 Alexandra J. Borst
Alexandra J. Borst Taizo A. Nakano
Taizo A. Nakano Francine Blei
Francine Blei Denise M. Adams
Denise M. Adams Jessica Duis2*
Jessica Duis2*- 1Vascular Anomalies Program, Monroe Carrell Jr. Children's Hospital, Vanderbilt University Medical Center, Nashville, TN, United States
- 2Vascular Anomalies Center, Children's Hospital Colorado, University of Colorado School of Medicine, Aurora, CO, United States
- 3Vascular Anomalies Program, Lenox Hill Hospital, Northwell Health, New York, NY, United States
- 4Vascular Anomalies Center, Children's Hospital of Philadelphia, University of Pennsylvania, Philadelphia, PA, United States
The field of vascular anomalies has grown tremendously in the last few decades with the identification of key molecular pathways and genetic mutations that drive the formation and progression of vascular anomalies. Understanding these pathways is critical for the classification of vascular anomalies, patient care, and development of novel therapeutics. The goal of this review is to provide a basic understanding of the classification of vascular anomalies and knowledge of their underlying molecular pathways. Here we provide an organizational framework for phenotype/genotype correlation and subsequent development of a diagnostic and treatment roadmap. With the increasing importance of genetics in the diagnosis and treatment of vascular anomalies, we highlight the importance of clinical geneticists as part of a comprehensive multidisciplinary vascular anomalies team.
Introduction
Vascular anomalies encompass both vascular malformations and vascular tumors (1). Over the past few decades, our knowledge of the genetic changes underlying these tumors and malformations has grown tremendously. This has led to updates in the classification of some lesions as well as new therapeutic options. The goal of this review is to provide teams taking care of children and adults with vascular anomalies an overview of the molecular pathways involved in disease pathogenesis and an organizational framework based on the current classification system that encompasses knowledge of their genetic and molecular etiologies. We also aim to provide a roadmap for evaluation of vascular anomalies based on both clinical and genetic features and to highlight the critical role of genetic diagnosis in the classification and treatment of vascular anomalies.
Developmental Origin and Classification of Vascular Anomalies
Vascular malformations form due to errors in vascular morphogenesis, which occurs during early embryonic life, sometime between 4 and 10 weeks of gestation. Mesenchymal cells form primitive groups, called hemangioblasts, that differentiate into the four types of circulatory vessels (arterial, venous, capillary, lymphatic) through a series of remodeling, pruning, and continued growth. This is a process referred to as angiogenesis. The molecular mechanisms that govern angiogenesis are complex, with a series of genes controlling each critical step, from blood island formation, to endothelial cell differentiation, sprouting, migration, and remodeling (2, 3). Identifying and understanding these molecular mechanisms is key to the development of targeted therapeutics for children with vascular anomalies.
The care of patients with vascular anomalies has been plagued by misdiagnosis, which can delay appropriate treatment and misinform patients and their families. Comprehensive diagnostic evaluation requires careful attention to patient and family history, clinical and radiologic features of the lesion, and thoughtful classification of the lesion. Many centers utilize a multidisciplinary team, with both medical and surgical providers with experience and expertise in vascular anomalies, to provide comprehensive care for these patients. Increasingly, geneticists are becoming key members of such multidisciplinary teams. The important role of genetics in the care of patients with vascular anomalies is highlighted by updates to the classification system. In 1996, the International Society for the Study of Vascular Anomalies (ISSVA) developed a standardized classification guideline for children and adults with vascular Anomalies (4–6). In 2018, the ISSVA classification system was updated to capture our expanding knowledge base and report known genetic causes of vascular anomalies (6, 7).
Inherited vs. Sporadic Vascular Anomalies
Understanding the types of genetic changes that drive the formation of vascular anomalies is key to appropriate testing and diagnosis. These pathogenic genetic variants can be categorized as either germline (familial or heritable mutations) or somatic (acquired mutation in somatic “body” tissues). The majority of vascular anomalies arise from somatic mutations, in which there is a post-zygotic mutation in a single cell that then perpetuates itself and is present in groups of cells throughout the body (called mosaicism). In this case, the mutation is not present in every cell. Genetic testing in patients with high suspicion for somatic driver mutations is presently best completed on directly affected tissue, which often requires surgical or skin biopsy from the vascular anomaly (Figure 1). Conversely, germline mutations are present in all of the individual's cells including the egg and sperm. Genetic testing in patients with high suspicion for germline driver mutations can be completed on any tissue, skin, blood, hair, or saliva.
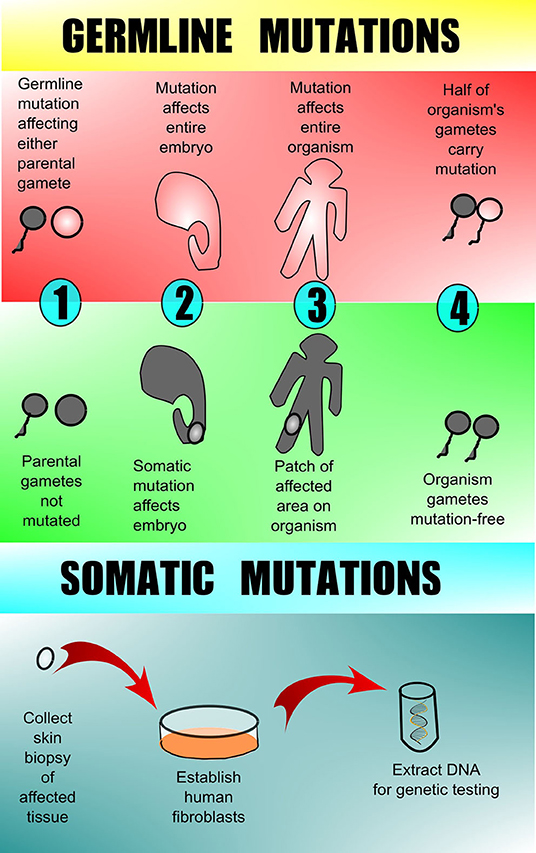
Figure 1. Germline vs. somatic mutations. Germline mutations are found in one or more of the parental gametes and thus subsequently affect all of the cells of the offspring. Somatic mutations arise in a non-gamete cell of the affected offspring and therefore are present in only 1 area or tissue type and are not in the offspring's gametes. Testing for germline mutations can be done on peripheral blood, but testing for somatic mutations requires biopsy of the affected tissue with subsequent DNA analysis. Peripheral blood and/or unaffected tissue may sometimes be sent along with affected tissue for comparison when searching for somatic mutation.
Familial or inherited vascular malformations or syndromes are less common but provide unique insight regarding the molecular mechanisms that control vascular morphogenesis. The majority are inherited in an autosomal dominant fashion and involve mutations that lead to a loss-of-function of the affected gene. There is generally not 100% penetrance of familial vascular disorders and some of the phenotypes may not be readily apparent until later in life, suggesting that a second hit may be needed for tumor or malformation to present. In addition, a wide spectrum may be noted with respect to type, location, and presence of the malformation. When evaluating a patient with a vascular anomaly, family history including a 3-generation pedigree, may determine whether one might consider a germline mutation. Family and personal history of cutaneous findings, macrocephaly, history of malignancies, and developmental or learning disabilities; for example, implicates PTEN Hamartoma Tumor Syndrome (PHTS) and prompts germline testing to confirm. This has immediate implications for management with surveillance for thyroid cancer starting in childhood and additional malignancies in adulthood (8).
Historically, a low frequency of identified germline mutations led clinicians to seek tissue biopsy and many key somatic mutations were identified in this manner. Identification of tissue specific somatic mutations expands our understanding of the phenotypic spectrum of these disorders and our knowledge of their pathophysiology. As we continue to define the specific molecular changes and learn the breadth of clinical findings, we improve the inaccuracies in clinical classification and may be able to pursue genotype-phenotype correlation (9). Molecular diagnosis provides objective classification and serves to direct personalized therapies based on molecular phenotype (10). The identification of genes involved in these developmental defects of the vasculature is critical to our understanding of their etiology, as well as providing targeted therapeutic interventions (3, 11, 12).
