in particular, is well-characterized (6).
Pituitary gland development starts at an equivalent human gestational age of 4–6 weeks and occurs in 4 stages: (i) the pituitary placode, (ii) the rudimentary Rathke's pouch, (iii) the definitive Rathke's pouch, and (iv) the mature pituitary gland.
In the mouse the pituitary placode appears at embryonic (E) day 7.5, located ventrally to the anterior neural ridge and next to the future hypothalamo-infundibular region, which will give rise to the roof of the oral cavity. Initial pituitary development consists of a thickening of the roof of the oral ectoderm at E8.5. By E9.0 it invaginates dorsally and the rudimentary Rathke's pouch is formed (7). The definitive Rathke's pouch, formed by E10.5, gives rise to the anterior and intermediate lobes. The posterior lobe is derived from the posterior part of the developing diencephalon. By E12.5, the precursors of the hormone-secreting cells start to proliferate ventrally from the pouch to constitute the future AP.
Normal pituitary organogenesis requires apposition between the rudimentary Rathke's pouch and the diencephalon. This is critical as the induction and correct formation of the pouch requires at least two sequential inductive signals from the diencephalon (8, 9). Bone Morphogenetic Protein 4 (Bmp4) is the first secreted signaling molecule. Fibroblast Growth Factor 8 (Fgf8) that is the second signal activates two key regulatory genes, LIM homeobox 3 (Lhx3) and LIM homeobox 4 (Lhx4) that play a critical role in the development of the rudimentary pouch into a definitive pouch (10–12).
These signaling molecules are derived from different embryogenic origins: the ventral diencephalon (Bmp4, Fgf8, Fgf4, Nkx2.1, Wnt5α), the oral ectoderm (Sonic Hedgehog, Shh, the surrounding mesenchyme (Bmp2, Chordin) and the pouch (Bmp2, Wnt4) (13, 14).
Transcription factors expressed early in pituitary organogenesis include Hesx1, Lhx3, Lhx4, Sox2, Sox3, Gli2, and Otx2. Prop1 and Pou1f1 (previously known as Pit1) are implicated in the later stages.
Further cell determination and specification rely on the expression and interaction of multiple signaling molecules and transcription factors (15) and these will be expanded upon in the following section.
Correlation Between Phenotype-Genotype
The variety of phenotypes seen in patients with CPHD is a reflection of the close developmental relationships seen during organogenesis of the pituitary gland, eye, optic nerve, ear, nose, and cranial nerve ganglia.
As a general rule, genetic mutations in genes involved in early development (HESX1, LHX3, LHX4, SOX2, SOX3, GLI2, and OTX2) tend to be part of a syndrome that includes extra-pituitary defects and midline abnormalities such as cleft lip and/or palate, as well as CH (Table 1). In contrast, mutations in the genes implicated in later stages (PROP1 and POU1F1) result in variable phenotypes of CPHD without any extra-pituitary defects (16) (Table 2).
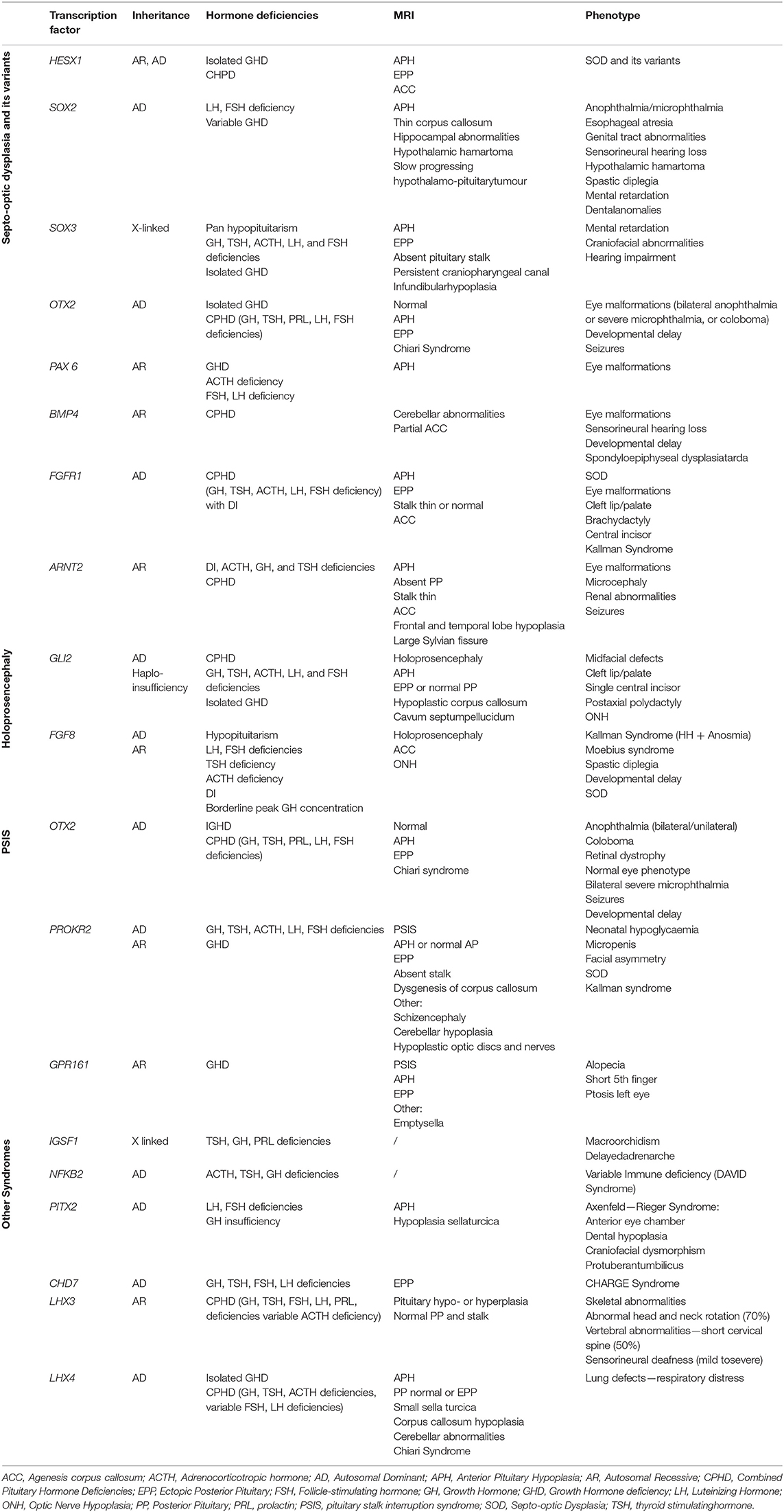
Table 1. Mutations and characteristics of genes involved in syndromic hypopituitarism.
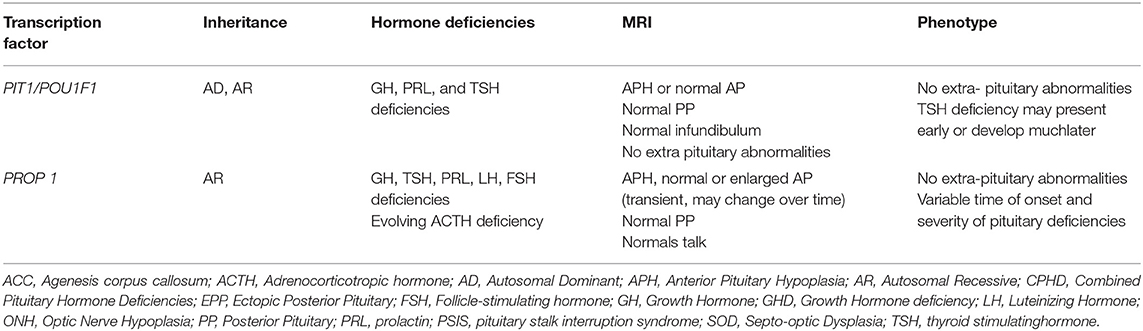
Table 2. Mutations and characteristics of genes involved in non-syndromic Hypopituitarism.
Different gene mutations can result in the same phenotype and different phenotypes can be secondary to the same single genetic mutation. Therefore, the clinical phenotype and associated morphological findings are crucial in the investigation of the underlying genetic mutations of cases of congenital hypopituitarism.
Syndromic Hypopituitarism
Syndromic forms of CH are mainly due to mutations in transcription factors implicated during early pituitary development as listed in Table 1 and described in detail below.
Septo-Optic Dysplasia and Its Variants
Septo-optic dysplasia (SOD; de Morsier Syndrome) is an extremely heterogeneous and complex disorder defined by the presence of at least 2 of the following: (i) optic nerve hypoplasia (ONH), (ii) midline abnormalities seen in brain and pituitary MRI [mainly agenesis of the corpus callosum (ACC) and absence of the septum pellucidum], (iii) pituitary hypoplasia with hypopituitarism. Its estimated prevalence is 1 in 10,000 live births (17–19).
To date, transcription factors, such as HESX1, SOX2, SOX3, and OTX2, are the most common genes implicated in the etiology of SOD. Genetic mutations implicated in Kallmann syndrome (KS), such as KAL1, FGFR1, PROKR2, and FGF8, have also been recently identified in patients with SOD (16, 20, 21).
HESX1
The transcription factor HESX1 is a member of the paired-like class of homeodomain proteins, the initial activation of which may be dependent upon LHX1 and OTX2. Hesx1 is one of the earliest markers of the pituitary primordium and can be detected in the anterior forebrain from E7.5 to E8.5 and in the Rathke's pouch from E8.5 to E135 (22). From E12, it is rapidly downregulated and becomes undetectable by E13.5 (23).
Hesx1 is a transcriptional repressor and its down-regulation activates other downstream genes such as Prop1, suggesting that both function as opposing transcription factors.
Targeted disruption of Hesx1 in mice results in anophthalmia or microphthalmia and midline neurological defects (such as absent septum pellucidum and pituitary hypoplasia), reminiscent of SOD (24).
The first homozygous missense mutation reported in HESX1 (p.R160C) was described in two siblings with SOD born to consanguineous parents who presented with ACC, ONH, a hypoplastic AP gland and complete panhypopituitarism (25–27).
Since then, several homozygous and heterozygous HESX1 mutations have been described. There is no clear genotype-phenotype correlation, and clinical features range from idiopathic GHD to CPHD, associated in some cases with anomalies such as SOD and pituitary malformations (28–33). MRI findings are variable, including a hypoplastic or aplastic AP and an ectopic posterior pituitary (EPP).
The phenotype of those patients with heterozygous mutations in HESX1 tends to be milder presenting with isolated GHD and an ectopic or undescended posterior PP, although midline forebrain abnormalities can also be seen.
The majority of the cases are sporadic and just around 1% of the patients with SOD present with genetic mutations in HESX1 (34, 35). In some patients, the penetrance is variable, suggesting the impact of additional genetic, or environmental factors.
SOX2
SOX2 is a transcription factor member of the SRY-related HMG box (SOX) family. It is expressed at 4.5–9 weeks of human pituitary development within Rathke's pouch and is maintained throughout AP development as well as in the diencephalon.
It is expressed throughout the developing central nervous system as well as in sensory placodes, inner ear, cochlea and in the developing lens, retina, and optic nerve (36–44).
SOX2 is extremely important in the maintenance of pituitary progenitor cells and its differentiation into all hormone-producing lineages (45).
The pituitary phenotype associated with murine Sox2 loss of function mutations usually includes GH, TSH, and gonadotrophin deficiencies (46).
Heterozygous de novo mutations in humans have been observed in several patients with hypogonadotropic hypogonadism, bilateral, often severe, anophthalmia/microphthalmia, small corpus callosum, hippocampal abnormalities, and variable mental retardation (47–51). Esophageal atresia has also been reported (52, 53). The pituitary phenotype occasionally includes GH deficiency (GHD).
