 Brahim Tabarki
Brahim Tabarki Wejdan Hakami1
Wejdan Hakami1 Nader Alkhuraish
Nader Alkhuraish Majid Alfadhel
Majid Alfadhel- 1Division of Pediatric Neurology, Department of Pediatrics, Prince Sultan Military Medical City, Riyadh, Saudi Arabia
- 2Division of Neuroradiology, Department of Radiology, Prince Sultan Military Medical City, Riyadh, Saudi Arabia
- 3Medical Genomics Research Department, King Abdullah International Medical Research Center, King Saud Bin Abdulaziz University for Health Sciences, King Abdulaziz Medical City, Ministry of National Guard-Health Affairs, Riyadh, Saudi Arabia
- 4Genetics and Precision Medicine Department, King Abdulaziz Medical City, Ministry of National Guard Health Affairs, King Abdullah Specialist Children's Hospital, King Saud Bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia
Previous reviews have described the features of brain involvement in pediatric-onset metabolic disorders with Mendelian and mitochondrial inheritance, but only a few have focused on spinal cord abnormalities. An increasing number of metabolic disorders with Mendelian and mitochondrial inheritance in children with predominant spinal cord involvement has been recognized. Spinal cord involvement may be isolated or may occur more frequently with brain involvement. Timely diagnosis and occasional genetic counseling are needed for timely therapy. Therefore, clinicians must be aware of the clinical, laboratory, and radiographic features of these disorders. In this review, we describe pediatric-onset metabolic disorders with Mendelian and mitochondrial inheritance and predominant spinal cord involvement. Furthermore, we provide an overview of these conditions, including background information and examples that require rapid identification, focusing on treatable conditions; that would be catastrophic if they are not recognized.
Introduction
Among the most-affected systems in metabolic disorders with Mendelian and mitochondrial inheritance, the central nervous system, including the brain, is the most involved (1, 2). Owing to advances in neuroimaging and molecular genetic and/or biochemical testing, an increasing number of genetic and metabolic diseases of childhood with predominant spinal cord involvement is being recognized. Spinal cord involvement in Mendelian and mitochondrial diseases most frequently occurs with brain involvement. However, it might also be the disease manifestation as well (3–8). Prompt recognition of metabolic disorders with Mendelian and mitochondrial inheritance as a potential cause for a phenotype involving the spinal cord is critical for accurate patient management, as some of these diseases are potentially treatable and evoke broader genetic counseling implications for the affected families. In this review, we discuss common pediatric-onset metabolic disorders with Mendelian and mitochondrial inheritance and associated spinal cord abnormalities.
Methods
Written informed consent was obtained from each minor's legal guardian for the publication of any potentially identifiable images or data included in this article.
The online database MEDLINE was used to perform a literature search for papers published between January 1985 and December 2019 without any date or language restrictions. We used a combination of relevant search terms, such as “children AND leukodystrophy AND spinal cord” (83 papers), “children AND leukoencephalopathy AND spinal cord” (385 papers), “children AND mitochondrial diseases AND spinal cord” (122 papers), “inherited metabolic disorders AND spinal cord AND children” (741 papers), and “vitamin AND spinal cord AND children” (126 papers). We independently reviewed articles to identify metabolic disorders with Mendelian and mitochondrial inheritance in pediatric patients with spinal cord involvement and systematically screened titles, abstracts, and full texts of the collected articles. Reviews and editorials were excluded (238 papers).
Pediatric-onset metabolic disorders with Mendelian and mitochondrial inheritance and spinal cord involvement were etiologically classified into three groups: (1) leukodystrophies, (2) mitochondrial diseases, and (3) deficiency-related metabolic diseases. The common pediatric-onset metabolic disorders with Mendelian and mitochondrial inheritance and spinal cord involvement are outlined in Tables 1, 2 (4–42).