Role of Genetics
Genetic testing is now a key component of comprehensive care for patients with vascular anomalies. Accurate identification of a genetic variant can aid in definitive diagnosis, allow for necessary preventive screening, and directly impact therapeutic decision making. Elucidation of the genetic changes underlying vascular anomalies has also played an invaluable role in understanding normal vascular developmental programming and biology. The landscape has changed dramatically with the recognition of an expanding repertoire of germline and somatic mutations recognized to drive the development of vascular malformations and tumors. The majority of identified mutations in vascular anomalies occur within two key intracellular signaling pathways—the RAS/MAPK and PI3K/AKT/mTOR pathways (Figure 2). The PI3K/AKT/mTOR pathway is crucial for many cellular processes, including cell cycle regulation, proliferation, and migration, and is often termed the “anti-apoptosis pathway.” Several activating mutations within this pathway are associated with vascular anomalies and complex vascular syndromes. The RAS/MAPK pathway is also crucial for cell cycle regulation, proliferation, and migration, and is often referred to as the “proliferation pathway.” Several vascular anomalies are associated with mutations in this pathway and frequently termed “rasopathies.” There is also crosstalk between these 2 main pathways. Finally, TGF-β signaling, which is ubiquitous and key for regulation of numerous biological processes, has been implicated in the pathophysiology of hereditary hemorrhagic telangiectasia. As we learn more about the mutations involved in the development of various vascular malformations and tumors, it becomes clear that activating mutations in these key intracellular signaling pathways are frequently the cause of endothelial cell dysfunction and malformation development (13). The types of malformations or tumors that result are dependent on the specific mutation, tissue mosaicism, impact on protein function, and interaction with other genes. For example, mammalian target of rapamycin (mTOR) signaling is initiated in the phosphoinositide 3-kinase (PI3K)/AKT pathway involved in cell cycle regulation and often implicated in patients with vascular anomalies and overgrowth who have somatic mutations in PIK3CA. This finding has led to targeting of the overactive PI3K/AKT/mTOR pathway with sirolimus (an mTOR inhibitor), which has demonstrated clinical benefit in several vascular malformations and tumors (10, 11). As we continue to identify the specific driver mutations that cause mosaic or sporadic vascular anomalies, our understanding of the pathophysiology will continue to grow and offer insight into therapeutic strategies. It is very likely that additional gene mutations will be identified and become future targets for therapy. We offer here the current information regarding protein function for the known genetic mutations in vascular malformations and tumors. However, this information is growing rapidly and we are still at the very early stages of understanding this biology and identifying future targets for therapy that will continue to shape the management of vascular anomalies.
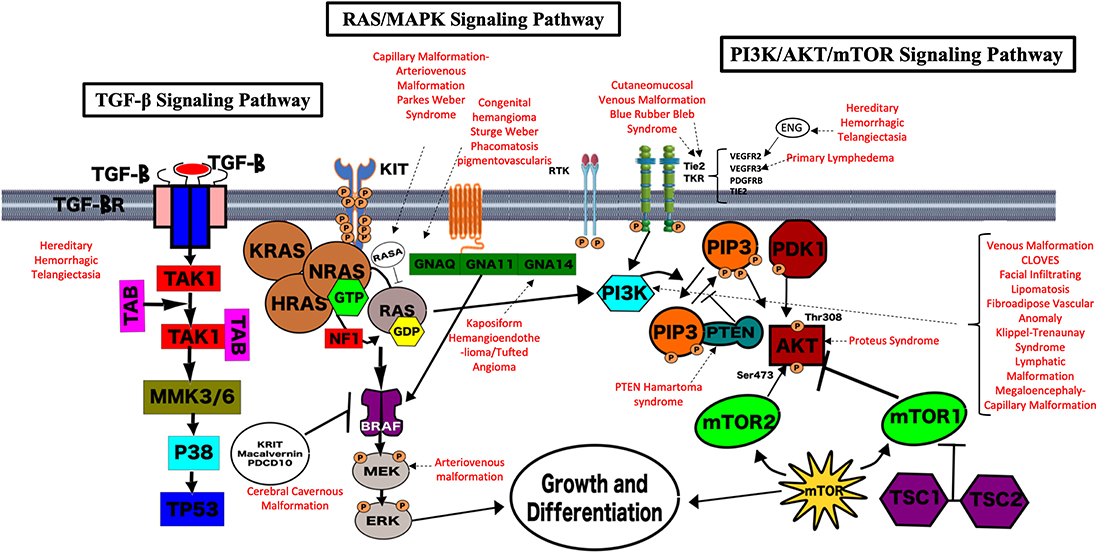
Figure 2. Genetic pathways implicated in vascular anomalies. The majority of identified mutations in vascular anomalies occur within two key intracellular signaling pathways—the RAS/MAPK and PI3K/AKT/mTOR pathways. The PI3K/AKT/mTOR pathway is crucial for many cellular processes, including cell cycle regulation, proliferation, and migration, and is often termed the “anti-apoptosis pathway.” Several activating mutations within this pathway are associated with vascular anomalies and complex vascular syndromes. The RAS/MAPK pathway is also crucial for cell cycle regulation, proliferation and migration, and is often referred to as the “proliferation pathway.” Several vascular anomalies are associated with mutations in this pathway and frequently termed “rasopathies.” There is also crosstalk between these 2 main pathways. The TGF-β Signaling Pathway is also key for regulation of numerous biological processes, has been implicated in the pathophysiology of hereditary hemorrhagic telangiectasia. Each pathway and their overlap are demonstrated pictorially here. The known associations with vascular malformations and syndromes are highlighted in red.
Vascular Malformations
Using the 2018 International Society for the Study of Vascular Anomalies classification of vascular malformation by simple subtype (6, 7), we provide an outline of the inherited/germline and somatic mutations identified in various malformations and recommended clinical evaluation for each malformation or syndrome. Examples of some of the key vascular malformations, syndromes, and tumors are found in Figure 3. We also provide a pathway for initial evaluation and recommended “next steps” for each type of vascular malformation or tumor (Figure 4). When guided by a careful history and physical exam, we believe this tool can be beneficial to clinicians as they navigate initial evaluation and management, recognizing that ultimately a multidisciplinary approach and opinion will be invaluable.
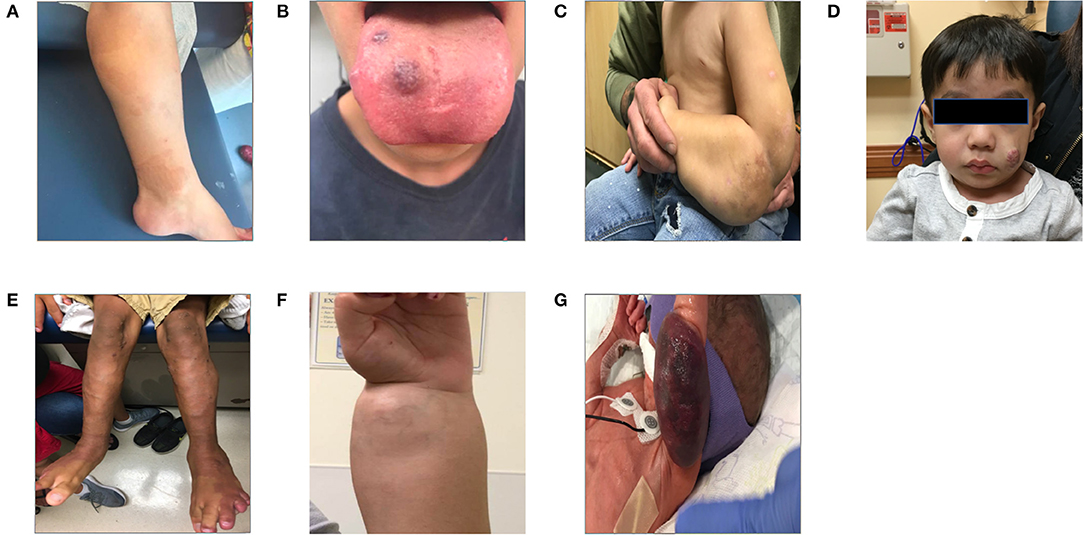
Figure 3. Patients with vascular anomalies. This panel of patient photographs shows a sampling of some of the physical exam findings in a few key disorders. (A) Shows a patient with the typical capillary malformations seen in patients with RASA1 mutation found on the lower extremity. (B) Shows a patient with Blue Rubber Bleb Nevus syndrome and the typical venous malformations seen here on the tongue. (C) Shows a patient with a large lymphatic malformation of the left upper extremity. The patient is several months into treatment with sirolimus. (D) Shows a young child with an infantile hemangioma of the cheek. (E) Shows the somatic overgrowth, capillary malformation, prominent superficial veins, and lymphangiomas in a patient with CLOVES syndrome. (F) Shows a patient with PTEN hamartoma syndrome and an intramuscular vascular malformation of the forearm/wrist. (G) Shows an infant with a Kaposiform hemangioendothelioma who presented with Kasabach-Merritt syndrome.