Imaging of the hypothalamo-pituitary region can show morphological anomalies such as hippocampal abnormalities, hypoplasia of the corpus callosum, hypothalamic hamartoma, and pituitary enlargement that is reminiscent of tumors (54).
SOX3
SOX3 is also a member of the SRY-related HMG box (SOX) family. It is located on the X chromosome (Xp27.1) and is expressed along the full length of the central nervous system including the brain and spinal cord. SOX3 dosage is critical for normal hypothalamic-pituitary development and both under- and over- dosage of the gene can lead to hypopituitarism (4, 55, 56).
Male patients present with variable hypopituitarism (CPHD or idiopathic GHD) and infundibular hypoplasia, an ectopic/undescended posterior pituitary (PP) and abnormalities of the corpus callosum. Intellectual disability is also frequently reported in these patients (4, 57, 58). Patients with duplication of SOX3 can present with GHD without other pituitary deficiencies (59). Loss of function polyalanine expansions and gene deletions are associated with hypopituitarism including GH, TSH, ACTH, and gonadotrophin deficiencies. In terms of neuroradiological features, AP hypoplasia, an absent pituitary stalk, and ectopic EPP are other findings associated with SOX3 sequence variants or whole gene deletions/duplications. Persistence of the craniopharyngeal canal has been reported in association with a SOX3 deletion (60).
OTX2
Orthodenticle homeobox 2 (Otx2) is a transcription factor gene involved in brain, eye, nose and ear development (61, 62). It is expressed from E10.5 to E14.5 in the ventral diencephalon, from E10.5 to E12.5 in Rathke's pouch, and then becomes undetectable at both sites from E16.5 (63).
OTX2 regulates various transcription factors implicated in brain, eye and pituitary development, including RX1, PAX6, SIX3, LHX2, MITF, GBX2, and HESX1 in order to coordinate cell determination and differentiation.
As OTX2 is essential in retinal development, many patients with OTX2 mutations and pituitary hormone deficiencies also present with a variety of ocular abnormalities. In humans, heterozygous OTX2 mutations or gene deletions have been implicated in the etiology of 2–3% of anophthalmia/microphthalmia syndromes (64–67).
There is no clear genotype-phenotype correlation, even among patients with the same mutation. The pituitary phenotype ranges from partial to complete GHD and brain MRI can show a normal or hypoplastic AP, normal or EPP, and Chiari malformation (68, 69). In those patients without ocular involvement, who have CPHD and a small AP with or without an undescended PP, mutations in OTX2 have been rarely reported (70). One of these rare cases is the p.N233S mutation where patients may not exhibit an ocular phenotype (71).
PAX6
PAX6 is an early dorsal marker of early AP gland and its expression is required for somatotrope, lactotrope, and thyrotrope development. It is also an important regulator of eye development, and heterozygous mutations in humans cause congenital eye anomalies (72). Recently, PAX6 mutations have been reported to be associated with impaired pituitary function (ACTH deficiency, hypogonadotropic hypogonadism, and GHD) (73–75).
BMP4
Bone morphogenetic protein 4 (Bmp4) is the first secreted molecule detected in the prospective infundibulum at E8.5. It is essential for Rathke's pouch formation and maintenance. It is expressed in the optic vehicle, in the diencephalic floor and in the medial ganglionic eminence and in developing limbs. A recent study that included patients with eye abnormalities identified BMP4 mutations in a familial case of anophthalmia, retinal dystrophy, brain malformation, and poly/syndactyly (76). Deletions in BMP4 were associated with bilateral anophthalmia/microphthalmia, in association with hypothyroidism, deafness, developmental delay, and cerebellar and pituitary abnormalities.
FGFR1
FGF receptor 1 (FGFR1), a tyrosine kinase receptor for FGF, is the most important receptor involved in FGF8 signaling. Mutations in FGFR1 have previously been reported in patients with Kallmann syndrome; more recently, FGFR1 variants have been associated with CPHD, absent corpus callosum, SOD, and midline defects (77, 78).
ARNT2
Aryl hydrocarbon receptor nuclear translocator 2 (ARNT2) belongs to the HLH-PAS (Per-ARNT-Sim homology) subfamily of transcription factors. Arnt2 is find on the hypothalamus, eye (neural retina), and kidney and urinary tract in rodents, and this expression pattern recapitulates that observed in humans (76).
Holoprosencephaly: Gli2 and FGF8
GLI2, a mediator of SHH signaling, is expressed in the ventral diencephalon inducing BMP4 and FGF8 expression, and also in the oral ectoderm, inducing pituitary progenitors. GLI2 mutations are associated with holoprosencephaly (HPE) or HPE-like features with craniofacial anomalies, pituitary abnormalities and polydactyly (79–81).
Fibroblast growth factor 8 (Fgf8) is a member of the FGF family of signaling molecules that are involved in pituitary organogenesis. It is expressed in the infundibulum at E9.5, 1 day after the expression of Bmp4 (20, 82) and is important in midbrain development.
FGF8 mutations are associated with Kallmann syndrome and have more recently been described in association with recessive HPE, craniofacial defects, and hypothalamo-pituitary dysfunction (83).
Hypopituitarism With Spine Abnormalities: LHX3
Expression of the LIM homeobox 3 (LhX3) gene, a member of the LIM class of homeodomain proteins, is detected early during AP development at E9.5 (Ratkhe's pouch, ventral hindbrain, and spinal cord) and persists in the mature pituitary gland. It is one of the earliest markers implicated in the anterior and intermediate lobes development and its expression plays an important role for the formation of gonadotrophs, thyrotrophs, somatotrophs, and lactotrophs (84).
Mice with homozygous mutations of Lhx3 die soon after birth as a result of pituitary aplasia whereas those with heterozygous mutations have no abnormalities (85).
In humans, 14 homozygous (86–94) or compound heterozygous LHX3 mutations (10) and a heterozygous variant (95) have been reported to date.
Commonly, patients with LHX3 mutations present with GH, TSH, and FSH/LH deficiencies while ACTH deficiency is reported in 50% of cases (94).
The phenotype varies depending on which part of the gene is affected. If the mutation affects the entire gene or protein, the LIM domains or the homeodomain, patients will present with syndromes involving the nervous and skeletal systems, whereas if the mutation affects the carboxyl terminus of LHX3 protein alone, only pituitary dysfunction will be present. LHX3 is also required for inner ear development.
Extra-pituitary phenotypes can include a short neck with abnormal head and neck rotation (70% of cases), vertebral abnormalities (50% of cases) including a rigid cervical spine, flattened lumbar vertebrae, thoracic kyphosis, and progressive scoliosis, and variable degrees of sensorineural hearing loss (50% of cases). Developmental delay or learning difficulties have also been reported in nearly 40% of the patients. Two of the reported patients also had respiratory distress. Heterozygous family members are largely unaffected, although a recent publication has described a mild limitation of neck movement in a heterozygous carrier (10).
MRI findings can vary from a normal MRI (10% of the cases) to aplasia or hypoplasia of the AP, a hypointensity suggestive of microadenoma, and enlargement with a hyperintense signal (91).
Hypopituitarism With Cerebellar Abnormalities: LHX4
Lhx4 is closely related to Lhx3 and is expressed in the developing brain and spinal cord (96). It is also detected during early stages (E9.5 in Rathke's pouch and E12.5 in the anterior part of the pituitary) and is subsequently found in the future anterior lobe, with a decrease in expression by E15.5.
The AP gland in patients with LHX4 mutations is hypoplastic, containing all the differentiated cell types but in reduced numbers. Other brain abnormalities can also be present such as an EPP and a hypoplastic sella turcica as well as corpus callosum hypoplasia or Chiari syndrome (97–105).
In humans, the phenotype can range from isolated GHD to complete panhypopituitarism (106). Several sporadic or familial LHX4 mutations have been reported to date. Of note, four patients also presented with respiratory distress (76, 103, 107) and one presented with a cardiac defect (76). Several of the variants are variably penetrant, although the underlying mechanism remains to be established.
A lethal neonatal phenotype (severe panhypopituitarism associated with anterior pituitary aplasia and EPP, mild facial hypoplasia, undescended testes, and severe respiratory distress) has been recently described, secondary to a homozygous mutation (107).
Pituitary Stalk Interruption Syndrome (PSIS)
PSIS is a congenital defect of the pituitary gland characterized by the triad of (i) a thin pituitary stalk, (ii) an EPP gland, and (iii) hypoplasia or aplasia of the AP gland identified by MRI. Patients with PSIS may present with either isolated GHD or CPDH (22). Genetic alterations in HESX1, LHX4, OTX2, SOX3, and PROKR2 have been reported in patients with PSIS, amongst others.
PROKR2, a G protein-coupled receptor essential for proper neuronal migration and angiogenesis, is involved in sex development and olfactory bulb development in mice. In humans, patients with mutations in PROKR2 can present with hypogonadotropic hypogonadism or Kallmann syndrome. More recently, mutations have been associated with variably penetrant hypopituitarism including SOD (108–112). As described previously (section Septo-optic dysplasia and its variants), OTX2 gene defects were also reported in patients with no ocular abnormalities (71).
GPR161, an orphan member of the G protein–coupled receptor family, has also been recently identified in patients with PSIS. GPR161 is widely expressed in both mouse and human during the early stages of embryogenesis including the neural folds, the pituitary and the hypothalamus.
It is a key negative regulator of the SHH pathway, the pituitary target of which is GLI2. It has been suggested that gain-of-function mutations of GPR161 could lead to abnormal pituitary development by repressing the SHH pathway (22).
A homozygous missense mutation p.L19Q in GPR161 has been recently described in two female siblings with short stature due to GHD associated with AP hypoplasia and an empty sella with an EPP. They also had a short 5th finger, congenital alopecia, and ptosis of the left eye (113).
Central Hypothyroidism and Macroorchidism
IGSF1 is located at Xq26 and is expressed in Rathke's pouch, in the pituitary gland (present in GH, prolactin and TSH-secreting cells), and testis.
IGSF1 mutations (loss of function or deletions) cause an X-linked syndrome. Male patients with mutations in IGSF1 present with a characteristic phenotype that consists of congenital central hypothyroidism, delayed puberty, and adult macroorchidism. PRL and/or GH deficiencies have also been reported in some cases (114, 115). Some female patients with heterozygous mutations in IGFS1 present with central congenital hypothyrodism, PRL deficiency, and delayed puberty.
Deficient Anterior Pituitary Function With Variable Immune Deficiency (DAVID) Syndrome
NFKB2 belongs to the NF-κB family, which consists of a collection of evolutionarily conserved transcription factors involved primarily in development including the anterior pituitary gland, immunity, and oncogenesis.
Patients with NFKB2 mutation present with deficit in AP gland function and common variable immune deficiency, a novel disorder called DAVID syndrome (116). However, the precise mechanism underlying endocrine deficits remains largely unclear.
Axenfeld—Rieger Syndrome
Pitx2, a homeobox gene, is detected in the stomodeum at E8, Rathke's pouch at E10.5 and 2 days after in the anterior and intermediate lobes. Patients affected with PITX2 mutations present with Axenfeld–Rieger Syndrome which is characterized by eye, craniofacial, dental, cardiac, and umbilical anomalies (117).
CHARGE Syndrome and Pituitary Deficiencies
CHARGE syndrome is rare autosomal dominant disorder that affects multiple organs. It is characterized by ocular coloboma, cardiac defects, choanal atresia, growth, and developmental delay and ear abnormalities. EPP and hypopituitarism in patients with CHARGE syndrome has been recently reported in association with two novel CHD7 variants (118).