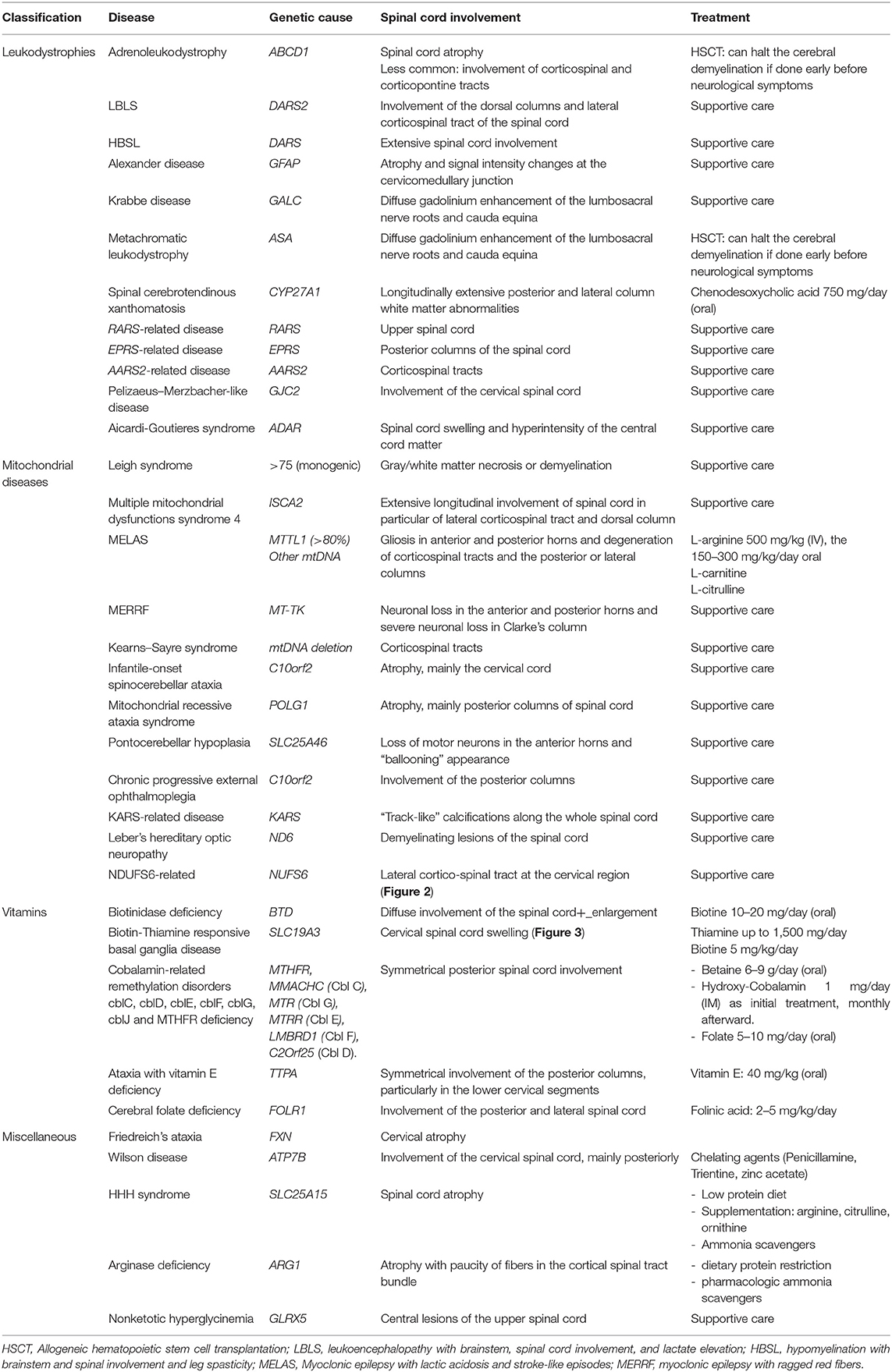
Table 1. Common pediatric-onset metabolic disorders with Mendelian and mitochondrial inheritance with predominant spinal cord involvement.
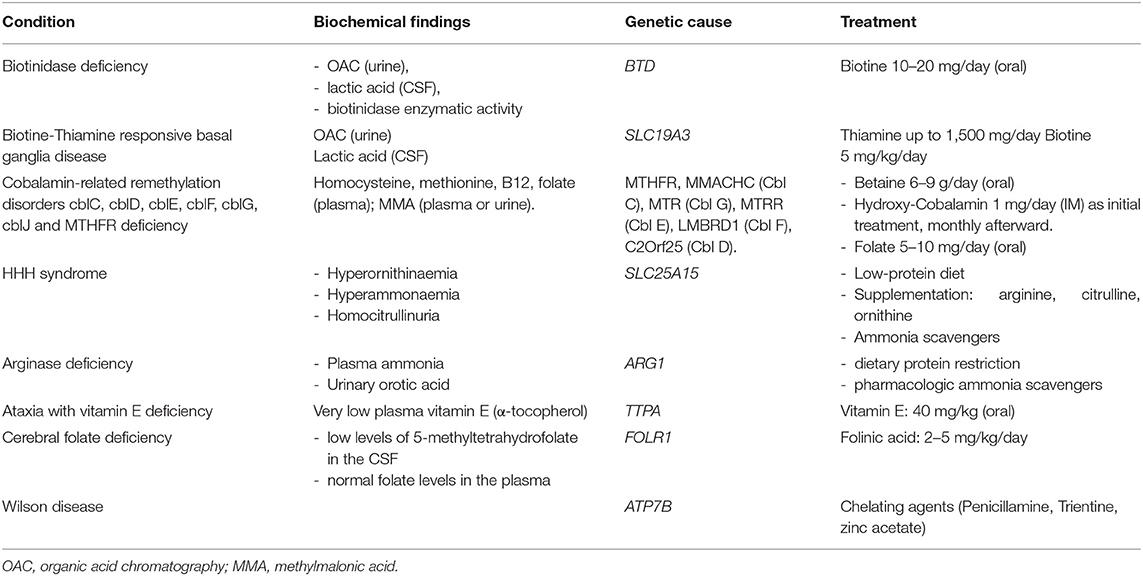
Table 2. Common treatable causes of metabolic myelopathy.
Leukodystrophies
Leukodystrophies are a heterogeneous group of heritable disorders that primarily affect the white matter of the central nervous system (8). They may affect both the brain and spinal cord (Table 1) (8–20). Their clinical manifestations often include ataxia, spasticity, and cognitive decline of variable severity.