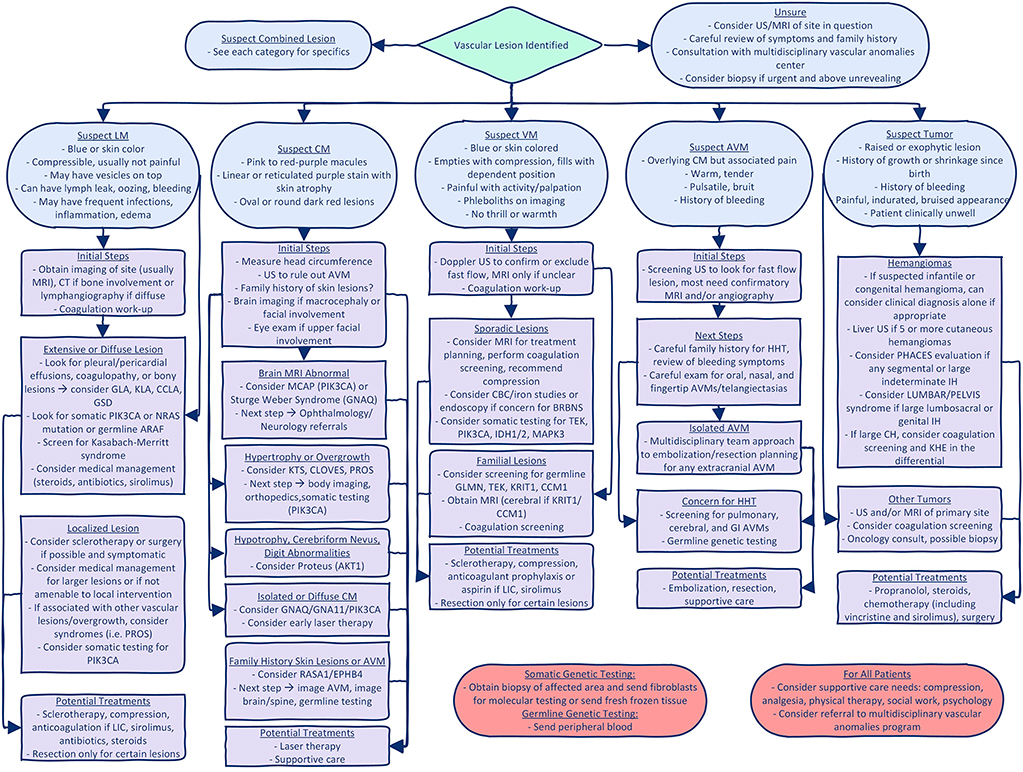
Figure 4. Diagnostic algorithm for vascular anomalies. This figure gives a suggested diagnostic algorithm for the initial evaluation and next steps when evaluating a patient with a vascular malformation or tumor. It is not all inclusive, but gives suggestions based on initial characteristics of the lesion and highlights key/critical steps that should not be missed. GLA, generalized lymphatic anomaly; KLA, Kaposiform lymphatic anomaly; CCLA, central conducting lymphatic anomaly; GSD, Gorham Stout Disease; LIC, localized intravascular coagulation; KTS, Klippel Trenaunay Syndrome; PROS, PIK3CA related overgrowth spectrum; BRBN, Blue Rubber Bleb Nevus; IH, infantile hemangioma; CH, congenital hemangioma.
Capillary Malformations
Capillary malformations (CMs), commonly known as port-wine stains, are common in the general population with an incidence of 0.3% (14). Pathologically they are defined by dilated venule-type channels in the superficial dermis with characteristically slow-flow on imaging. Capillary malformations are often associated with underlying soft tissue or bony hypertrophy, though whether this is causative or associated with the increased blood flow is unclear. They are typically present at the time of birth and may change in color over time. With age, vascular stasis in the abnormally formed capillaries results in vascular ectasia and soft tissue thickening. Although most isolated capillary malformations and associated syndromes are sporadic (Table 1), familial inheritance has been described (Table 2). Sturge-Weber syndrome (SWS) represents the most well-known cause of somatic capillary malformation. The capillary malformations seen in SWS and the more common isolated, uncomplicated capillary malformations are frequently found to have mutations in GNAQ or GNA11, which can lead to constitutive activation of the RAS/MAPK pathway (Figure 1) (24, 25). Inherited capillary malformation syndromes are most commonly due to mutations in the RAS/MAPK signaling pathway and are inherited in an autosomal dominant fashion with incomplete penetrance and variable expression.

Table 1. Somatic disorders involving capillary malformations.
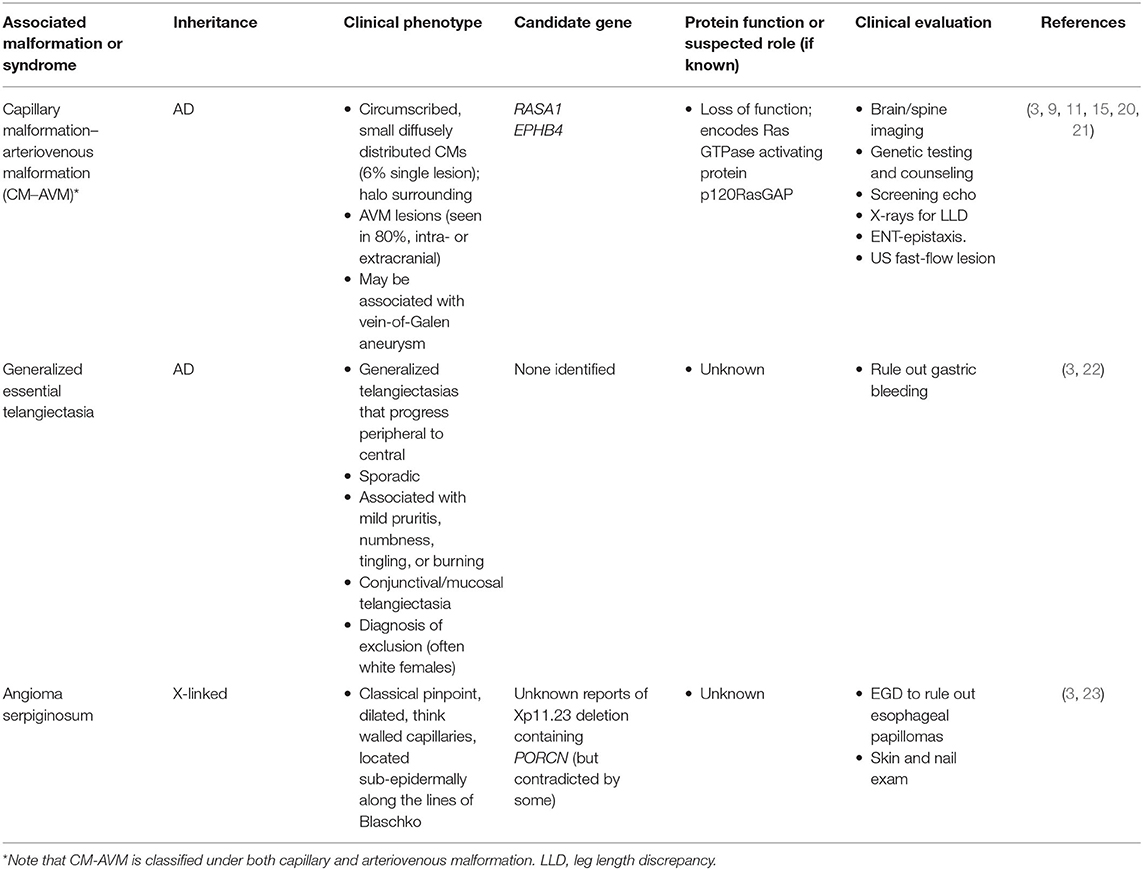
Table 2. Inherited/germline conditions involving capillary malformations (CM).