Isolated ACTH Deficiency
Congenital isolated ACTH deficiency is mainly due to recessive mutations in TBX19 (formerly TPIT) which are responsible of approximately 65% of the cases of isolated ACTH deficiency diagnosed during the first month of life (119). Neonates present with severe hypoglycaemia that can result in seizures and prolonged cholestatic jaundice. Biochemically this is characterized by low basal ACTH and cortisol concentrations and a poor ACTH response to corticotropin releasing hormone (CRH). It is extremely important to diagnose as this can be a potential cause of death during the neonatal period if no replacement treatment is started (120).
Non—syndromic Combined Pituitary Hormone Deficiencies
Mutations in PROP1 and POU1F1 constitute the main genetic cause found in patients with non-syndromic GHD or CPHD (Table 2).
Mutations in PROP1
Prop1 (prophet of PIT-1), a member of the paired-like family of homeodomain transcription factors, is the earliest expressed pituitary-specific transcription factor. It is detected within Rathke's pouch by E10, peaks at E12 and it disappears by E15.5 (121).
Prop1 can act as both a transcriptional repressor (for Hesx1 expression) or a transcriptional activator for Pou1f1 (121, 122). Mice with Prop1 over expression present with delayed puberty as a consequence of a delay in the differentiation of gonadotrophs (123).
The most frequent genetic cause of CPHD are recessive mutations in PROP1 (124–130). The most common of these is a 2 base pair deletion within exon 2, which results in a frameshift at codon 101 and introduction of a termination codon at position 109 (130).
Recessive PROP1 mutations are associated with GH, TSH, PRL and gonadotrophin deficiencies which vary in onset and severity. ACTH deficiency usually occurs later. They vary in the time of onset and severity and therefore it is important to have ongoing clinical surveillance.
GHD and growth delay is usually present during the first years of life in patients with PROP1 mutations, however, there is a case report of a patient who had normal growth and achieved a normal adult height without GH treatment (131).
Both TSH and gonadotropin deficiency can appear at birth or later in life (132–134). Some patients may present with micropenis and undescended testes at birth and some others with delayed puberty. Spontaneous puberty can also be seen (135–137).
The majority of the patients do not have ACTH or cortisol deficiency during the first years of life but this can evolve later and ongoing surveillance is therefore needed (135, 138).
MRI shows variable pituitary morphology. The commonest finding is a normal pituitary stalk and posterior lobe with a small or normal AP gland. An enlarged AP gland has also been described with posterior regression (137).
Mutations in POU1F1
POU1F1, a member of the POU family, is expressed later during the pituitary organogenesis (E14.5) and persists during adulthood (139). It plays a crucial role in the regulation of the genes encoding GH, PRL, and TSH-beta and the time of onset and severity also varies. Gonadotroph and corticotroph axes usually remain functional.
Patients tend to present first with GH and prolactin deficiencies during the first years of life whereas TSH deficiency tends to present later (140).
POU1F1 mutations are mainly recessive. However, dominant mutations have been recently described being the most frequent p.R271W mutation (141).
MRI reveals a normal or small AP gland; The PP and infundibulum are normal and no midline abnormalities have been reported (140, 142).
Clinical Presentation, Diagnosis, and Treatment
Given the crucial regulatory role of the pituitary gland, prompt recognition of those neonates at risk of CH is important, as a delay in replacement therapy can have devastating consequences. Identifying neonates with CH can be challenging because they often present with non-specific symptoms such as hypoglycaemia, prolonged jaundiced, poor weight gain, temperature dysregulation, electrolyte abnormalities, and haemodynamic instability (Table 3).

Table 3. Clinical presentation of hypopituitarism in a neonate.
Birth weight and length tend to be normal, although GHD can lead to a slight reduction in birth weight. The clinical presentation and its severity depend on the number of hormones affected. These patients can have associated genital abnormalities, eye malformations, and/or midline defects.
Neonates with ACTH deficiency can present with cholestasis during the first 2 weeks of life. To understand the association between cholestasis and ACTH deficiency it is important to remind that cortisol increases bile flow and therefore, its deficiency will cause abnormalities in the synthesis and transport of bile acid leading to cholestasis in some cases.
A rise in transaminase concentrations can be seen after 2–4 weeks but GGT remains normal.
Once cortisol replacement treatment is started, cholestasis tends to resolve in around 10 weeks' time. In those cases where a liver biopsy is performed due to a delay in the diagnosis of CH, this shows canalicular cholestasis, and histopathology reveals mild portal eosinophilic infiltration.
Investigations to diagnose CH include baseline pituitary function tests (+ dynamic tests if indicated) and brain MRI. Genetic testing should also be considered.
However, the sensitivity and specificity of laboratory tests are limited in the newborn, especially in premature infants due to hypothalamo-pituitary axis immaturity, and lack of normative values. Additionally, GH stimulation tests are contraindicated under the age of 1 year.
A high index of suspicion for CH and early treatment in these patients is vital to avoid clinical decompensation. Treatment involves the physiological replacement of the relevant hormone deficiencies and requires close lifelong monitoring.
Individual hormone deficiencies are discussed in detail in the following section.
ACTH Deficiency
Clinical Presentation
Neonates can present with failure to thrive, severe hypoglycaemia and cholestasis.
Diagnosis
As neonates do not have a circadian rhythm [reported to be established at around 2 months of age (143), or after 6 months of age (142), morning cortisol concentrations are not useful in evaluating ACTH deficiency in this population. Although cortisol deficiency related hypoglycaemia is severe, low cortisol concentrations at the time of hypoglycaemia have low specificity for the diagnosis of adrenal insufficiency and therefore should not be the only test for the diagnosis of ACTH deficiency (144). Many patients require a dynamic assessment (ACTH stimulation test using tetracosactide hexaacetate). The dose of tetracosactide hexaacetate used to diagnose central adrenal insufficiency, the timing for collection of blood samples for cortisol measurement, and the cut-off peak cortisol concentration for both the low-dose and standard ACTH test are the subject of much controversy. Stimulated cortisol concentrations ≥18 mg/dL (497 nmol/L) are indicative of a normal hypothalamo-pituitary-adrenal axis (145).
Treatment
In preterm infants, daily cortisol production is known to be ~7 mg/m2/day on the fifth day and ~6 mg/m2/day in the second week. When a neonate is diagnosed with ACTH deficiency, treatment needs to be started immediately. The treatment of choice is hydrocortisone due to its less potent side effects in terms of growth and bone health compared to other glucocorticoids. The starting dose of hydrocortisone is 9–12 mg/m2/day in 3–4 divided doses. This dose is higher compared to older infants because neonates have greater cortisol secretion rates. The dose can then be titrated with age. Prior to discharge, education for families about sick day rules and emergency dosing is important. In event of illness or stress, hydrocortisone doses should be doubled or even tripled. In event of an emergency, poor tolerance of oral hydrocortisone, or a suspected adrenal crisis, intramuscular hydrocortisone must be administered. The dose is age-dependent (<1 year 25 mg, 1–5 years 25–50 mg, >5 years 100 mg) and oral glucose should also be given to correct any associated hypoglycaemia. Those patients that cannot tolerate oral hydrocortisone require admission for intravenous hydrocortisone (1–2 mg/kg every 4–6 h). One they are able to tolerate oral hydrocortisone this is commenced at triple or double maintenance dose, and gradually weaned to maintenance depending on clinical improvement.
It is also important to highlight that cortisol deficiency can mask DI as cortisol is needed for water excretion. DI may develop after starting treatment with hydrocortisone and therefore close monitoring of fluid balance and electrolytes is important after starting glucocorticoid therapy (145).
Novel treatments such as continuous subcutaneous hydrocortisone infusion therapy, which may be difficult in neonates due to limited subcutaneous fat for insertion of the cannula, and sustained release hydrocortisone preparations aimed at mimicking physiological cortisol secretion remain to be established as potential therapies (146).
TSH Deficiency
Etiology
Defects in TRH or TSH signaling are responsible of isolated central congenital hypothyroidism. As mentioned before, the most frequent genetic cause of isolated central congenital hypothyroidism is IGSF1 gene mutation (147). Less common causes include genetic defects in TSH production, that is, mutations in the TRH receptor or TSH-B subunit (112, 148). More recently, mutations in TBL1X have been described in association with TSH deficiency.
Clinical Presentation
Newborns with TSH deficiency can present with prolonged physiological jaundice and low energy levels/sleepiness. Other findings such as temperature dysregulation, umbilical hernia, dry skin, bradycardia, macroglossia, and constipation may also be present.
X-linked central hypothyroidism due to IGSF1 mutation is also later associated with delayed puberty and adult macroorchidism (149).
Diagnosis
Thyroid hormone is critical for normal brain development within the first 3 years of life, and therefore a prompt diagnosis is essential so that treatment can be commenced rapidly. Central hypothyroidism is characterized by the biochemical picture of low free T4 and usually low TSH (although it can also be inappropriately normal or even slightly elevated).
Treatment
Levothyroxine (LT4) is the treatment of choice in newborns with TSH deficiency at a starting dose between 10 and 15 μg/kg/day (150). However, higher doses will be needed in newborns with cholestasis due to malabsorption. Iron, soy, calcium, and anticonvulsants can also affect LT4 absorption and thus should not be co-administered with them. LT4 should ideally be given on an empty stomach but this is not always practical in neonates and so may need to be given with a small amount of milk. LT4 solution or crushed tablets can be given with water, breastmilk or formula.
For those babies unable to tolerate enteral preparations, intravenous tri-iodothyronine (T3) is available. The recommended intravenous dose is 75% of the total oral LT4 dose (151).
Before starting treatment with LT4, it is extremely important to exclude cortisol deficiency. LT4 increases basal metabolic rate, enhancing cortisol clearance with the subsequent risk of precipitating an adrenal crisis.
Monitoring
T4 concentrations should be monitored every 2–4 weeks during initial period of dose tritration. Thereafter monitoring may reduce in frequency. The aim is to keep fT4 in the mid-upper half of the normal range (152). TSH is not useful for monitoring in these cases.
Gonadotrophin Deficiency
Clinical Presentation
Males present with micropenis, with or without undescended testes. Micropenis refers to a stretched penile length of −2.5 SD from the mean value: <1.5 cm at gestational age 30 weeks, 2 cm at 34 weeks, and <2.5 cm in term babies. Development of female genitalia is not affected by hypogonadotropic hypogonadism (HH) as it is independent of hormone secretion.
Diagnosis
Males
Mini puberty (raised LH and FSH) is seen between 15 days and 6 months old. Testosterone concentrations increase with a peak in the 4–10th week and start to decrease around the 6th month. LH concentrations <0.8 IU/L and testosterone <30 ng/mL between day 5 and 6 months of life are suggestive of the diagnosis. When an hCG test is done to assess testosterone production, penile growth and testicular descent may ensue and need to be documented. There are scant normative data pertaining to hCG tests in the first years of life. However, a study performed in adolescent males suggested that a peak LH concentration <2.8 IU/L after GnRH stimulation, with a testosterone peak of <3.6 nmol/L after 3 days of hCG injections and <9.5 nmol/L after 3 weeks of hCG injections are highly suggestive of hypogonadotrophic hypogonadism (153).
Females
Mini puberty is seen between 15 days and 2 years of age. FSH concentrations < 0.1 IU/L between 15 days and 2 years of life are diagnostic of probable hypogonadotrophic hypogonadism.