Adrenoleukodystrophy
X-linked adrenoleukodystrophy (X-ALD) is the most common peroxisomal disorder. It is caused by mutations in ABCD1, which encodes the peroxisomal membrane protein ALDP. The clinical spectrum of X-ALD ranges from isolated Addison's disease and slowly progressive myelopathy to brain involvement. Most patients with ALD develop adrenomyeloneuropathy in adulthood. Neuropathological studies have shown that distal non-inflammatory axonopathy mainly affects the corticospinal tracts and dorsal columns (8). Spinal cord imaging in ALD mainly reveals atrophy; however, other anomalies can be observed, including the involvement of the corticospinal and corticopontine tracts. Van de Stadt et al. showed that the cervical spinal cord in patients with ALD is smaller and flattened compared to that in controls, possibly due to atrophy of the dorsal columns, and can be a marker of disease severity (9). The treatment of X-ALD remains mainly supportive. Early hematopoietic stem-cell transplantation is a treatment option with variable results. However, treatment using Lorenzo's oil has not consistently resulted in a substantial improvement in affected individuals. No treatment is currently available for progressive myelopathy in X-ALD (8).
Leukoencephalopathy With Brainstem, Spinal Cord Involvement, and Lactate Elevation
This is an autosomal recessive disease caused by a homozygous or compound heterozygous mutation in the DARS2 gene, which encodes a mitochondrial aspartyl-tRNA synthetase. Its clinical manifestations include progressive pyramidal, cerebellar, and dorsal column dysfunction, variable cognitive deficit or epilepsy. The MRI pattern is characteristic of abnormalities in the cerebral white matter, selective involvement of the brainstem and spinal cord tracts, and lactate elevation. Imaging of the spinal cord shows the involvement of the dorsal columns and lateral corticospinal tracts (Figure 1) (10).
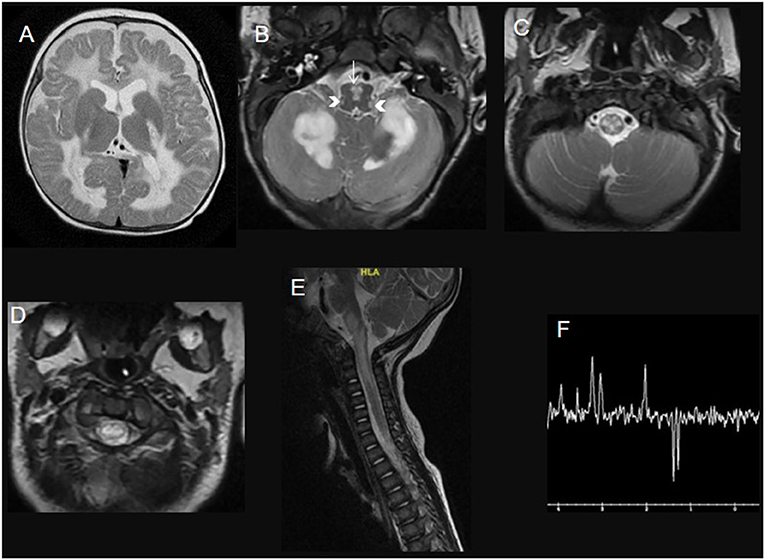
Figure 1. A 3-year-old child with leukoencephalopathy with the brainstem, spinal cord involvement, and lactate elevation due to DARS2 mutation. Brain (A–C) and spinal cord (D,E) MRI T2WI with Long Echotime MRS (F) hyperintensity of the deep white matter tracts in the brain (involving the posterior limb of internal capsules and sparing the subcortical U fibers), the deep cerebellar white matter, the pyramids in the medulla (white arrow) and the inferior cerebellar peduncles (head arrows), the dorsal column and lateral corticospinal tracts over the rostral spinal cord. Long echo time MRS (F) shows an inverted prominent lactate peak at 144 ms.
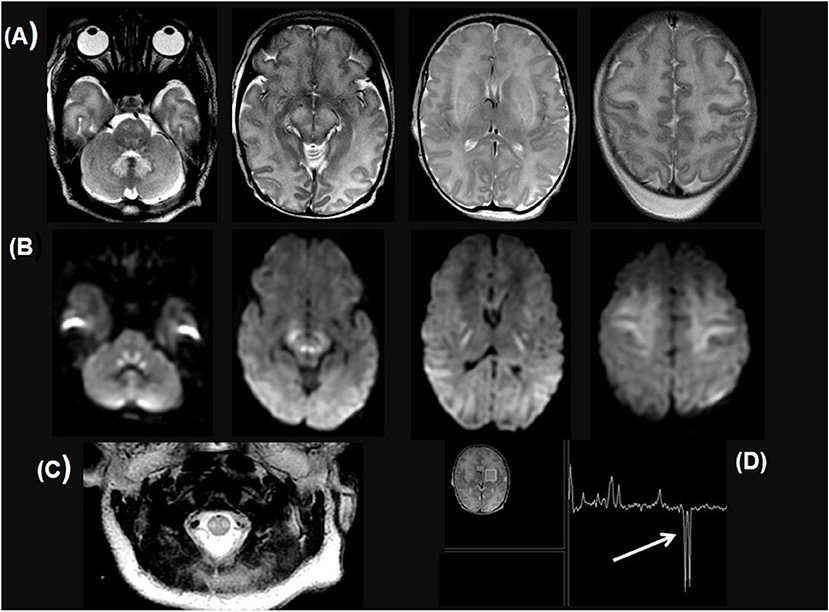
Figure 2. Ten-day-old newborn with NDUFS6 variant. Axial T2W brain MRI images (A), DWI images (B), and H-MR spectroscopy with TE 144 (C) show T2 hyperintensity of dentate nuclei, pontine tegmentum and corticospinal tract, dorsal mesencephalon and contiguous cerebral peduncles, internal capsules, basal ganglia, medial thalami, and supratentorial white matter, with symmetric restricted diffusion in the cerebellar peduncles, central tegmental and corticospinal tracts, perirolandic subcortical and deep white matter. (C) Axial T2W image of the cervical cord shows symmetrical abnormal dorso-lateral bright T2 signal including likely the corticospinal tract. (D) MR spectroscopy shows markedly elevated Lactate at 1.33 ppm (arrows).