Venous Malformations
Venous malformations (VMs) are slow-flow lesions that can either be mass-like within tissues and unrelated to named veins, or they can represent anomalous development of anatomic veins. They are often considered the most common subtype of vascular malformation, with an incidence of between 1 in 5,000 to 1 in 10,000 births. They can range in size from very small to extensive and are purple to bluish on color due to stagnant blood flow within the lesion, that contributes to pain, swelling, and thrombotic complications (4, 5, 26–28). The majority of lesions are noted at birth, but deeper lesions are sometimes asymptomatic until the time of puberty when they tend to increase in size and become symptomatic. More than 90% of venous malformations are sporadic and isolated in nature (Table 3), but may rarely present with a familial pattern (Table 4) (15). TIE2/TEK gene mutations are thought to be causative in most isolated venous malformations. However, more recently, somatic activating mutations in PIK3CA have also been identified in venous malformations (41). TIE2/TEK is an endothelial cell specific tyrosine kinase receptor that functions via the PI3K/AKT/mTOR signaling pathway (Figure 1) and plays a significant role in regulating angiogenesis, proliferation, cell migration, and vessels stability (3, 15). Blue rubber bleb nevus syndrome (BRBNS) is a rare, often severe disorder known to be caused by TIE2/TEK somatic mutations (30). Careful documentation of birthmarks on family pedigree can help identity hereditary cases. Glomuvenous malformation and mucocutaneous venous malformation (VMCM) have demonstrated autosomal dominant inheritance but are thought to require a second hit for lesion formation (26).
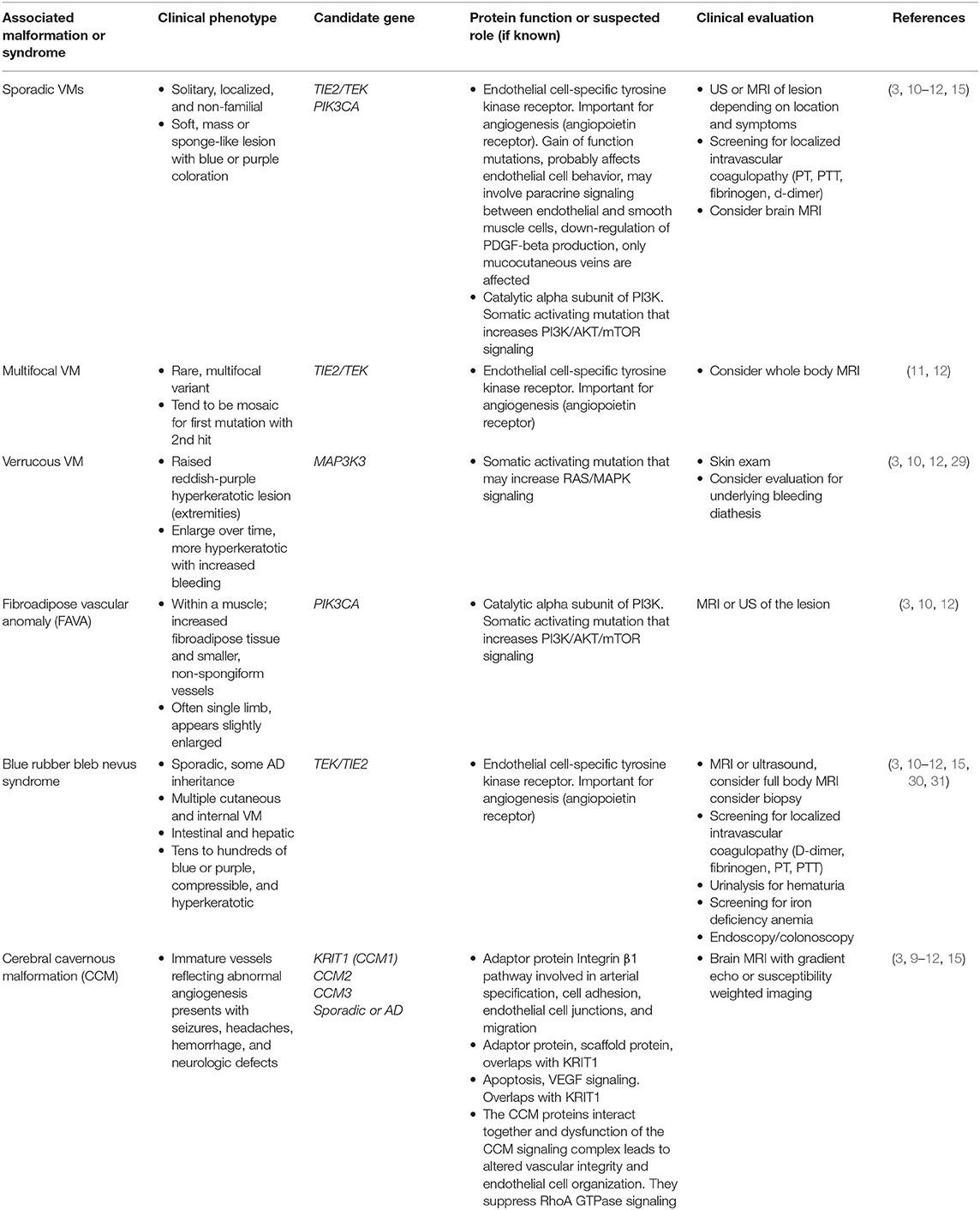
Table 3. Somatic disorders involving venous malformations.

Table 4. Inherited/germline conditions involving venous malformations.
Lymphatic Malformations
Lymphatic malformations (LM) encompass a range of developmental or functional defects affecting lymphatic vessels. These can be focal or diffuse disorders with marked heterogeneity (Table 5) (46). Lymphatic anomalies can be associated with significant comorbidities such as pain, infection, visceral, and bone involvement and disfigurement. Most lymphatic malformations are due to sporadic mutations in genes that regulate lymphangiogenesis. However, certain genetic syndromes are associated with developmental abnormalities of the lymphatics, including Down syndrome, Turner syndrome, Noonan syndrome and Cardiofaciocutaneous syndrome (47, 48). These are important to be aware of in the general evaluation of children with these disorders. Lymphedema is clinically distinct from lymphatic malformations in that it is due to functional defects in initial or collecting lymphatics. Some forms of lymphedema and related syndromes appear to have familial inheritance (Table 6) (3). Sporadic lymphatic malformations and related syndromes are believed to involve mutations in VEGFC/VEGFR3 and PI3K/AKT/mTOR pathways, but a clear pathogenetic mechanisms has not been defined (3, 15).