Treatment
In newborn male infants, the aim of the treatment is to ensure normal testicular descent, improve penile length and maximize fertility potential. Early treatment is recommended, ideally between 1 and 6 months of age. Testosterone can be given via intramuscular injections or topical gel (153–156). Testosterone injections (cypionate or enanthate) are commenced at a recommended dose of 25 mg every 4 weeks for 3 months. This is followed by clinical evaluation of the stretched penile length. Topical gel containing 5-α Dihydrotestosterone (DHT) is also useful and the recommended starting dose is 1 application (10 mg) every day for 3 months (153). The carer who is applying the testosterone gel should wash hands soon after the administration with soap and water and if the career is a female, the use of gloves is recommended. Cryptorchidism increases the risk of testicular neoplasia and also reduces fertility potential, therefore surgical correction (orchidopexy) is recommended during the first 2 years of life, ideally by 18 months of age (153, 157, 158). Treatment with LH and FSH during the neonatal period still remains under investigation (158–160).
GH Deficiency
Congenital isolated GH deficiency (GHD) has an incidence of 1 in 4,000–1 in 10,000 live births (33), and is the most commonly affected pituitary hormone in childhood.
Etiology
Most of the cases are sporadic but there are four genetic forms that account for 5–30% of cases (161, 162). Congenital isolated GHD can be secondary to genetic mutations in the genes encoding growth hormone (GH1) or the growth hormone releasing hormone receptor (GHRHR), or in the genes encoding transcription factors SOX3, HESX1, GLI2, OTX2, LHX3, LHX4, PROP1, and POU1F1 (4, 163). Mutations in GH1 and GHRHR may also lead to severe early growth failure with hypoglycaemia. Biallelic mutations in RNPC3 have also been recently described patients with severe IGHD and AP hypoplasia (23, 164).
Clinical Presentation
Key features of (GHD) include hypoglycaemia and micropenis. It is important to note that GHD does not significantly affect fetal growth, and therefore, affected newborns are usually of normal weight and length at birth, with subsequent post-natal growth failure.
Diagnosis
GH evaluation in a neonate differs from that in an older child. During the neonatal period GH concentrations are higher in the term neonate during the first week of life than throughout childhood but a rapid decrease is seen during the following weeks (165). In contrast, IGF-1 concentrations (stimulated by GH) cannot be used as a screening test in neonates as they remain low for at least the first 15–18 months of age (166). A random GH concentration of less than or equal to 5 ng/mL (5 mcg/L) during the first 7 days of life accompanied by other pituitary hormone deficiencies and/or the classical imaging triad (EPP with AP hypoplasia and an abnormal stalk) is sufficient to diagnose GHD (165). Binder et al. (167) suggested that a GH cut-off of 7 μg/L as measured on a neonatal screening card by a highly sensitive polyclonal ELISA gave 100% sensitivity and 98% specificity. GH stimulation tests are considered dangerous and are contraindicated during the neonatal period, and a low GH concentration at the time of hypoglycaemia in isolation is not enough to diagnose GHD.
Treatment
In the event of persisting hypoglycaemia, GH treatment can be commenced during the neonatal period with daily subcutaneous recombinant human GH (rhGH) injections in the evening to mimic physiological growth hormone release. The initial recommended dose is between 0.16 and 0.24 mg/kg per week (22–35 mcg/kg per day) (165). Lower doses (10–20 mcg/kg/day) can also lead to excellent responses at this age. GH treatment can contribute to hypoglycaemia recovery and may improve cholestasis during the neonatal period (168).
Monitoring
Subsequent dosing should be individualized by monitoring IGF-I concentrations (at least every 3 months at the beginning). Patients also should be monitored for hypothyroidism and adrenal insufficiency as GH treatment increases metabolism of thyroid hormone and cortisol and may unmask these conditions.
PRL Deficiency
Etiology
Prolactin deficiency is usually due to POU1F1, LHX3, OTX2, and IGSF-1 gene mutations. It is important to note that some medications can affect PRL concentrations such as dopamine, calcium channel blockers and ranitidine.
Clinical Presentation
Puerperal alactogenesis is the only specific physical finding.
Diagnosis
A random prolactin concentrations <31 ng/mL during the neonatal period supports a diagnosis of PRL deficiency, however, breast tissue should not be palpated prior to a blood sample being taken as the levels could be falsely elevated. Prolactin concentrations are often elevated in association with midline defects.
Treatment
There is no commercially available treatment for PRL deficiency.
Diabetes Insipidus (DI)
Etiology
In most cases of neonatal DI, anatomical defects or autosomal dominant or recessive genetic causes are present. DI is also observed in cases with SOD, corpus callosum agenesis and HPE. Renal concentrating mechanism can also be affected by other factors such as neonatal diabetes, hypercalcaemia, hypokalaemia. It is also important to note that mannitol, dextrose, saline fluids, and imaging contrast mediums can produce osmotic diuresis and secondary polyuria.
Clinical Presentation
The clinical features include polyhydramnios, polyuria, weight loss, irritability, dehydration, and hypernatremia.
Diagnosis
Diagnosis during the neonatal period is challenging as the capacity to concentrate urine is not as efficient as in older children and a water deprivation test is not recommended. Polyuria in DI during the neonatal period is defined as >5 mls/kg/h. Urine osmolality <300 mOsm/kg with a paired serum osmolality >300 mOsm/kg is suggestive of the diagnosis. If the urine osmolality is >600 mOsm/kg, DI is unlikely. The vasopressin test is useful to distinguish between central (CDI) and nephrogenic forms of DI but this can be hazardous during the neonatal period.
Treatment
Fluid therapy alone, without DDAVP, is the recommended management during the neonatal period as it can maintain euvolaemia. However, when CDI is extremely severe, a neonate may not respond to fluid therapy alone and DDAVP might be needed. In some cases of severe CDI, a thiazide diuretic may also be used. DDAVP can result in rapid fluid retention, hyponatremia and secondary cerebral oedema or even death in this vulnerable cohort of patients. Over-treatment is more dangerous than under-treatment and this is why a low starting dose of DDAVP (e.g., 1 μg) is recommended. Close monitoring (electrolytes, paired plasma and urine osmolalities, weight, and clinical examination for signs of fluid retention) is crucial, and dose adjustment will depend on the response to treatment (169). It is important to ensure breakthrough urine output prior to the next dose of DDAVP, in order to avoid severe fluid retention and hyponatraemia. It must also be highlighted that DDAVP may need to be withheld in neonates with concomitant ACTH deficiency who are unable to tolerate or absorb hydrocortisone when unwell (e.g., if vomiting), until appropriate steroid cover is provided. This is because cortisol is required for free water excretion and ongoing therapy with DDAVP without appropriate steroid replacement puts the neonate at risk of water intoxication.
Imaging: Brain and Pituitary MRI
MRI of the brain and pituitary gland is recommended in all patients with suspected or confirmed CH. Abnormal brain and pituitary MRI findings do correlate with the severity and evolution of the disease (170).
The pituitary gland in newborns tends to be convex showing high signal intensity on T1-weighted images. As discussed previously, patients with CH usually have abnormal MRI findings ranging from a small AP gland to severe hypoplastic pituitary gland with EPP or undescended PP and an interrupted or hypoplastic pituitary stalk. A “bright spot” identifies the PP gland, however it can be absent in 10% of healthy individuals (170).
Pituitary/brain MRI studies should include qualitative description and dimensions of the AP; location and size of the PP gland, description of the pituitary stalk and comments about extra pituitary structures such as the optic chiasm, septum pellucidum, and corpus callosum. In order to have a proper description, the best technique is 2-mm to 3-mm thick, high-resolution T1-weighted, and T2-weighted images in coronal and sagittal planes.
The majority of newborns with severe CH show an EPP, abnormal pituitary stalk, and/or AP hypoplasia on MRI. This triad is known as “Pituitary Stalk Interruption Syndrome” (PSIS). Patients with IGHD and PSIS need to be closely monitored for evolving endocrinopathies as they can progress to CPHD.
Other midline brain abnormalities (absent/hypoplastic corpus callosum, absent septum pellucidum, schizencephaly, heterotopia) and ONH may be associated (33, 171).
Conclusion
CH can be a life-threatening condition. A high index of suspicion is required for its early identification and treatment. However, early diagnosis during the neonatal period is challenging due to the variable and non-specific presenting symptoms. Red flag symptoms of CH include hypoglycaemia at birth and a micropenis.
In neonates with confirmed or suspected CH, a brain MRI with pituitary views is essential to exclude structural abnormalities. Ophthalmological review is also recommended to evaluate the optic nerves as many cases can have associated ocular abnormalities.
CH is an evolving and lifelong condition and therefore neonates with CH will require long term follow-up in order to detect early evolving endocrinopathies and optimize treatment. For those cases with a positive genetic finding, counseling is recommended (172). Currently, genetic analysis is successful in identifying an aetiological basis only in around 20% of cases (173). However, rapid advances in next-generation sequencing technology will help and improve our understanding of the complex mechanisms involved in congenital hypopituitarism (174). This technological progress is likely to have a positive impact on the clinical care of patients in the future (175).
Author Contributions
Edited by LB. Reviewed by HK and MD. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Davis SW, Castinetti F, Carvalho LR, Ellsworth BS, Potok MA, Lyons RH, et al. Molecular mechanisms of pituitary organogenesis: in search of novel regulatory genes. Mol Cell Endocrinol. (2010) 323:4–19. doi: 10.1016/j.mce.2009.12.012
2. Davis SW, Ellsworth BS, Perez Millan MI, Gergics P, Schade V, Foyouzi N, et al. Pituitary gland development and disease: from stem cell to hormone production. Curr Top Dev Biol. (2013) 106:1–47. doi: 10.1016/B978-0-12-416021-7.00001-8
3. Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT. Genetic regulation of pituitary gland development in human and mouse. Endocr Rev. (2009) 30:790–829. doi: 10.1210/er.2009-0008
4. Alatzoglou KS, Dattani MT. Genetic forms of hypopituitarism their manifestation in the neonatal period. Early Hum Dev. (2009) 11:705–12. doi: 10.1016/j.earlhumdev.2009.08.057
5. Sheng HZ, Moriyama K, Yamashita T, Li H, Potter SS, Mahon KA, et al. Multistep control of pituitary organogenesis. Science. (1997) 278:1809–12. doi: 10.1126/science.278.5344.1809
6. Rizzoti K, Lovell-Badge R. Early development of the pituitary gland: induction and shaping of Rathke's pouch. Rev Endocr Metab Disord. (2005) 6:161–72. doi: 10.1007/s11154-005-3047-7
7. Treier M, Rosenfeld MG. The hypothalamic–pituitary axis: co-development of two organs. Curr Opin Cell Biol. (1996) 8:833–43. doi: 10.1016/S0955-0674(96)80085-8
8. Takuma N, Sheng HZ, Furuta Y, Ward JM, Sharma K, Hogan BL, et al. Formation of Rathke's pouch requires dual induction from the diencephalon. Development. (1998) 125:4835–40.