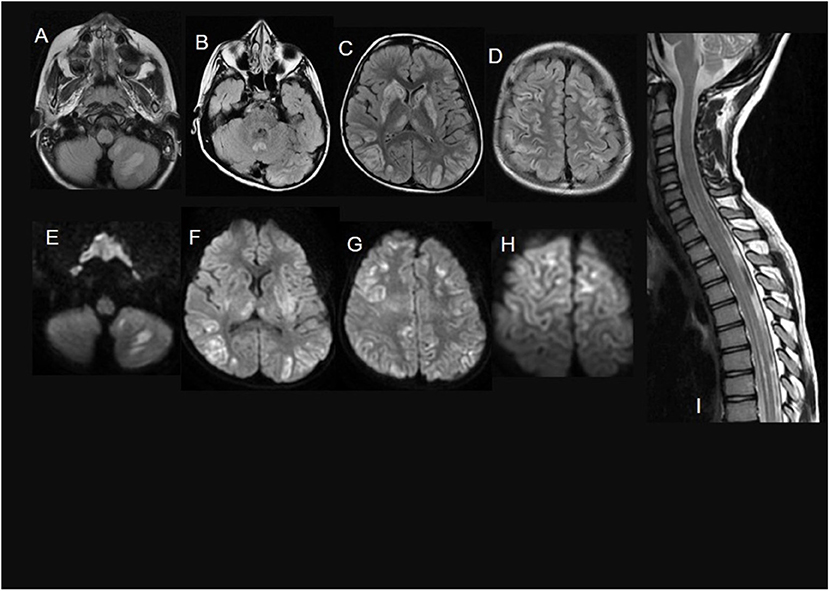
Figure 3. A 7-year-old child with Biotin thiamine responsive basal ganglia disease due to SLC19A3 mutation. (A–D) axial FLAIR sections show multiple bilateral infra and supratentorial FLAIR hyperintense lesions involving the striatum, the medial thalami, the vermis, and the cortical/juxtacortical fronto-parietal and temporal areas. In (E–H), diffusion restriction in many of these lesions is noted. Sagittal T2WI (I) shows an intramedullary T2 hyperintense lesion along with the lower cervical and dorsal cord till level opposite to 10th thoracic vertebra.
Alexander Disease
Alexander disease, caused by GFAP variants, is characterized by defects in the synthesis or maintenance of the myelin sheath (12). The clinical manifestations of early-onset Alexander disease include megalencephaly, difficulty in swallowing, cognitive defects, seizures, and spasticity. MRI findings typically show bilateral white-matter lesions with frontal predominance. Patients with late-onset Alexander disease typically present with brainstem-spinal cord junction dysfunction and rarely have seizures or megalencephaly. Late-onset Alexander disease imaging demonstrates more involvement of the hindbrain structures, including the cerebellum, brainstem, and cervical spinal cord, and less involvement of the forebrain. Periventricular hyperintensities are also described. The hindbrain lesions may evolve to so-called tadpole-like atrophy of the medulla oblongata and cervical spinal cord (13).
Krabbe Disease
Krabbe disease is an autosomal recessive disorder caused by a deficiency in galactocerebrosidase (galactosylceramide beta-galactosidase) activity. It has been classified into early-onset (<6 months) and late-onset (>6 months) variants. Clinical features of early-onset disease include severe progressive cognitive and motor deficits, visual loss, and seizures leading to death, usually by the age of 2 years. The typical presentation in late-onset disease is psychomotor regression, spastic paraparesis, and visual problems. Spinal MRI abnormalities include corticospinal tract involvement, posterior predominance, cauda equina enhancement, or atrophy of the cervical spinal cord (14).
Mitochondrial Diseases
Mitochondrial diseases are clinically heterogeneous disorders caused by a wide spectrum of mutations in genes encoded mostly by the nuclear or mitochondrial genome in small fractions (2). These diseases usually affect multiple organs, frequently involving the central nervous system. Mitochondrial diseases mainly involve the brain, but the spinal cord is an increasingly recognized site of involvement. Spinal cord involvement in mitochondrial diseases most frequently occurs with brain involvement, but it may also be the only disease manifestation (4, 5). Early recognition of spinal cord involvement in mitochondrial diseases is essential because it may influence management. The list of mitochondrial diseases with spinal cord involvement is expanding (Table 1) (21–31) and is frequently reported in ISCA2-related multiple mitochondrial dysfunction syndrome 4 (24); Leigh syndrome; leukoencephalopathy with the brain stem, spinal cord involvement, and lactate elevation (10); myoclonic epilepsy with ragged-red fibers (22);
Kearns-Sayre syndrome (23); infantile-onset spinocerebellar ataxia; mitochondrial recessive ataxia syndrome (25); mitochondrial multiorgan dysfunction syndrome (4), more rarely in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) (21); chronic progressive external ophthalmoplegia (28); KARS-related disease (29); and Leber's hereditary optic neuropathy (30). Spinal cord involvement presents as weakness, sensory loss, autonomic disturbances, and bowel/bladder dysfunction. These abnormalities may be the initial manifestations of mitochondrial diseases, such as in Leigh syndrome, or may overlap with seizures, ataxia, headache, or movement disorders in cases of brain involvement (4, 5). With the progression of mitochondrial diseases, spinal cord involvement may advance as well.