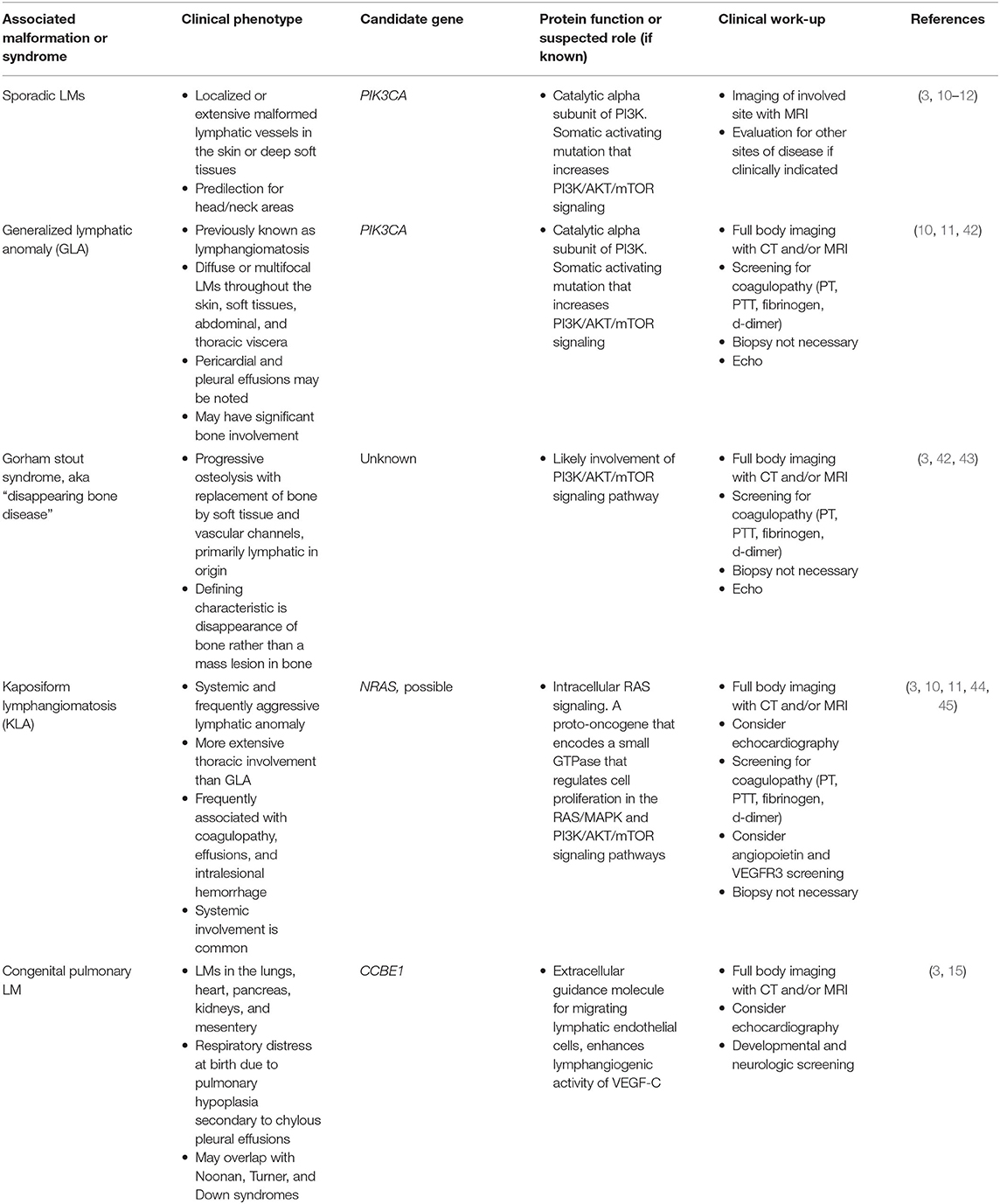
Table 5. Somatic disorders involving lymphatic malformations.

Table 6. Inherited/germline conditions involving lymphatic malformations.
Arteriovenous Malformations
Arteriovenous malformations (AVMs) occur due to a direct connection between an artery and vein, bypassing the normal capillary bed, leading to arterialization of the veins and a high-flow lesion with propensity to grow and bleed (54). Sporadic AVMs (Table 7) are commonly associated with mutations in the RAS/MAPK pathway (Figure 1). RASA1 mutations are associated with CM-AVM and may be causative in some sporadic AVMs (3, 15). Other known molecular changes found in sporadic AVMs are found in MAP2K/MEK and several groups are starting to use MEK inhibitors for off-label management (29, 57). The most common inherited AVM syndrome (Table 8) is hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome. HHT is a disorder of vascular dysplasia characterized by multiple AVMs. Small telangiectasias may manifest as superficial skin and mucosal membrane lesions with potential to rupture presenting as epistaxis or gastrointestinal bleeding. Larger AVMs can be found within the lungs, central nervous system, and liver. HHT is inherited in an autosomal dominant manner with clinical heterogeneity and an age-dependent natural history. Pathologic mutations to genes involved in TGF-beta signaling pathway (Figure 1), important for angiogenesis, have been described including ENG, ALK1/ACVRL1, GDF2, SMAD4/MADH4. An appropriate pediatric HHT screening evaluation must include family history, physical examination, and a low threshold to proceed with non-invasive screening and imaging (58, 59). Heterozygous ENG and ALK mutations, such as found in HHT, have also been found in sporadic AVMs.
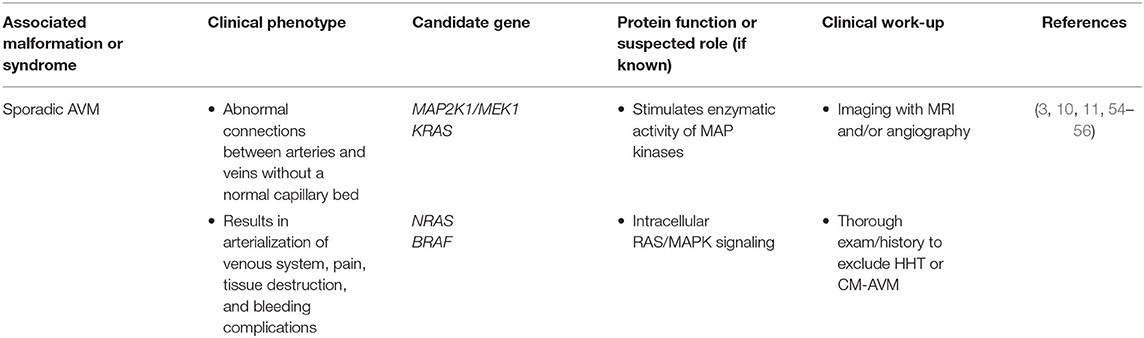
Table 7. Somatic disorders involving arteriovenous malformations.

Table 8. Inherited/germline conditions involving arteriovenous malformations.
Complex Syndromes Associated With Vascular Anomalies
There are several complex syndromes that include overgrowth of soft tissues in combination with vascular malformations. The majority of these syndromes are caused by somatic mutations, primarily isolated to the PI3K/AKT/mTOR signaling pathway (Figure 1) (11, 15). These syndromes and the candidate genes involved in their development are summarized in Table 9. Recognition of these syndromes is important due to the association of other complications, such as malignancy in CLOVES and Maffuci syndromes, and thrombosis in Klippel-Trenaunay and Proteus syndromes.