9. Sheng HZ, Westphal H. Early steps in pituitary organogenesis. Trends Genet. (1999) 15:236–40. doi: 10.1016/S0168-9525(99)01742-4
10. Mullen RD, Colvin SC, Hunter CS, Savage JJ, Walvoord EC, Bhangoo AP, et al. Roles of the LHX3 and LHX4 LIM-homeodomain factors in pituitary development. Mol Cell Endocrinol. (2007). 265–6:190–5. doi: 10.1016/j.mce.2006.12.019
11. Zhu X, Wang J, Ju BG, Rosenfeld MG. Signaling and epigenetic regulation of pituitary development. Curr Opin Cell Biol. (2007) 19:605–11. doi: 10.1016/j.ceb.2007.09.011
12. Zhu X, Gleiberman AS, Rosenfeld MG. Molecular physiology of pituitary development: signaling and transcriptional networks. Physiol Rev. (2007) 87:933–63. doi: 10.1152/physrev.00006.2006
13. Kelberman D, Dattani MT. The role of transcription factors implicated in anterior pituitary development in the aetiology of congenital hypopituitarism. Ann Med. (2006) 38:560–77. doi: 10.1080/07853890600994963
14. Cohen LE, Radovick S. Molecular basis of combined pituitary hormone deficiencies. Endocr Rev. (2002) 23:431–42. doi: 10.1210/er.2001-0030
15. Cohen LE. Genetic disorders of the pituitary. Curr Opin Endocrinol Diabetes Obes. (2012) 19:33–9. doi: 10.1097/MED.0b013e32834ed639
16. Gregory LC, Dattani MT. The molecular basis of congenital hypopituitarism and related disorders. J Clin Endocrinol Metab. (2020) 105:184. doi: 10.1210/clinem/dgz184
17. McCabe MJ, Hu Y, Gregory LC, Gaston-Massuet C, Alatzoglou KS, Saldanha JW, et al. Novel application of luciferase assay for the in vitro functional assessment of KAL1 variants in three females with septo-optic dysplasia (SOD). Mol Cell Endocrinol. (2015) 417:63–72. doi: 10.1016/j.mce.2015.09.010
18. Morishima A, Aranoff GS. Syndrome of septo-optic-pituitary dysplasia: the clinical spectrum. Brain Dev. (1986) 8:233–9. doi: 10.1016/S0387-7604(86)80075-4
19. Webb EA, Dattani MT. Septo-optic dysplasia. Eur J Hum Genet. (2010) 18:393–7. doi: 10.1038/ejhg.2009.125
20. McCabe MJ, Gaston-Massuet C, Tziaferi V, Gregory LC, Alatzoglou KS, Signore M, et al. Novel FGF8 mutations associated with recessive holoprosencephaly, craniofacial defects, and hypothalamo-pituitary dysfunction. J Clin Endocrinol Metab. (2011) 96:E1709–18. doi: 10.1210/jc.2011-0454
21. Xatzipsalti M, Voutetakis A, Stamoyannou L, Chrousos GP, Kanaka-Gantenbein C. Congenital hypopituitarism: various genes, various phenotypes. Horm Metab Res. (2019) 51:81–90. doi: 10.1055/a-0822-3637
22. Dasen JS, Barbera JP, Herman TS, Connell SO, Olson L, Ju B, et al. Temporal regulation of a paired-like homeodomain repressor/TLE corepressor complex and a related activator is required for pituitary organogenesis. Genes Dev. (2001) 15:3193–207. doi: 10.1101/gad.932601
23. Alatzoglou KS, Gregory LC, Dattani MT. Development of the pituitary gland. Compre Physiol. (2020) 10:389–413. doi: 10.1002/cphy.c150043
24. Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Martensson IL, et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet. (1998) 19:125–33. doi: 10.1038/477
25. Andoniadou CL, Signore M, Sajedi E, Gaston-Massuet C, Kelberman D, Burns AJ, et al. Lack of the murine homeobox gene Hesx1 leads to a posterior transformation of the anterior forebrain. Development. (2007) 134:1499–508. doi: 10.1242/dev.02829
26. Sajedi E, Gaston-Massuet C, Signore M, Andoniadou CL, Kelberman D, Castro S, et al. Analysis of mouse models carrying the I26T and R160C substitutions in the transcriptional repressor HESX1 as models for septo-optic dysplasia and hypopituitarism. Dis Model Mech. (2008) 1:241–54. doi: 10.1242/dmm.000711
27. Thomas PQ, Dattani MT, Brickman JM, McNay D, Warne G, Zacharin M, et al. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum Mol Genet. (2001) 10:39–45. doi: 10.1093/hmg/10.1.39
28. Brickman JM, Clements M, Tyrell R, McNay D, Woods K, Warner J, et al. Molecular effects of novel mutations in Hesx1/HESX1 associated with human pituitary disorders. Development. (2001) 128:5189–99.
29. Vivenza D, Godi M, Faienza MF, Mellone S, Moia S, Rapa A, et al. A novel HESX1 splice mutation causes isolated GH deficiency by interfering with mRNA processing. Eur J Endocrinol. (2011) 164:705–13. doi: 10.1530/EJE-11-0047
30. Takagi M, Takahashi M, Ohtsu Y, Sato T, Narumi S, Arakawa H, et al. A novel mutation in HESX1 causes combined pituitary hormone deficiency without septo optic dysplasia phenotypes. Endocr J. (2016) 63:405–10. doi: 10.1507/endocrj.EJ15-0409
31. Fang Q, Benedetti AF, Ma Q, Gregory L, Li JZ, Dattani M, et al. HESX1 mutations in patients with congenital hypopituitarism: variable phenotypes with the same genotype. Clin Endocrinol. (2016) 85:408–14. doi: 10.1111/cen.13067
32. McNay DE, Turton JP, Kelberman D, Woods KS, Brauner R, Papadimitriou, et al. HESX1 mutations are an uncommon cause of septo-optic dysplasia and hypopituitarism. J Clin Endocrinol Metab. (2007) 92:691–7. doi: 10.1210/jc.2006-1609
33. Parks JS. Congenital Hypopituitarism. Clin Perinatol. (2018) 45:75–91. doi: 10.1016/j.clp.2017.11.001
34. Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. (2003) 17:126–40. doi: 10.1101/gad.224503
35. Collignon J, Sockanathan S, Hacker A, Cohen-Tannoudji M, Norris D, Rastan S, et al. A comparison of the properties of Sox-3 with Sry and two related genes, Sox-1 and Sox-2. Development. (1996) 122:509–20.
36. Dabdoub A, Puligilla C, Jones JM, Fritzsch B, Cheah KS, Pevny LH, et al. Sox2 signaling in prosensory domain specification and subsequent hair cell differentiation in the developing cochlea. Proc Natl Acad Sci USA. (2008) 105:18396–401. doi: 10.1073/pnas.0808175105
37. Episkopou V. SOX2 functions in adult neural stem cells. Trends Neurosci. (2005) 28:219–21. doi: 10.1016/j.tins.2005.03.003
38. Hume CR, Bratt DL, Oesterle EC. Expression of LHX3 and SOX2 during mouse inner ear development. Gene Expr Patterns. (2007) 7:798–807. doi: 10.1016/j.modgep.2007.05.002
39. Kamachi Y, Uchikawa M, Collignon J, Lovell-Badge R, Kondoh H. Involvement of Sox1, 2 and 3 in the early and subsequent molecular events of lens induction. Development. (1998) 125:2521–32.
40. Kiernan AE, Pelling AL, Leung KK, Tang AS, Bell DM, Tease C, et al. Sox2 is required for sensory organ development in the mammalian inner ear. Nature. (2005) 434:1031–35. doi: 10.1038/nature03487
41. Taranova OV, Magness ST, Fagan BM, Wu Y, Surzenko N, Hutton SR, Pevny LH. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. (2006) 20:1187–202. doi: 10.1101/gad.1407906
42. Wood HB, Episkopou V. Comparative expression of the mouse Sox1, Sox2 and Sox3 genes from pre-gastrulation to early somite stages. Mech Dev. (1999) 86:197–201. doi: 10.1016/S0925-4773(99)00116-1
43. Andoniadou CL, Matsushima D, Mousavy Gharavy SN, Signore M, Mackintosh AI, Schaeffer M, et al. Sox2(+) stem/progenitor cells in the adult mouse pituitary support organ homeostasis and have tumor-inducing potential. Cell Stem Cell. (2013) 13:433–45. doi: 10.1016/j.stem.2013.07.004
44. Macchiaroli A, Kelberman D, Auriemma RS, Drury S, Islam L, Giangiobbe S, et al. A novel heterozygous SOX2 mutation causing congenital bilateral anophthalmia, hypogonadotropic hypogonadism and growth hormone deficiency. Gene. (2014) 534:282–5. doi: 10.1016/j.gene.2013.10.043
45. Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, et al. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest. (2006) 116:2442–55. doi: 10.1172/JCI28658
46. Kelberman D, de Castro SC, Huang S, Crolla JA, Palmer R, Gregory JW, et al. SOX2 plays a critical role in the pituitary, forebrain, and eye during human embryonic development. J Clin Endocrinol Metab. (2008) 93:1865–73. doi: 10.1210/jc.2007-2337
47. Sato N, Kamachi Y, Kondoh H, Shima Y, Morohashi K, Horikawa R, et al. Hypogonadotropic hypogonadism in an adult female with a heterozygous hypomorphic mutation of SOX2. Eur J Endocrinol. (2007) 156:167–71. doi: 10.1530/EJE-06-0606
48. Stark Z, Storen R, Bennetts B, Savarirayan R, Jamieson RV. Isolated hypogonadotropic hypogonadism with SOX2 mutation and anophthalmia/microphthalmia in offspring. Eur J Hum Genet. (2011) 19:753–6. doi: 10.1038/ejhg.2011.11
49. Errichiello E, Gorgone C, Giuliano L, Iadarola B, Cosentino E, Rossato M, et al. SOX2: not always eye malformations. Severe genital but no major ocular anomalies in a female patient with the recurrent c.70del20 variant. Eur J Med Genet. (2018) 61:335–40. doi: 10.1016/j.ejmg.2018.01.011
50. Williamson KA, Hver AM, Rainger J, Rogers RC, Magee A, Fiedler Z, et al. Mutations in SOX2 cause anophthalmia-esophageal-genital (AEG) syndrome. Hum Mol Genet. (2006) 15:1413–22. doi: 10.1093/hmg/ddl064
51. Hagstrom SA, Pauer GJ, Reid J, Simpson E, Crowe S, Maumenee IH, et al. SOX2 mutation causes anophthalmia, hearing loss, and brain anomalies. Am J Med Genet. (2005) 138:95–8. doi: 10.1002/ajmg.a.30803
52. Alatzoglou KS, Andoniadou CL, Kelberman D, Buchanan CR, Crolla J, Arriazu MC, et al. SOX2 haploinsufficiency is associated with slow progressing hypothalamo-pituitary tumours. Hum Mutat. (2011) 32:1376–80. doi: 10.1002/humu.21606
53. Woods KS, Cundall M, Turton J, Rizzoti K, Mehta A, Palmer R, et al. Over- and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am J Hum Genet. (2005) 76:833–49. doi: 10.1086/430134
54. Solomon NM, Ross SA, Morgan T, Belsky JL, Hol FA, Karnes PS, et al. Array comparative genomic hybridisation analysis of boys with X linked hypopituitarism identifies a 3.9 Mb duplicated critical region at Xq27 containing SOX3. J Med Genet. (2004) 41:669–78. doi: 10.1136/jmg.2003.016949
55. Laumonnier F, Ronce N, Hamel BC, Thomas P, Lespinasse J, Raynaud M, et al. Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am J Hum Genet. (2002) 71:1450–5. doi: 10.1086/344661