Based on imaging or pathological findings, the most frequently involved regions in the spinal cord in mitochondrial diseases are the dorsal columns, corticospinal tracts, and spinocerebellar tracts. In ISCA2-related mitochondrial disease, imaging findings show extensive longitudinal involvement of the spinal cord, particularly the lateral corticospinal tract and dorsal column (Figure 4) (24). In MELAS, neuronal loss of the anterior and posterior horns and degeneration of the corticospinal tracts are observed (21). In KARS-related mitochondrial disease, it was described as “track-like” calcifications along the entire spinal cord (29).
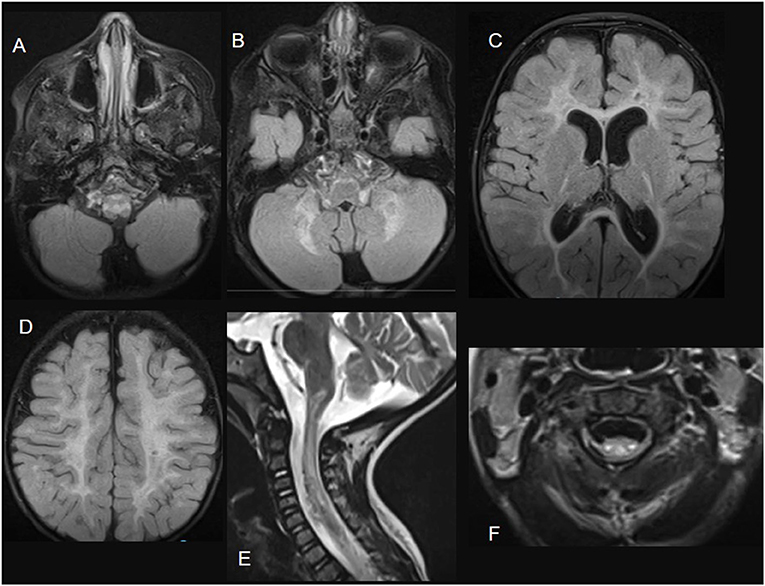
Figure 4. ISCA2-related disease. Brain MRI images; (A–D) axial FLAIR sections showing extensive diffuse bilateral supra and infratentorial signal abnormalities involving subcortical, deep, and subcortical white matter. Sagittal (E) and axial (F) T2-weighted images demonstrate spinal cord white matter involvement of the dorsal columns and the lateral corticospinal tracts.
Deficiency-Related Metabolic Diseases
Cobalamin-Related Remethylation Disorders cblC, cblD, cblE, cblF, cblG, cblJ, and MTHFR Deficiency
Genetic defects that affect the remethylation pathway cause hyperhomocysteinemia (32, 33). The clinical presentation of remethylation disorders varies; however, it mainly includes neurological manifestations (seizures, movement disorders, cognitive/behavior disorders, and microcephaly) and hematological features (megaloblastic anemia). Myelopathy is also a common feature of remethylation disorders and can be the presenting symptom in late-onset or untreated patients. Clinical manifestations include progressive abnormal gait, numbness, and sphincter dysfunction. Imaging changes always involve the posterior and lateral columns of the spinal cord, with the neuropathologic change showing vacuolation of the white matter, conceived as spongiform demyelination (32, 33). Early treatment of remethylation disorders is crucial and based on an appropriate diet and parenteral hydroxocobalamin, folate, and betaine administration in individuals with MTHFR deficiency.
Biotinidase Deficiency
Biotinidase deficiency is an autosomal recessive disorder that manifests during the first few months of life with seizures, hypotonia, hearing, visual loss, skin rashes, and alopecia. Spinal cord involvement is usually a manifestation of late-onset biotinidase deficiency. Clinically, patients with biotinidase deficiency exhibit limb weakness (spastic paraplegia/tetraplegia) with or without vision problems. Imaging findings show long-segment spinal cord involvement (34). Postmortem studies have shown loss of myelination and necrotizing lesions in the deep cerebral gray matter, brainstem, and spinal cord. The diagnosis of biotinidase deficiency is established by analyzing serum levels of the biotinidase enzyme and the BTD gene. Early treatment with biotin (10–20 mg/day) markedly improves or completely resolves the myelopathy in biotinidase deficiency, but if the treatment is delayed, the symptoms may be irreversible.
Ataxia With Vitamin E Deficiency
This rare autosomal recessive disease is caused by a mutation in the alpha-tocopherol transfer protein gene. Pathological studies of postmortem examinations have shown neuronal atrophy, axonal spheroids with a predilection for dorsal columns, and sensory demyelination. Neurologic phenotypes vary from progressive cerebellar ataxia and lower limb areflexia in early-onset disease to subacute combined degeneration with more pronounced peripheral neuropathy in late-onset disease. Spinal imaging findings are non-specific and may show changes in the posterior columns, particularly in the lower cervical segments. These abnormalities are associated with cerebellar atrophy in half of the affected cases (36). Early treatment with high-dose vitamin E supplementation has resulted in good outcomes in the treatment of ataxia with vitamin E deficiency, commonly seen in the Mediterranean region.