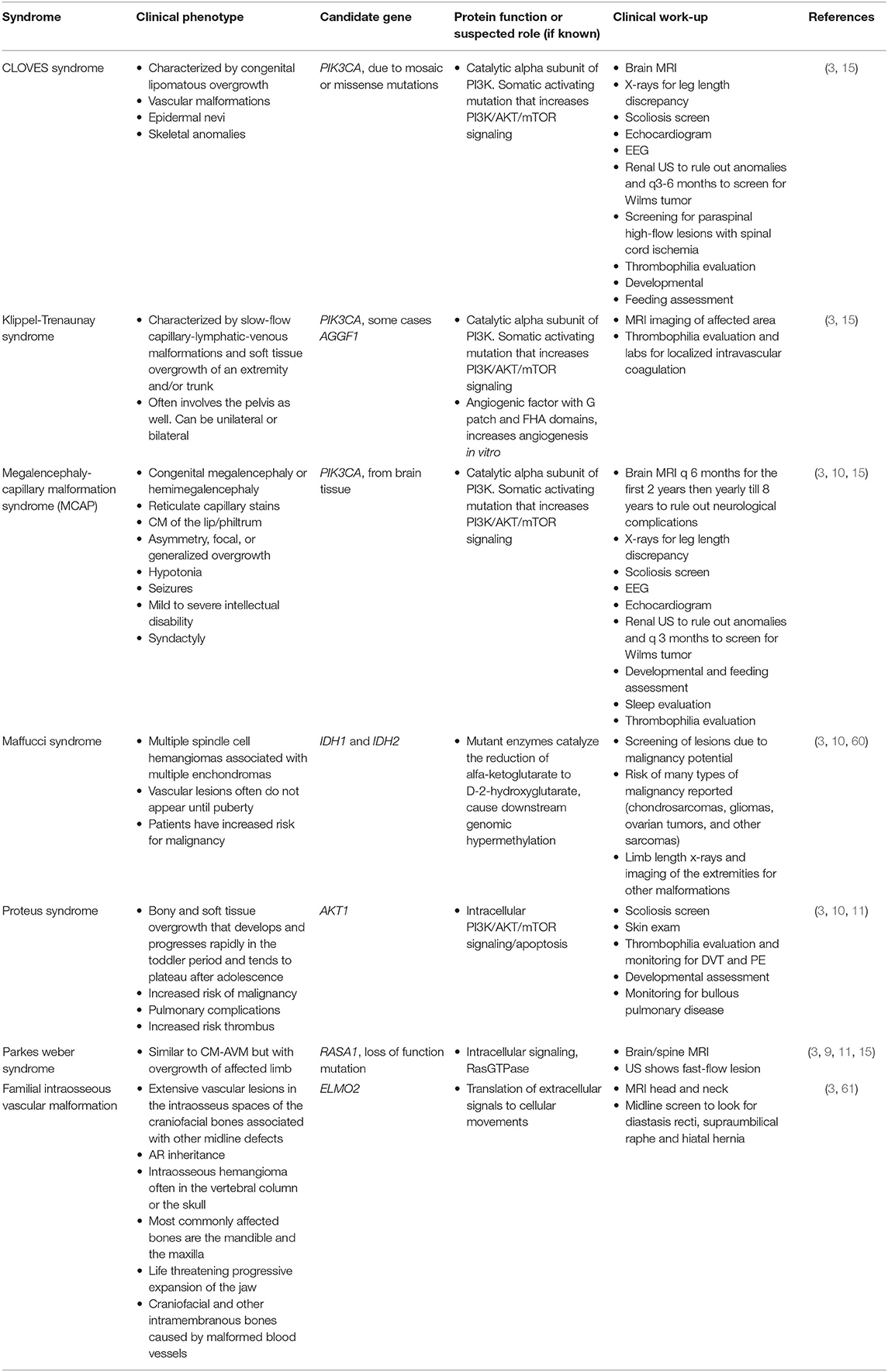
Table 9. Syndromes associated with vascular anomalies.
Vascular Tumors
Vascular tumors include both benign, locally aggressive, and malignant tumors comprised of one or more vascular components (7, 62). The molecular etiology of many of these tumors is unknown, but some candidate genes have recently been identified (Table 10). Infantile hemangiomas are the most common vascular tumor, and are generally benign, but growth can be disruptive of function or cosmesis, and occasionally life-threatening. The role of the pediatrician is key to determining which lesions require referral for additional management. Vascular tumors other than hemangiomas often require multi-modal therapy, including chemotherapy, and consultation with a pediatric hematologist-oncologist is indicated.
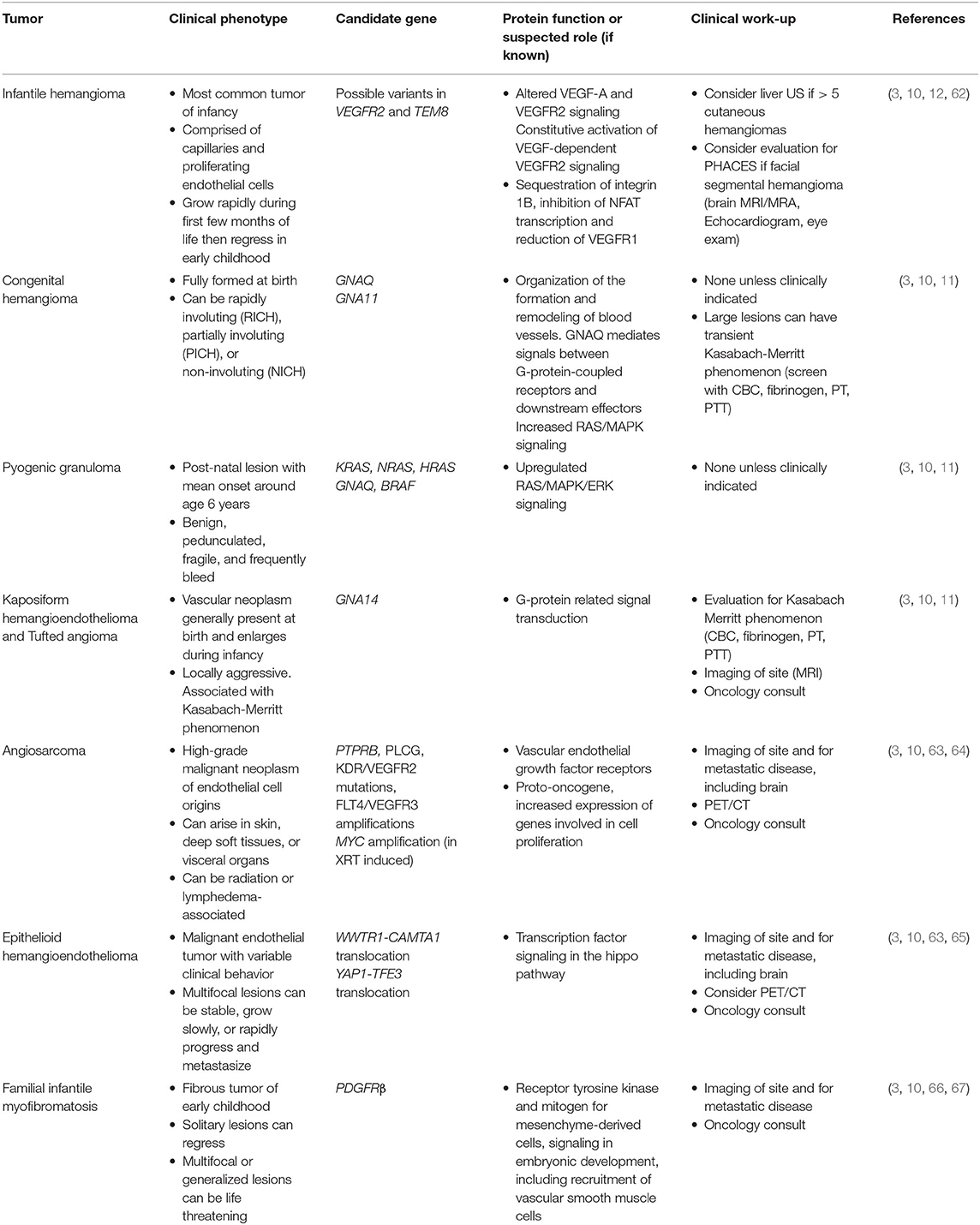
Table 10. Vascular tumors.
Discussion
It is critical for teams treating patients with vascular anomalies to have a basic understanding of the genetic changes underlying vascular anomalies. Genetic results have implications for patient management, screening, and ultimately treatment.
As we begin to understand more about the genetic underpinnings of vascular anomalies, we identify potential therapeutic targets that will ultimately change treatment paradigms. Prior to the discovery of the underlying molecular pathways involved in the pathogenesis of vascular malformations and tumors, medical management was based primarily on symptom management with antiplatelet and anticoagulant agents to control localized intravascular coagulation and platelet activation, and pain medications to alleviate symptoms (1, 5, 8, 9). Over the years, multiple medications, primarily based on adaptation of oncologic agents, have been utilized in patients with complex and life-threatening vascular anomalies. This includes drugs such as bevacizumab, interferon-alpha, and cyclophosphamide, that have come with their own significant risks and side effects and generally can be considered sledgehammer rather than target approaches (68–70). Even propranolol's efficacy in infantile hemangiomas was identified serendipitously in children who received the drug for the cardiac complications they developed due to their massive infantile hemangiomas (71). Despite its successful use, the mechanism and relationship of propranolol to vascular growth pathways remains to be elucidated (72–74).