56. Stankiewicz P, Thiele H, Schlicker M. Duplication of Xq26.2-q27.1, including SOX3, in a mother and daughter with short stature and dyslalia. Am J Med Genet A. (2005) 138:11–7. doi: 10.1002/ajmg.a.30910
57. Takagi M, Ishii T, Torii C, Kosaki K, Hasegawa T. A novel mutation in SOX3 polyalanine tract: a case of Kabuki syndrome with combined pituitary hormone deficiency harboring double mutations in MLL2 and SOX3. Pituitary. (2014) 17:569–74. doi: 10.1007/s11102-013-0546-5
58. Alatzoglou KS, Azriyanti A, Rogers N, Ryan F, Curry N, Noakes C, et al. SOX3 deletion in mouse and human is associated with persistence of the craniopharyngeal canal. J Clin Endocr Metab. (2014) 99:E2702–8. doi: 10.1210/jc.2014-1160
59. Kurokawa D, Kiyonari H, Nakayama R, Kimura-Yoshida C, Matsuo I, Aizawa S. Regulation of Otx2 expression and its functions in mouse forebrain and midbrain. Development. (2004) 131:3319–31. doi: 10.1242/dev.01220
60. Boncinelli E, Morgan R. Downstream of Otx2, or how to get a head. Trends Genet. (2001) 17:633–6. doi: 10.1016/S0168-9525(01)02418-0
61. Mortensen AH, MacDonald JW, Ghosh D, Camper SA. Candidate genes for panhypopituitarism identified by gene expression profiling. Physiol Genomics. (2011) 43:1105–16. doi: 10.1152/physiolgenomics.00080.2011
62. Hever AM, Williamson KA, van Heyningen V. Developmental malformations of the eye: the role of PAX6, SOX2 and OTX2. Clin Genet. (2006) 69:459–70. doi: 10.1111/j.1399-0004.2006.00619.x
63. Ragge NK, Brown AG, Poloschek CM, Lorenz B, Henderson RA, Clarke MP, et al. Heterozygous mutations of OTX2 cause severe ocular malformations. Am J Hum Genet. (2005) 76:1008–22. doi: 10.1086/430721
64. Jones GE, Robertson L, Warman P, Craft EV, Cresswell L, Vasudevan PC. 14q22.3 Microdeletion encompassing OTX2 in a five-generation family with microphthalmia, pituitary abnormalities, and intellectual disability. Ophthalmic Genet. (2016) 37:352–3. doi: 10.3109/13816810.2015.1059463
65. Wyatt A, Bakrania P, Bunyan DJ, Osborne RJ, Crolla JA, Salt A, et al. Novel heterozygous OTX2 mutations and whole gene deletions in anophthalmia, microphthalmia and coloboma. Hum Mutat. (2008). 29:E278–83. doi: 10.1002/humu.20869
66. Tajima T, Ohtake A, Hoshino M, Amemiya S, Sasaki N, Ishizu K, et al. OTX2 loss of function mutation causes anophthalmia and combined pituitary hormone deficiency with a small anterior and ectopic posterior pituitary. J Clin Endocrinol Metab. (2009) 94:314–9. doi: 10.1210/jc.2008-1219
67. Dateki S, Fukami M, Sato N, Muroya K, Adachi M, Ogata T. OTX2 mutation in a patient with anophthalmia, short stature, and partial growth hormone deficiency: functional studies using the IRBP, HESX1, and POU1F1 promoters. J Clin Endocrinol Metab. (2008) 93:3697–702. doi: 10.1210/jc.2008-0720
68. Diaczok D, Romero C, Zunich J, Marshall I, Radovick S. A novel dominant negative mutation of OTX2 associated with combined pituitary hormone deficiency. J Clin Endocrinol Metab. (2008) 93:4351–9. doi: 10.1210/jc.2008-1189
69. Dateki S, Kosaka K, Hasegawa K, Tanaka H, Azuma N, Yokoya S, et al. Heterozygous orthodenticle homeobox 2 mutations are associated with variable pituitary phenotype. J Clin Endocrinol Metab. (2010) 95:756–64. doi: 10.1210/jc.2009-1334
70. Kioussi C, O'Connell S, St-Onge L, Treier M, Gleiberman AS, Gruss P, et al. Pax6 is essential for establishing ventral-dorsal cell boundaries in pituitary gland development. Proc Natl Acad Sci USA. (1999) 96:14378–82. doi: 10.1073/pnas.96.25.14378
71. Hergott-Faure L, Borot S, Kleinclauss C, Abitbol M, Penfornis A. Pituitary function and glucose tolerance in a family with a PAX6 mutation. Ann Endocrinol Paris. (2012) 73:510–4. doi: 10.1016/j.ando.2012.10.001
72. Shimo N, Yasuda T, Kitamura T, Matsushita K, Osawa S, Yamamoto Y, et al. Aniridia with a heterozygous PAX6 mutation in which the pituitary function was partially impaired. Intern Med. (2014) 53:39–42. doi: 10.2169/internalmedicine.53.1184
73. Takagi M, Nagasaki K, Fujiwara I, Ishii T, Amano N, Asakura Y, et al. Heterozygous defects in PAX6 gene and congenital hypopituitarism. Eur J Endocrinol. (2015) 172:37–45. doi: 10.1530/EJE-14-0255
74. Bakrania P, Efthymiou M, Klein JC, Salt A, Bunyan DJ, Wyatt A, et al. Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am J Hum Genet. (2008) 82:304–19. doi: 10.1016/j.ajhg.2007.09.023
75. Koika V, Varnavas P, Valavani H, Sidis Y, Plummer L, Dwyer A, et al. Comparative functional analysis of two fibroblast growth factor receptor 1. (FGFR1) mutations affecting the same residue. (R254W and R254Q) in isolated hypogonadotropic hypogonadism (IHH). Gene. (2013) 516:146–51. doi: 10.1016/j.gene.2012.12.041
76. Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J, et al. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab. (2012) 97:E694–9. doi: 10.1210/jc.2011-2938
77. Webb EA, AlMutair A, Kelberman D, Bacchelli C, Chanudet E, Lescai F, et al. ARNT2 mutation causes hypopituitarism, post-natal microcephaly, visual and renal anomalies. Brain. (2013) 136:3096–105. doi: 10.1093/brain/awt218
78. Roessler E, Du YZ, Mullor JL, Casas E, Allen WP, Gillessen-Kaesbach G, et al. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci USA. (2003) 100:13424–9. doi: 10.1073/pnas.2235734100
79. Bear KA, Solomon BD, Antonini S, Arnhold IJ, França MM, Gerkes EH, et al. Pathogenic mutations in GLI2 cause a specific phenotype that is distinct from holoprosencephaly. J Med Genet. (2014) 51:413–8. doi: 10.1136/jmedgenet-2013-102249
80. Treier M, Gleiberman AS, O'Connell SM, Szeto DP, McMahon JA, McMahon AP, et al. Multistep signaling requirements for pituitary organogenesis in vivo. Genes Dev. (1998) 12:1691–704. doi: 10.1101/gad.12.11.1691
81. Treier M, O'Connell S, Gleiberman A, Price J, Szeto DP, Burgess R, et al. Hedgehog signaling is required for pituitary gland development. Development. (2001) 128:377–86.
82. Ellsworth BS, Butts DL, Camper SA. Mechanisms underlying pituitary hypoplasia and failed cell specification in Lhx3-deficient mice. Dev Biol. (2008) 313:118–29. doi: 10.1016/j.ydbio.2007.10.006
83. Sheng HZ, Zhadanov AB, Mosinger B, Fujii T, Bertuzzi S, Grinberg A, et al. Specification of pituitary cell lineages by the LIM homeobox gene Lhx3. Science. (1996) 272:1004–7. doi: 10.1126/science.272.5264.1004
84. Pfaeffle RW, Savage JJ, Hunter CS, Palme C, Ahlmann M, Kumar P, et al. Four novel mutations of the LHX3 gene cause combined pituitary hormone deficiencies with or without limited neck rotation. J Clin Endocrinol Metab. (2007) 92:1909–19. doi: 10.1210/jc.2006-2177
85. Savage JJ, Hunter CS, Clark-Sturm SL, Jacob TM, Pfaeffle RW, Rhodes SJ. Mutations in the LHX3 gene cause dysregulation of pituitary and neural target genes that reflect patient phenotypes. Gene. (2007) 400:44–51. doi: 10.1016/j.gene.2007.05.017
86. Savage JJ, Hunter CS, Clark-Sturm SL, Jacob TM, Pfaeffle RW, Rhodes SJ. Clinical case seminar: a novel LHX3 mutation presenting as combined pituitary hormonal deficiency. J Clin Endocrinol Metab. (2006) 91:747–53. doi: 10.1210/jc.2005-2360
87. Sloop KW, Parker GE, Hanna KR, Wright HA, Rhodes SJ. LHX3 transcription factor mutations associated with combined pituitary hormone deficiency impair the activation of pituitary target genes. Gene. (2001) 265:61–9. doi: 10.1016/S0378-1119(01)00369-9
88. Howard PW, Maurer RA. A point mutation in the LIM domain of Lhx3 reduces activation of the glycoprotein hormone alpha-subunit promoter. J Biol Chem. (2001) 276:19020–6. doi: 10.1074/jbc.M101782200
89. Netchine I, Sobrier ML, Krude H, Schnabel D, Maghnie M, Marcos E, et al. Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nat Genet. (2000) 25:182–6. doi: 10.1038/76041
90. Kriström B, Zdunek AM, Rydh A, Jonsson H, Sehlin P, Escher SA. A novel mutation in the LIM homeobox 3 gene is responsible for combined pituitary hormone deficiency, hearing impairment, and vertebral malformations. J Clin Endocrinol Metab. (2009) 94:1154–61. doi: 10.1210/jc.2008-0325
91. Bonfig W, Krude H, Schmidt H. A novel mutation of LHX3 is associated with combined pituitary hormone deficiency including ACTH deficiency, sensorineural hearing loss, and short neck-A case report and review of the literature. Eur J Pediatr. (2011) 170:1017–21. doi: 10.1007/s00431-011-1393-x
92. Rajab A, Kelberman D, de Castro SC, Biebermann H, Shaikh H, Pearce K, et al. Novel mutations in LHX3 are associated with hypopituitarism and sensorineural hearing loss. Hum Mol Genet. (2008) 17:2150–9. doi: 10.1093/hmg/ddn114
93. Sobrier ML, Brachet C, Vié-Luton MP, Perez C, Copin B, Legendre M, et al. Symptomatic heterozygotes and prenatal diagnoses in a nonconsanguineous family with syndromic combined pituitary hormone deficiency resulting from two novel LHX3 mutations. J Clin Endocrinol Metab. (2012) 97:E503–9. doi: 10.1210/jc.2011-2095
94. Jullien N, Romanet P, Philippon M, Quentien MH, Beck-Peccoz P, Bergada I, et al. Heterozygous LHX3 mutations may lead to a mild phenotype of combined pituitary hormone deficiency. Eur J Hum Genet. (2018) 27:216–25. doi: 10.1038/s41431-018-0264-6
95. Ahern S, Daniels M, Bhangoo A. LHX3 deficiency presenting in the United States with severe developmental delay in a child of Syrian refugee parents. Endocrinol Diabetes Metab Case Rep. (2018) 2018:18–0079. doi: 10.1530/EDM-18-0079
96. Dateki S, Fukami M, Uematsu A, Kaji M, Iso M, Ono M, et al. Mutation and gene copy number analyses of six pituitary transcription factor genes in 71 patients with combined pituitary hormone deficiency: identification of a single patient with LHX4 deletion. J Clin Endocrinol Metab. (2010) 95:4043–7. doi: 10.1210/jc.2010-0150
97. Takagi M, Ishii T, Inokuchi M, Amano N, Narumi S, Asakura Y, et al. Gradual loss of ACTH due to a novel mutation in LHX4: comprehensive mutation screening in Japanese patients with congenital hypopituitarism. PLoS ONE. (2012) 7:e46008. doi: 10.1371/journal.pone.0046008