Discussion
Spinal cord involvement, either clinical and/or radiological, in pediatric-onset metabolic disorders with Mendelian and mitochondrial inheritance is likely more frequent than anticipated and requires special attention. In inherited metabolic diseases, spinal cord involvement is most frequently reported in adrenoleukodystrophy, biotinidase deficiency, arginase deficiency, and cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ, and MTHFR deficiency (9, 32–34, 41). In mitochondrial disorders, spinal cord involvement is most frequently reported in leukoencephalopathy with brainstem, spinal cord involvement and lactate elevation, Leigh syndrome, and multiple mitochondrial dysfunction syndrome (4). Spinal cord involvement in pediatric-onset metabolic disorders with Mendelian and mitochondrial inheritance must be considered a differential diagnosis of abnormalities more often found in acquired spinal cord diseases such as inflammation, demyelination, infection, or neoplastic, vascular, or toxic diseases (3–6).
The combination of careful clinical evaluation, brain and spine MRI, and metabolic/genetic investigations will help assess the frequency and pattern of spinal cord involvement in various inherited metabolic and mitochondrial myelopathies. Clinical manifestations of spinal cord involvement include weakness, sensory and autonomic disturbances, and sphincter dysfunction. These abnormalities may overlap with cerebral manifestations in cases with cerebral involvement. In some cases, an affected spinal cord may be the initial manifestation, such as in Leigh syndrome. Compared to acquired spinal cord diseases, heritable diseases in patients who exhibit myelopathic signs are more likely to have an insidious onset, progressive disease course, involvement of other systems (such as the eye, heart, skin, and kidneys), and a positive family history of a related disease. Several “red flag” MRI characteristics should prompt careful consideration of specific genetic metabolic or mitochondrial disorders. Although it is true that there is no uniform pattern of spinal cord involvement in metabolic disorders with Mendelian and mitochondrial inheritance, the dorsal columns, corticospinal tracts, or spinocerebellar tracts are most frequently involved, and gadolinium enhancement and significant spinal cord atrophy are absent (4, 7). However, some important exceptions to these general rules need to be mentioned: a selective spinal signal change in the dorsal columns could be found in patients with acquired dorsal root ganglion alteration (i.e., Sjogren syndrome), and gadolinium enhancement has been reported in some genetic (DARS mutation, Alexander disease, biotinidase deficiency) and acquired (B12 deficiency and methotrexate toxicity) conditions, which can mimic inflammatory conditions (8).
Advanced metabolic and genetic investigations to rule out metabolic disorders with Mendelian and mitochondrial inheritance are mainly based on the clinical phenotype and neuroimaging findings. In cases of high suspicion, targeted biomarker analysis or single gene analysis may facilitate the diagnostic process of treatable disorders and the initiation of disease-specific treatment. If the clinical and neuroimaging findings are non-specific and no specific cause is suspected, next-generation sequencing is a useful tool. In recent years, advanced genetic testing techniques have become efficient strategies to diagnose rare disease-causing mutations in children with metabolic disorders with Mendelian and mitochondrial inheritance. Timely diagnosis and management of such conditions is essential because early identification and treatment may limit the morbidity and mortality associated with metabolic disorders with Mendelian and mitochondrial inheritance. It is also important to anticipate the natural history of the newly diagnosed disorder and provide accurate genetic counseling.
Ethics Statement
Written informed consent was obtained from the parent of the participant for the publication of any potentially identifiable images or data included in this article.
Author Contributions
All authors participated in gathering the data, designing the article, and discussing and editing the manuscript.
Funding
This work was funded by King Abdullah International Medical Research Centre (KAIMRC) (project number: RC18/017/R). The funding authorities had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, et al. The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet. (2015) 97:199–215. doi: 10.1016/j.ajhg.2015.06.009
2. Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, et al. Mitochondrial diseases. Nat Rev Dis Primers. (2016) 2:16080. doi: 10.1038/nrdp.2016.80
3. Hedera P. Hereditary and metabolic myelopathies. Handb Clin Neurol. (2016) 136:769–85. doi: 10.1016/B978-0-444-53486-6.00038-7