In the early 2000s, several groups began to report on the importance of PI3K/AKT/MAPK, and TGF-β signaling in the pathogenesis of vascular malformations (Figure 1) (75–78). Based on the efficacy of sirolimus, an mTOR inhibitor, in angiomyolipomas associated with tuberous sclerosis complex (79, 80) and Kaposi sarcoma (81), sirolimus was first used in a child with Kaposiform hemangioendothelioma in 2010 (82). Shortly after, a small case series of children with complex vascular malformations and tumors was published, in which all patients demonstrated a significant response and improvement with sirolimus (68). This prompted the first prospective clinical trial that was published in 2016 and confirmed both the efficacy and safety of Sirolimus in the treatment of complex vascular anomalies (83). With the identification of the PI3K/AKT/mTOR pathway as an important driver of somatic overgrowth syndromes (84) and the identification of somatic PI3K mutations within certain vascular malformations (41, 85, 86), PI3K inhibition has become the next major therapeutic target for vascular anomalies (87–89). Future targets will likely be aimed at the RAS/MAPK pathway, where somatic mutations are known to be important in AVMs, capillary malformations, and other aggressive vascular syndromes (10, 44, 55). A list of targeted therapies currently under investigation or in use for vascular anomalies is in Table 11. These discoveries will likely continue to grow at a rapid pace and ultimately inform our classification and management of patients in the clinic. For this reason, it is key for burgeoning vascular anomalies clinical teams to include geneticists as key team members for discovery as well as patient management.
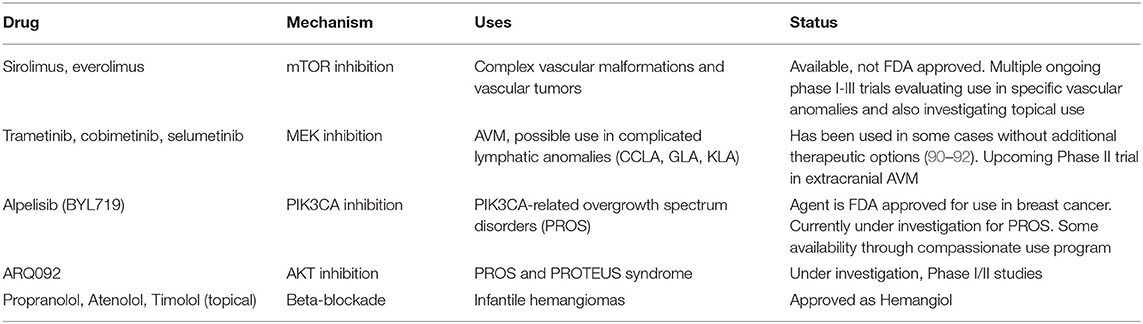
Table 11. Current targeted therapies in-use or under investigation for vascular anomalies.
Trained geneticists and genetic counselors are crucial members of a comprehensive multidisciplinary vascular anomalies team. Detailed discussion of family history, consideration of genetic testing, and review of results is an important part of the comprehensive care of patients with vascular malformations and tumors. Understanding which lesions are likely to be associated with somatic vs. germline changes is key to developing a thoughtful approach to genetic testing. A family history suggestive of an inherited mutation will prompt consideration of germline testing, which can readily be performed by routine phlebotomy. Somatic mutation testing requires sampling the affected tissue, which for most patients requires a skin biopsy with appropriate tissue handling after biopsy is performed. The decision about which genes to screen for and which laboratory to use is made based on a combination of experience, cost, and insurance approval. Current technology available in the field of cancer genomics is likely to be useful as we attempt to further define the genetics of vascular anomalies.(93). Members of the genetics and genetic counseling teams are key to providing input on which testing to send as well as interpretation of results. Family should be prepared to receive either definitive positive or negative results, or commonly a variant of uncertain significance with a more nuanced interpretation. Help with interpretation and contextualization of variants of uncertain significance by a geneticist is key as we begin to understand more of the molecular mechanisms and genetic changes in these malformations and tumors. The role of the geneticist and the importance of genetic testing will become increasingly important as the field of vascular anomalies continues to grow, offering new insights into the etiology of these malformations and tumors, and new avenues for improvement in patient care.
Author Contributions
JD conceived and wrote the manuscript together with AB. DA, FB, and TN contributed critical revision of the article, patient photos for the figure, and final approval of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the patients and families from whom we have gained this knowledge as a community of physicians. We also thank the patients who gave permission for publication of their photographs.
References
1. Mulliken JB, Glowacki J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg. (1982) 69:412–22. doi: 10.1097/00006534-198203000-00003
2. Cox JA, Bartlett E, Lee EI. Vascular malformations: a review. Semin Plast Surg. (2014) 28:58–63. doi: 10.1055/s-0034-1376263
3. Mulliken JB, Burrows PE, Fishman SJ. Mulliken and Young's Vascular Anomalies Hemangiomas and Malformations. 2nd ed. New York, NY: University Press (2013). doi: 10.1093/med/9780195145052.001.0001
4. Enjolras O. Classification and management of the various superficial vascular anomalies: hemangiomas and vascular malformations. J Dermatol. (1997) 24:701–10. doi: 10.1111/j.1346-8138.1997.tb02522.x
5. Enjolras O, Mulliken JB. Vascular tumors and vascular malformations (new issues). Adv Dermatol. (1997) 13:375–423.
6. ISSVA Classification of Vascular Anomalies ©2018. International Society for the Study of Vascular Anomalies (2018). Available online at: “issva.org/classification” (accessed April 23, 2020).
7. Wassef M, Blei F, Adams D, Alomari A, Baselga E, Berenstein A, et al. Vascular anomalies classification: recommendations from the international society for the study of vascular anomalies. Pediatrics. (2015) 136:e203–14. doi: 10.1542/peds.2014-3673
8. Anusic S, Clemens RKJ, Meier TO, Amann-Vesti BR. Assessment of PTEN-associated vascular malformations in a patient with Bannayan-Riley-Ruvalcaba syndrome. Case Rep. (2016) 2016:bcr2016215188. doi: 10.1136/bcr-2016-215188
9. Limaye N, Boon LM, Vikkula M. From germline towards somatic mutations in the pathophysiology of vascular anomalies. Hum Mol Genet. (2009) 18:R65–R74. doi: 10.1093/hmg/ddp002
10. Greene AK, Goss JA. Vascular anomalies: from a clinicohistologic to a genetic framework. Plast Reconstr Surg. (2018) 141:709e–17e. doi: 10.1097/PRS.0000000000004294
11. Adams DM. Practical genetic and biologic therapeutic considerations in vascular anomalies. Tech Vasc Interv Radiol. (2019) 22:100629. doi: 10.1016/j.tvir.2019.100629
12. Queisser A, Boon LM, Vikkula M. Etiology and genetics of congenital vascular lesions. Otolaryngol Clin North Am. (2018) 51:41–53. doi: 10.1016/j.otc.2017.09.006
13. Nguyen H-L, Boon LM, Vikkula M. Vascular anomalies caused by abnormal signaling within endothelial cells: targets for novel therapies. Semin Intervent Radiol. (2017) 34:233–8. doi: 10.1055/s-0037-1604296
14. Couto RA, Greene AK. Capillary Malformations. In: Rahbar R, Rodriguez-Galindo C, Meara JG, Smith ER, Perez-Atayde AR, editors. Pediatric Head and Neck Tumors: A-Z Guide to Presentation and Multimodality Management. New York, NY: Springer. (2014). p. 73–9.