98. Castinetti F, Saveanu A, Reynaud R, Quentien MH, Buffin A, Brauner R, et al. A novel dysfunctional LHX4 mutation with high phenotypical variability in patients with hypopituitarism. J Clin Endocrinol Metab. (2008) 93:2790–9. doi: 10.1210/jc.2007-2389
99. Rochette C, Jullien N, Saveanu A, Caldagues E, Bergada I, Braslavsky D, et al. Identifying the deleterious effect of rare LHX4 allelic variants, a challenging issue. PLoS ONE. (2015) 10:e012664821. doi: 10.1371/journal.pone.0126648
100. Pfaeffle RW, Hunter CS, Savage JJ, Duran-Prado M, Mullen RD, Neeb ZP, et al. Three novel missense mutations within the LHX4 gene are associated with variable pituitary hormone deficiencies. J Clin Endocrinol Metab. (2008) 93:1062–71. doi: 10.1210/jc.2007-1525
101. Tajima T, Hattori T, Nakajima T, Okuhara K, Tsubaki J, Fujieda K. A novel missense mutation. (P366T) of the LHX4 gene causes severe combined pituitary hormone deficiency with pituitary hypoplasia, ectopic posterior lobe and a poorly developed sella turcica. Endocr J. (2007) 54:637–41. doi: 10.1507/endocrj.K06-200
102. Machinis K, Amselem S. Functional relationship between LHX4 and POU1F1 in light of the LHX4 mutation identified in patients with pituitary defects. J Clin Endocrinol Metab. (2005) 90:5456–62. doi: 10.1210/jc.2004-2332
103. Machinis K, Pantel J, Netchine I, Leger J, Camand OJ, Sobrier ML, et al. Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J Hum Genet. (2001) 69:961–8. doi: 10.1086/323764
104. Tajima T, Yorifuji T, Ishizu K, Fujieda K. A novel mutation (V101A) of the LHX4 gene in a Japanese patient with combined pituitary hormone deficiency. Exp Clin Endocrinol Diabetes. (2010) 118:405–9. doi: 10.1055/s-0029-1225612
105. Filges I, Bischof-Renner A, Röthlisberger B, Potthoff C, Glanzmann R, Günthard J, et al. Panhypopituitarism presenting as life-threatening heart failure caused by an inherited microdeletion in 1q25 including LHX4. Pediatrics. (2012) 129:e529–34. doi: 10.1542/peds.2010-3849
106. Gregory LC, Humayun KN, Turton JP, McCabe MJ, Rhodes SJ, Dattani MT. Lethal form of congenital hypopituitarism associated with the first recessive LHX4 mutation. J Clin Endocrinol Metab. (2015) 100:2158–64. doi: 10.1210/jc.2014-4484
107. Reynaud R, Jayakody SA, Monnier C, Saveanu A, Bouligand J, Guedj AM, et al. PROKR2 variants in multiple hypopituitarism with pituitary stalk interruption. J Clin Endocrinol Metab. (2012) 97:E1068–73. doi: 10.1210/jc.2011-3056
108. Reynaud R, Albarel F, Saveanu A, Kaffel N, Castinetti F, Lecomte P, et al. Pituitary stalk interruption syndrome in 83 patients: novel HESX1 mutation and severe hormonal prognosis in malformative forms. Eur J Endocrinol. (2011) 164:457–65. doi: 10.1530/EJE-10-0892
109. McCabe MJ, Gaston-Massuet C, Gregory LC, Alatzoglou KS, Tziaferi V, Sbai O, et al. Variations in PROKR2, but not PROK2, are associated with hypopituitarism and septo-optic dysplasia. J Clin Endocrinol Metab. (2013) 98:E547–57. doi: 10.1210/jc.2012-3067
110. Correa FA, Trarbach EB, Tusset C, Latronico AC, Montenegro LR, Carvalho LR, et al. FGFR1 and PROKR2 rare variants found in patients with combined pituitary hormone deficiencies. Endocr Connect. (2015) 4:100–7. doi: 10.1530/EC-15-0015
111. Karaca E, Buyukkaya R, Pehlivan D, Charng WL, Yaykasli KO, Bayram Y, et al. Whole-exome sequencing identifies homozygous GPR161 mutation in a family with pituitary stalk interruption syndrome. J Clin Endocrinol Metab. (2015) 100:E140–7. doi: 10.1210/jc.2014-1984
112. Sun Y, Bak B, Schoenmakers N, van Trotsenburg AS, Oostdijk W, Voshol P, et al. Loss-of-function mutations in IGSF1 cause an X-linked syndrome of central hypothyroidism and testicular enlargement. Nat Genet. (2012) 44:1375–81. doi: 10.1038/ng.2453
113. Joustra SD, Schoenmakers N, Persani L, Campi I, Bonomi M, Radetti G, et al. The IGSF1 deficiency syndrome: characteristics of male and female patients. J Clin Endocrinol Metab. (2013) 98:4942–52. doi: 10.1210/jc.2013-2743
114. Quentien MH, Delemer B, Papadimitriou DT, Souchon PF, Jaussaud R, Pagnier A, et al. Deficit in anterior pituitary function and variable immune deficiency (DAVID) in children presenting with adrenocorticotropin deficiency and severe infections. J Clin Endocrinol Metab. (2012) 97:E121–8. doi: 10.1210/jc.2011-0407
115. Hassed SJ, Li S, Xu W, Taylor AC. Mutation in PITX2 in a Patient with Axenfeld-Rieger Syndrome. Mol Syndromol. (2017) 8:107–9. doi: 10.1159/000454963
116. Vallette-Kasic S, Brue T, Pulichino AM, Gueydan M, Barlier A, David M, et al. Congenital isolated adrenocorticotropin deficiency: an underestimated cause of neonatal death, explained by TPIT gene mutations. J Clin Endocrinol Metab. (2005) 90:1323–31. doi: 10.1210/jc.2004-1300
117. Lamolet B, Pulichino AM, Lamonerie T, Gauthier Y, Brue T, Enjalbert A, Drouin J, et al. A pituitary cell restricted T box factor, Tpit, activates POMC transcription in cooperation with Pitx homeoproteins. Cell. (2001) 104:849–59. doi: 10.1016/S0092-8674(01)00282-3
118. Gregory LC, Gevers EF, Baker J, Kasia T, Chong K, Josifova DJ, et al. Structural pituitary abnormalities associated with CHARGE syndrome. J Clin Endocrinol Metab. (2013) 98:E737–43. doi: 10.1210/jc.2012-3467
119. Olson LE, Tollkuhn J, Scafoglio C, Krones A, Zhang J, Ohgi KA, et al. Homeodomain mediated beta-catenin-dependent switching events dictate cell-lineage determination. Cell. (2006) 125:593–605. doi: 10.1016/j.cell.2006.02.046
120. Couture C, Saveanu A, Barlier A, Carel JC, Fassnacht M, Flück CE, et al. Phenotypic homogeneity and genotypic variability in a large series of congenital isolated ACTH deficiency patients with TPIT gene mutations. J Clin Endocrinol Metab. (2012) 97:E486–95. doi: 10.1210/jc.2011-1659
121. Vesper AH, Raetzman LT, Camper SA. Role of prophet of Pit1 (PROP1) in gonadotrope differentiation and puberty. Endocrinology. (2006) 147:1654–63. doi: 10.1210/en.2005-1080
122. De Rienzo F, Mellone S, Bellone S, Babu D, Fusco I, Prodam F, et al. Frequency of genetic defects in combined pituitary hormone deficiency: a systematic review and analysis of a multicentre Italian cohort. Clin Endocrinol. (2015) 83:849–60. doi: 10.1111/cen.12849
123. Nose O, Tatsumi K, Nakano Y, Amino N. Congenital combined pituitary hormone deficiency attributable to a novel PROP1 mutation (467insT). J Pediatr Endocrinol Metab. (2006) 19:491–8. doi: 10.1515/jpem.2006.19.4.491
124. Abr ao MG, Leite MV, Carvalho LR, Billerbeck AE, Nishi MY, Barbosa AS, et al. Combined pituitary hormone deficiency (CPHD) due to a complete PROP1 deletion. Clin Endocrinol. (2006) 65:294–300. doi: 10.1111/j.1365-2265.2006.02592.x
125. Lebl J, Vosáhlo J, Pfaeffle RW. Auxological and endocrine phenotype in a population-based cohort of patients with PROP1 gene defects. Eur J Endocrinol. (2005) 153:389–96. doi: 10.1530/eje.1.01989
126. Tatsumi KI, Kikuchi K, Tsumura K, Amino N. A novel PROP1 gene mutation. (157delA) in Japanese siblings with combined anterior pituitary hormone deficiency. Clin Endocrinol. (2004) 61:635–40. doi: 10.1111/j.1365-2265.2004.02147.x
127. Reynaud R, Chadli-Chaieb M, Vallette-Kasic S, Barlier A, Sarles J, Pellegrini-Bouiller I, et al. A familial form of congenital hypopituitarism due to a PROP1 mutation in a large kindred: phenotypic and in vitro functional studies. J Clin Endocrinol Metab. (2004) 89:5779–86. doi: 10.1210/jc.2003-032124
128. Paracchini R, Giordano M, Corrias A, Mellone S, Matarazzo P, Bellone J, et al. Two new PROP1 gene mutations responsible for compound pituitary hormone deficiency. Clin Genet. (2003) 64:142–7. doi: 10.1034/j.1399-0004.2003.00106.x
129. Wu W, Cogan JD, Pfaffle RW, Dasen JS, Frisch H, O'Connell SM, et al. Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet. (1998) 18:147–9. doi: 10.1038/ng0298-147
130. Cogan JD, Wu W, Phillips JA III, Arnhold IJ, Agapito A, Fofanova OV, et al. The PROP1 2-base pair deletion is a common cause of combined pituitary hormone deficiency. J Clin Endocrinol Metab. (1998) 83:3346–9. doi: 10.1210/jc.83.9.3346
131. Arroyo A, Pernasetti F, Vasilyev VV. A unique case of combined pituitary hormone deficiency caused by a PROP1 gene mutation (R120C) associated with normal height and absent puberty. Clin Endocrinol. (2002) 57:283–91. doi: 10.1046/j.1365-2265.2002.01550.x
132. Voutetakis A, Maniati-Christidi M, Kanaka-Gantenbein C, Dracopoulou M, Argyropoulou M, Livadas S, et al. A Prolonged jaundice and hypothyroidism as the presenting symptoms in a neonate with a novel Prop1 gene mutation. (Q83X). Eur J Endocrinol. (2004) 150:257–64. doi: 10.1530/eje.0.1500257
133. Deladoëy J, Flück C, Büyükgebiz A, Kuhlmann BV, Eblé A, Hindmarsh PC, et al. “Hot spot” in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab. (1999) 84:1645–50. doi: 10.1210/jc.84.5.1645
134. Asteria C, Oliveira JH, Abucham J, Beck-Peccoz P. Central hypocortisolism as part of combined pituitary hormone deficiency due to mutations of PROP-1 gene. Eur J Endocrinol. (2000) 143:347–52. doi: 10.1530/eje.0.1430347
135. Pernasetti F, Toledo SP, Vasilyev VV, Hayashida CY, Cogan JD, Ferrari C, et al. Impaired adrenocorticotropin-adrenal axis in combined pituitary hormone deficiency caused by a two-base pair deletion (301–302delAG) in the prophet of Pit-1 gene. J Clin Endocrinol Metab. (2000) 85:390–7. doi: 10.1210/jc.85.1.390
136. Voutetakis A, Sertedaki A, Livadas S, Xekouki P, Bossis I, Dacou-Voutetakis C, et al. Pituitary size fluctuation in long-term MR studies of PROP1 deficient patients: a persistent pathophysiological mechanism? J Endocrinol Invest. (2006) 29:462–6. doi: 10.1007/BF03344131