4. Finsterer J, Zarrouk-Mahjoub S. Involvement of the spinal cord in mitochondrial disorders. J Neurosci Rural Pract. (2018) 9:245–51. doi: 10.4103/jnrp.jnrp_446_17
5. Finsterer J. Spinal cord involvement in Leigh syndrome. Brain Dev. (2018) 40:733–4. doi: 10.1016/j.braindev.2018.04.003
6. Hedera P. Hereditary myelopathies. Continuum. (2018) 24:523–50. doi: 10.1212/CON.0000000000000580
7. Marelli C, Salsano E, Politi LS, Labauge P. Spinal cord involvement in adult-onset metabolic and genetic diseases. J Neurol Neurosurg Psychiatry. (2019) 90:211–8. doi: 10.1136/jnnp-2018-318666
8. Ashrafi MR, Amanat M, Garshasbi M, Kameli R, Nilipour Y, Heidari M, et al. An update on clinical, pathological, diagnostic, and therapeutic perspectives of childhood leukodystrophies. Expert Rev Neurother. (2020) 20:65–84. doi: 10.1080/14737175.2020.1699060
9. van de Stadt SIW, van Ballegoij WJC, Labounek R, Huffnagel IC, Kemp S, Nestrasil I, et al. Spinal cord atrophy as a measure of severity of myelopathy in adrenoleukodystrophy. J Inherit Metab Dis. (2020) 43:852–60. doi: 10.1002/jimd.12226
10. van Berge L, Hamilton EM, Linnankivi T, Uziel G, Steenweg ME, Isohanni P, et al. Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation: clinical and genetic characterization and target for therapy. Brain. (2014) 137(Pt 4):1019–29. doi: 10.1093/brain/awu026
11. Taft RJ, Vanderver A, Leventer RJ, Damiani SA, Simons C, Grimmond SM, et al. Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am J Hum Genet. (2013) 92:774–80. doi: 10.1016/j.ajhg.2013.04.006
12. Brackmann F, Coras R, Rössler K, Kraus C, Rompel O, Trollmann R. Histopathological proof of the pathogenicity of a rare GFAP mutation in a patient with flaccid paraparesis. Brain Dev. (2018) 40:330–3. doi: 10.1016/j.braindev.2017.11.005
13. Namekawa M, Takiyama Y, Honda J, Shimazaki H, Sakoe K, Nakano I. Adult-onset Alexander disease with typical “tadpole” brainstem atrophy and unusual bilateral basal ganglia involvement: a case report and review of the literature. BMC Neurol. (2010) 10:21. doi: 10.1186/1471-2377-10-21
14. Michael SN, Madaan P, Jauhari P, Chakrabarty B, Kumar A, Gulati S. Selective pyramidal tract involvement in late-onset Krabbe disease. Indian J Pediatr. (2019) 86:970–1. doi: 10.1007/s12098-019-02972-x
15. Toldo I, Carollo C, Battistella PA, Laverda AM. Spinal cord and cauda equina MRI findings in metachromatic leukodystrophy: case report. Neuroradiology. (2005) 47:572–5. doi: 10.1007/s00234-005-1369-5
16. Gelzo M, Di Taranto MD, Bisecco A, D'Amico A, Capuano R, Giacobbe C, et al. A case of Cerebrotendinous Xanthomatosis with spinal cord involvement and without tendon xanthomas: identification of a new mutation of the CYP27A1 gene. Acta Neurol Belg. (2019). doi: 10.1007/s13760-019-01267-4. [Epub ahead of print].
17. Rezaei Z, Hosseinpour S, Ashrafi MR, Mahdieh N, Alizadeh H, Mohammadpour M, et al. Hypomyelinating leukodystrophy with spinal cord involvement caused by a novel variant in RARS: report of two unrelated patients. Neuropediatrics. (2019) 50:130–4. doi: 10.1055/s-0039-1679911
18. Mendes MI, Gutierrez Salazar M, Guerrero K, Thiffault I, Salomons GS, Gauquelin L, et al. Bi-allelic mutations in EPRS, encoding the glutamyl-prolyl-aminoacyl-tRNA synthetase, cause a hypomyelinating leukodystrophy. Am J Hum Genet. (2018) 102:676–84. doi: 10.1016/j.ajhg.2018.02.011
19. Biancheri R, Rosano C, Denegri L, Lamantea E, Pinto F, Lanza F, et al. Expanded spectrum of Pelizaeus-Merzbacher-like disease: literature revision and description of a novel GJC2 mutation in an unusually severe form. Eur J Hum Genet. (2013) 21:34–9. doi: 10.1038/ejhg.2012.93
20. Samanta D, Ramakrishnaiah R. Recurrent encephalopathy with spinal cord involvement: an atypical manifestation of Aicardi-Goutières syndrome. Ann Indian Acad Neurol. (2019) 22:111–5. doi: 10.4103/aian.AIAN_12_18
21. Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC, et al. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord. (1992) 2:125–35. doi: 10.1016/0960-8966(92)90045-8
22. Shen C, Xian W, Zhou H, Li X, Liang X, Chen L. Overlapping Leigh syndrome/myoclonic epilepsy with ragged red fibres in an adolescent patient with a mitochondrial DNA A8344G mutation. Front Neurol. (2018) 9:724. doi: 10.3389/fneur.2018.00724
23. Luca P, Alessia G, Camilla RM, Antonio N, Diego M, Federica D, et al. Spinal cord involvement in Kearns-Sayre syndrome: a neuroimaging study. Neuroradiology. (2020) 62:1315–21. doi: 10.1007/s00234-020-02501-0
24. Toldo I, Nosadini M, Boscardin C, Talenti G, Manara R, Lamantea E, et al. Neonatal mitochondrial leukoencephalopathy with brain and spinal involvement and high lactate: expanding the phenotype of ISCA2 gene mutations. Metab Brain Dis. (2018) 33:805–12. doi: 10.1007/s11011-017-0181-3
25. Nikali K, Suomalainen A, Saharinen J, Kuokkanen M, Spelbrink JN, Lönnqvist T, et al. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum Mol Genet. (2005) 14:2981–90. doi: 10.1093/hmg/ddi328
26. McKelvie P, Marotta R, Thorburn DR, Chin J, Punchihewa S, Collins S. A case of myelopathy, myopathy, peripheral neuropathy and subcortical grey matter degeneration associated with recessive compound heterozygous POLG1 mutations. Neuromuscul Disord. (2012) 22:401–5. doi: 10.1016/j.nmd.2011.10.017
27. Wan J, Steffen J, Yourshaw M, Mamsa H, Andersen E, Rudnik-Schöneborn S, et al. Loss of function of SLC25A46 causes lethal congenital pontocerebellar hypoplasia. Brain. (2016) 139:2877–90. doi: 10.1093/brain/aww212
28. Galassi G, Maggi L, Lamantea E, Ariatti A, Malagoli M. C10ORF2 mutation associated with progressive external ophthalmoplegia and clinically isolated syndrome. Acta Neurol Belg. (2017) 117:947–9. doi: 10.1007/s13760-017-0793-8
29. Ardissone A, Tonduti D, Legati A, Lamantea E, Barone R, Dorboz I, et al. KARS-related diseases: progressive leukoencephalopathy with brainstem and spinal cord calcifications as new phenotype and a review of literature. Orphanet J Rare Dis. (2018) 13:45. doi: 10.1186/s13023-018-0788-4
30. La Russa A, Cittadella R, Andreoli V, Valentino P, Trecroci F, Caracciolo M, et al. Leber's hereditary optic neuropathy associated with a multiple-sclerosis-like picture in a man. Mult Scler. (2011) 17:763–6. doi: 10.1177/1352458511404033
31. Rouzier C, Chaussenot A, Fragaki K, Serre V, Ait-El-Mkadem S, Richelme C, et al. NDUFS6 related Leigh syndrome: a case report and review of the literature. J Hum Genet. (2019) 64:637–45. doi: 10.1038/s10038-019-0594-4
32. Wang SJ, Yan CZ, Liu YM, Zhao YY. Late-onset cobalamin C deficiency Chinese sibling patients with neuropsychiatric presentations. Metab Brain Dis. (2018) 33:829–35. doi: 10.1007/s11011-018-0189-3
33. Huemer M, Mulder-Bleile R, Burda P, Froese DS, Suormala T, Zeev BB, et al. Clinical pattern, mutations and in vitro residual activity in 33 patients with severe 5, 10 methylenetetrahydrofolate reductase (MTHFR) deficiency. J Inherit Metab Dis. (2016) 39:115–24. doi: 10.1007/s10545-015-9860-6
34. Girard B, Bonnemains C, Schmitt E, Raffo E, Bilbault C. Biotinidase deficiency mimicking neuromyelitis optica beginning at the age of 4: a treatable disease. Mult Scler. (2017) 23:119–22. doi: 10.1177/1352458516646087
35. Tabarki B, Al-Hashem A, Alfadhel M. Biotin-Thiamine-responsive basal Ganglia disease. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (1993–2020).
36. Larnaout A, Belal S, Zouari M, Fki M, Ben Hamida C, Goebel HH, et al. Friedreich's ataxia with isolated vitamin E deficiency: a neuropathological study of a Tunisian patient. Acta Neuropathol. (1997) 93:633–7. doi: 10.1007/s004010050662
37. Ramaekers V, Sequeira JM, Quadros EV. Clinical recognition and aspects of the cerebral folate deficiency syndromes. Clin Chem Lab Med. (2013) 51:497–511. doi: 10.1515/cclm-2012-0543
38. Dogan I, Romanzetti S, Didszun C, Mirzazade S, Timmann D, Saft C, et al. Structural characteristics of the central nervous system in Friedreich ataxia: an in vivo spinal cord and brain MRI study. J Neurol Neurosurg Psychiatry. (2019) 90:615–7. doi: 10.1136/jnnp-2018-318422
39. Silva-Júnior FP, Machado AA, Lucato LT, Barbosa ER. Resolution of MRI findings of copper deficiency myeloneuropathy in a patient with Wilson's disease. Arq Neuropsiquiatr. (2014) 72:255–6. doi: 10.1590/0004-282X20130226
40. Olivieri G, Pro S, Diodato D, Di Capua M, Longo D, Martinelli D, et al. Corticospinal tract damage in HHH syndrome: a metabolic cause of hereditary spastic paraplegia. Orphanet J Rare Dis. (2019) 14:208. doi: 10.1186/s13023-019-1181-7
41. Liu XB, Haney JR, Cantero G, Lambert JR, Otero-Garcia M, Truong B, et al. Hepatic arginase deficiency fosters dysmyelination during postnatal CNS development. JCI Insight. (2019) 4:e130260. doi: 10.1172/jci.insight.130260
Keywords: spinal cord, Mendelian disease, mitochondrial disease, MRI, children
Citation: Tabarki B, Hakami W, Alkhuraish N, Tlili-Graies K and Alfadhel M (2021) Spinal Cord Involvement in Pediatric-Onset Metabolic Disorders With Mendelian and Mitochondrial Inheritance. Front. Pediatr. 8:599861. doi: 10.3389/fped.2020.599861
Received: 28 August 2020; Accepted: 24 November 2020;
Published: 14 January 2021.
Edited by:
Kirsten A. Donald, University of Cape Town, South AfricaReviewed by:
Manoj Menezes, The Children's Hospital at Westmead, AustraliaMaurizio Elia, Oasi Research Institute (IRCCS), Italy
Copyright © 2021 Tabarki, Hakami, Alkhuraish, Tlili-Graies and Alfadhel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brahim Tabarki, YnRhYmFya2lAaG90bWFpbC5jb20=