137. Andersen B, Rosenfeld MG. POU domain factors in the neuroendocrine system: lessons from developmental biology provide insights into human disease. Endocr Rev. (2001) 22:2–35. doi: 10.1210/edrv.22.1.0421
138. Vallette-Kasic S, Barlier A, Teinturier C, Diaz A, Manavela M, Berthezène F, et al. PROP1 gene screening in patients with multiple pituitary hormone deficiency reveals two sites of hypermutability and a high incidence of corticotroph deficiency. J Clin Endocrinol Metab. (2001) 86:4529–35. doi: 10.1210/jcem.86.9.7811
139. Turton JP, Reynaud R, Mehta A, Torpiano J, Saveanu A, Woods KS, et al. Novel mutations within the POU1F1 gene associated with variable combined pituitary hormone deficiency. J Clin Endocrinol Metab. (2005) 90:4762–70. doi: 10.1210/jc.2005-0570
140. Cohen LE, Wondisford FE, Salvatoni A, Maghnie M, Brucker-Davis F, Weintraub BD, et al. A “hot spot” in the Pit-1 gene responsible for combined pituitary hormone deficiency: clinical and molecular correlates. J Clin Endocrinol Metab. (1995) 80:679–84. doi: 10.1210/jcem.80.2.7852536
141. Pfaffle RW, DiMattia GE, Parks JS, Brown MR, Wit JM, Jansen M, et al. Mutation of the POU-specific domain of Pit-1 and hypopituitarism without pituitary hypoplasia. Science. (1992) 257:1118–21. doi: 10.1126/science.257.5073.1118
142. Cherella C, Cohen L. Congenital Hypopituitarism in Neonates. NeoReviews. (2018) 19:e742–52. doi: 10.1542/neo.19-12-e742
143. Kelly A, Tang R, Becker S, Stanley CA. Poor specificity of low growth hormone and cortisol levels during fasting hypoglycemia for the diagnoses of growth hormone deficiency and adrenal insufficiency. Pediatrics. (2008) 122:e522–8. doi: 10.1542/peds.2008-0806
144. Shulman DI, Palmert MR, Kemp SF, Lawson Wilkins Drug and Therapeutics Committee. Adrenal insufficiency: still a cause of morbidity and death in childhood. Pediatrics. (2007) 119:e484–94. doi: 10.1542/peds.2006-1612
145. Oprea A, Bonnet NCG, Pollé O, Lysy PA. Novel insights into glucocorticoid replacement therapy for pediatric and adult adrenal insufficiency. Ther Adv Endocrinol Metab. (2019) 10:2042018818821294. doi: 10.1177/2042018818821294
146. Cherella CE, Wassner AJ. Congenital hypothyroidism: insights into pathogenesis and treatment. Int J Pediatr Endocrinol. (2017) 2017:11. doi: 10.1186/s13633-017-0051-0
147. Collu R, Tang J, Castagné J, Lagacé G, Masson N, Huot C, et al. A novel mechanism for isolated central hypothyroidism: inactivating mutations in the thyrotropin releasing hormone receptor gene. J Clin Endocrinol Metab. (1997) 82:1561–5. doi: 10.1210/jc.82.5.1561
148. Nicholas AK, Jaleel S, Lyons G, Schoenmakers E, Dattani MT, Crowne E, et al. Molecular spectrum of TSHb subunit gene defects in central hypothyroidism in the UK and Ireland. Clin Endocrinol. (2017) 86:410–8. doi: 10.1111/cen.13149
149. Rose SR, Brown RS, Foley T, Kaplowitz PB, Kaye CI, Sundararajan S, et al. American Academy of Pediatrics; Section on Endocrinology and Committee on Genetics, American Thyroid Association; Public Health Committee, Lawson Wilkins Pediatric Endocrine Society. Update of newborn screening and therapy for congenital hypothyroidism. Pediatrics. (2006) 117:2290–303. doi: 10.1542/peds.2006-0915
150. Jonklaas J, Bianco AC, Bauer AJ, Burman KD, Cappola AR, Celi FS, et al. American Thyroid Association Task Force on Thyroid Hormone Replacement. Guidelines for the treatment of hypothyroidism: prepared by the american thyroid association task force on thyroid hormone replacement. Thyroid. (2014) 24:1670–751. doi: 10.1089/thy.2014.0028
151. Schoenmakers N, Alatzoglou KS, Chatterjee VK, Dattani MT. Recent advances in central congenital hypothyroidism. J Endocrinol. (2015) 227:R51–71. doi: 10.1530/JOE-15-0341
152. Segal TY, Mehta A, Anazodo A, Hindmarsh PC, Dattani MT. Role of gonadotropin-releasing hormone and human chorionic gonadotropin stimulation tests in differentiating patients with hypogonadotropic hypogonadism from those with constitutional delay of growth and puberty. J Clin Endocrinol Metab. (2009) 94:780–5. doi: 10.1210/jc.2008-0302
153. Hatipoglu N, Kurtoglu S. Micropenis: etiology, diagnosis and treatment approaches. J Clin Res Pediatr Endocrinol. (2013) 5:217–23. doi: 10.4274/Jcrpe.1135
154. Bouvattier C, Maione L, Bouligand J, Dodé C, Guiochon-Mantel A, Young J. Neonatal gonadotropin therapy in male congenital hypogonadotropic hypogonadism. Nat Rev Endocrinol. (2011) 8:172–82. doi: 10.1038/nrendo.2011.164
155. Main KM, Schmidt IM, Toppari J, Skakkebaek NE. Early postnatal treatment of hypogonadotropic hypogonadism with recombinant human FSH and LH. Eur J Endocrinol. (2002) 146:75–9. doi: 10.1530/eje.0.1460075
156. Mullis PE. Genetic control of growth. Eur J Endocrinol. (2005) 152:11–31. doi: 10.1530/eje.1.01797
157. Kolon TF, Herndon CD, Baker LA, Baskin LS, Baxter CG, Cheng EY, et al. Evaluation and treatment of cryptorchidism: AUA guideline. J Urol. (2014) 192:337–45. doi: 10.1016/j.juro.2014.05.005
158. Ludwikowski B, Gonzalez R. The controversy regarding the need for hormonal treatment in boys with unilateral cryptorchidism goes on: a review of the literature. Eur J Pediatr. (2013) 172:5–8. doi: 10.1007/s00431-012-1711-y
159. Elder JS. Surgical management of the undescended testis: recent advances and controversies. Eur J Pediatr Surg. (2016) 26:418–26. doi: 10.1055/s-0036-1592197
160. Boehm U, Bouloux PM, Dattani MT, de Roux N, Dode C, Dunkel L, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. (2015) 11:547–64. doi: 10.1038/nrendo.2015.112
161. Hernandez LM, Lee PD, Camacho-Hubner C. Isolated growth hormone deficiency. Pituitary. (2007) 10:351–7. doi: 10.1007/s11102-007-0073-3
162. Alatzoglou KS, Turton JP, Kelberman D, Clayton PE, Mehta A, Buchanan C, et al. Expanding the spectrum of mutations in GH1 and GHRHR: genetic screening in a large cohort of patients with congenital isolated growth hormone deficiency. J Clin Endocrinol Metab. (2009) 94:191–9. doi: 10.1210/jc.2008-2783
163. Grimberg A, DiVall SA, Polychronakos C, Allen DB, Cohen LE, Quintos JB, et al. Drug and Therapeutics Committee and Ethics Committee of the Pediatric Endocrine Society. Guidelines for growth hormone and insulin-like growth factor-I treatment in children and adolescents: growth hormone deficiency, idiopathic short stature, and primary insulin like growth factor-I deficiency. Horm Res Paediatr. (2008) 86:361–97. doi: 10.1159/000452150
164. Argente J, Flores R, Gutiérrez-Arumí A, Verma B, Martos-Moreno GÁ, Cuscó I, et al. Defective minor spliceosome mRNA processing results in isolated familial growth hormone deficiency. EMBO Mol Med. (2014) 6:299–306. doi: 10.1002/emmm.201303573
165. Ogilvy-Stuart AL. Growth hormone deficiency. (GHD) from birth to 2 years of age: diagnostic specifics of GHD during the early phase of life. Horm Res. (2003) 60(Suppl. 1):2–9. doi: 10.1159/000071219
166. Binder G, Weidenkeller M, Blumenstock G, Langkamp M, Weber K, Franz AR. Rational Approach to the Diagnosis of Severe Growth Hormone Deficiency in the Newborn. J Clinic Endocrinol Metab. (2010) 2219–26. doi: 10.1210/jc.2009-2692
167. Ueda Y, Aoyagi H, Tajima T. A newborn with combined pituitary hormone deficiency developing shock and sludge. J Pediatr Endocrinol Metab. (2017) 30:1333–6. doi: 10.1515/jpem-2017-0203
168. Van der Kaay DC, Van Heel WJ, Dudink J, van den Akker EL. Transient diabetes insipidus in a preterm neonate and the challenge of desmopressin dosing. J Pediatr Endocrinol Metab. (2014) 27:769–71. doi: 10.1515/jpem-2013-0305
169. Smego AR, Backeljauw P, Gutmark-Little I. Buccally Administered Intranasal Desmopressin Acetate For The Treatment Of Neurogenic Diabetes Insipidus In Infancy. J Clin Endocrinol Metabo. (2016) 101:2084–8. doi: 10.1210/jc.2016-1157
170. Mehta A, Hindmarsh PC, Mehta H, Turton JP, Russell-Eggitt I, Taylor D, et al. Congenital hypopituitarism: clinical, molecular and neuroradiological correlates. Clin Endocrinol. (2009) 71:376–82. doi: 10.1111/j.1365-2265.2009.03572.x
171. Di Iorgi N, Allegri AE, Napoli F, Bertelli E, Olivieri I, Rossi A, et al. The use of neuroimaging for assessing disorders of pituitary development. Clin Endocrinol. (2012) 76:161–76. doi: 10.1111/j.1365-2265.2011.04238.x
172. Cerbone M, Katugampola H, Dattani MT. Chapter 42: Hypothalamic and Pituitary Disorders Affecting the Fetus and Neonate. Elsevier (2020). P. 3–28. doi: 10.1016/B978-0-12-814823-5.00042-8
173. Crisafulli G, Aversa T, Zirilli G, De Luca F, Gallizzi R, Wasniewska M. Congenital hypopituitarism: how to select the patients for genetic analyses. Ital J Pediatr. (2018) 44:47. doi: 10.1186/s13052-018-0484-y
174. Roessler E, Ermilov AN, Grange DK, Wang A, Grachtchouk M, Dlugosz AA, et al. A previously unidentified amino-terminal domainregulates transcriptional activity of wild-type and disease-associated human GLI2. Hum Mol Genet. (2005) 14:2181–8. doi: 10.1093/hmg/ddi222
Keywords: hypopituitarism, newborn, hypoglycaemia, pituitary gland, hormone deficiencies, septo-optic dysplasia, growth hormone, micropenis
Citation: Bosch i Ara L, Katugampola H and Dattani MT (2021) Congenital Hypopituitarism During the Neonatal Period: Epidemiology, Pathogenesis, Therapeutic Options, and Outcome. Front. Pediatr. 8:600962. doi: 10.3389/fped.2020.600962
Received: 31 August 2020; Accepted: 31 December 2020;
Published: 02 February 2021.
Edited by:
Amanda Lesley Ogilvy-Stuart, Cambridge University Hospitals NHS Foundation Trust, United KingdomReviewed by:
Malgorzata Gabriela Wasniewska, University of Messina, ItalyStefano Zucchini, Sant'Orsola-Malpighi Polyclinic, Italy
Copyright © 2021 Bosch i Ara, Katugampola and Dattani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Bosch i Ara, bGF1cmFib3NjaDE1MUBob3RtYWlsLmNvbQ==; Mehul T. Dattani, bWVodWwuZGF0dGFuaUBnb3NoLm5ocy51aw==