15. Nguyen H-L, Boon LM, Vikkula M. Genetics of vascular malformations. Semin Pediatr Surg. (2014) 23:2216. doi: 10.1053/j.sempedsurg.2014.06.014
16. Dedania VS, Moinuddin O, Lagrou LM, Sathrasala S, Cord Medina FM, Del Monte MA, et al. Ocular manifestations of cutis marmorata telangiectatica congenita. Ophthalmol Retina. (2019) 3:791–801. doi: 10.1016/j.oret.2019.03.025
18. Thomas AC, Zeng Z, Rivière JB, O'Shaughnessy R, Al-Olabi L, St-Onge J, et al. Mosaic activating mutations in gna11 and gnaq are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J Invest Dermatol. (2016) 136:770–8. doi: 10.1016/j.jid.2015.11.027
19. Villarreal DJV, Leal F. Phacomatosis pigmentovascularis of cesioflammea type. An Bras Dermatol. (2016) 91(5 Suppl. 1):54–6. doi: 10.1590/abd1806-4841.20164516
20. Revencu N, Fastre E, Ravoet M, Helaers R, Brouillard P, Bisdorff-Bresson A, et al. RASA1 mosaic mutations in patients with capillary malformation-arteriovenous malformation. J Med Genet. (2020) 57:48–52. doi: 10.1136/jmedgenet-2019-106024
21. Wooderchak-Donahue WL, Johnson P, McDonald J, Blei F, Berenstein A, Sorscher M, et al. Expanding the clinical and molecular findings in RASA1 capillary malformation-arteriovenous malformation. Eur J Hum Genet. (2018) 26:1521–36. doi: 10.1038/s41431-018-0196-1
22. Gordon Spratt EA, DeFelice T, O'Reilly K, Robinson M, Patel RR, Sanchez M. Generalized essential telangiectasia. Dermatol Online J. (2012) 18:13. doi: 10.1172/JCI98589
23. Anjaneyan G, Kaliyadan F. Angioma Serpiginosum. Treasure Island, FL: StatPearls Publishing (2020).
24. Wellman RJ, Cho SB, Singh P, Tune M, Pardo CA, Comi AM, et al. Gαq and hyper-phosphorylated ERK expression in sturge-weber syndrome leptomeningeal blood vessel endothelial cells. Vasc Med. (2019) 24:72–5. doi: 10.1177/1358863X18786068
25. Sundaram SK, Michelhaugh SK, Klinger NV, Kupsky WJ, Sood S, Chugani HT, et al. GNAQ mutation in the venous vascular malformation and underlying brain tissue in sturge-weber syndrome. Neuropediatrics. (2017) 48:385–9. doi: 10.1055/s-0037-1603515
26. Boon LM, Mulliken JB, Enjolras O, Vikkula M. Glomuvenous malformation (glomangioma) and venous malformation: distinct clinicopathologic and genetic entities. Arch Dermatol. (2004) 140:971–6. doi: 10.1001/archderm.140.8.971
27. Da Costa L, O' Donohue MF, van Dooijeweert B, Albrecht K, Unal S. Molecular approaches to diagnose diamond-blackfan anemia: the EuroDBA experience. Eur J Med Genet. (2018) 61:664–73. doi: 10.1016/j.ejmg.2017.10.017
28. Vikkula M, Boon LM, Mulliken JB. Molecular genetics of vascular malformations. Matrix Biol. (2001) 20:327–35. doi: 10.1016/S0945-053X(01)00150-0
29. Couto JA, Vivero MP, Kozakewich HP, Taghinia AH, Mulliken JB, Warman ML, et al. A somatic MAP3K3 mutation is associated with verrucous venous malformation. Am J Hum Genet. (2015) 96:480. doi: 10.1016/j.ajhg.2015.01.007
30. Soblet J, Kangas J, Nätynki M, Mendola A, Helaers R, Uebelhoer M, et al. Blue Rubber Bleb Nevus (BRBN) syndrome is caused by somatic TEK (TIE2) mutations. J Invest Dermatol. (2017) 137:207–16. doi: 10.1016/j.jid.2016.07.034
32. Tan WH, Baris HN, Burrows PE, Robson CD, Alomari AI, Mulliken JB, et al. The spectrum of vascular anomalies in patients with PTEN mutations: implications for diagnosis and management. J Med Genet. (2007) 44:594–602. doi: 10.1136/jmg.2007.048934
33. Eng C. PTEN hamartoma tumor syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, and Amemiya A. editors. GeneReviews®. University of Washington, Seattle (1993).
34. Pilarski R. PTEN hamartoma tumor syndrome: a clinical overview. Cancers. (2019) 11:844. doi: 10.3390/cancers11060844
35. Limaye N, Wouters V, Uebelhoer M, Tuominen M, Wirkkala R, Mulliken JB, et al. Somatic mutations in the angiopoietin-receptor TIE2 can cause both solitary and multiple sporadic venous malformations. Nat Genet. (2009) 41:118–24. doi: 10.1038/ng.272
36. Morrison L, Akers A. Cerebral cavernous malformation, familial. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, and Amemiya A. editors. GeneReviews®. Seattle, WA: University of Washington (1993)
37. Brouillard P, Boon LM, Mulliken JB, Enjolras O, Ghassibé M, Warman ML, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am J Hum Genet. (2002) 70:866–74. doi: 10.1086/339492
38. Eerola I, Plate KH, Spiegel R, Boon LM, Mulliken JB, Vikkula M. KRIT1 is mutated in hyperkeratotic cutaneous capillary-venous malformation associated with cerebral capillary malformation. Hum Mol Genet. (2000) 9:1351–5. doi: 10.1093/hmg/9.9.1351
39. Sirvente J, Enjolras O, Wassef M, Tournier-Lasserve E, Labauge P. Frequency and phenotypes of cutaneous vascular malformations in a consecutive series of 417 patients with familial cerebral cavernous malformations. J Eur Acad Dermatol Venereol. (2009) 23:1066–72. doi: 10.1111/j.1468-3083.2009.03263.x
40. Mellor RH, Brice G, Stanton AW, French J, Smith A, Jeffery S, et al. Mutations in FOXC2 are strongly associated with primary valve failure in veins of the lower limb. Circulation. (2007) 115:1912–20. doi: 10.1161/CIRCULATIONAHA.106.675348
41. Limaye N, Kangas J, Mendola A, Godfraind C, Schlögel MJ, Helaers R, et al. Somatic activating PIK3CA mutations cause venous malformation. Am J Hum Genet. (2015) 97:914–21. doi: 10.1016/j.ajhg.2015.11.011
42. Rodriguez-Laguna L, Agra N, Ibañez K, Oliva-Molina G, Gordo G, Khurana N, et al. Somatic activating mutations in PIK3CA cause generalized lymphatic anomaly. J Exp Med. (2019) 216:407–18. doi: 10.1084/jem.20181353
43. Lala S, Mulliken JB, Alomari AI, Fishman SJ, Kozakewich HP, Chaudry G. Gorham-Stout disease and generalized lymphatic anomaly–clinical, radiologic, and histologic differentiation. Skeletal Radiol. (2013) 42:917–24. doi: 10.1007/s00256-012-1565-4
44. Barclay SF, Inman KW, Luks VL, McIntyre JB, Al-Ibraheemi A, Church AJ, et al. A somatic activating NRAS variant associated with kaposiform lymphangiomatosis. Genet Med. (2019) 21:1517–24. doi: 10.1038/s41436-018-0390-0
45. Ozeki M, Nozawa A, Kawamoto N, Fujino A, Hirakawa S, Fukao T. Potential biomarkers of kaposiform lymphangiomatosis. Pediatr Blood Cancer. (2019) 66:e27878. doi: 10.1002/pbc.27878
46. Brouillard P, Boon L, Vikkula M. Genetics of lymphatic anomalies. J Clin Invest. (2014) 124:898–904. doi: 10.1172/JCI71614
47. Joyce S, Gordon K, Brice G, Ostergaard P, Nagaraja R, Short J, et al. The lymphatic phenotype in noonan and cardiofaciocutaneous syndrome. Eur J Hum Genet. (2016) 24:690–6. doi: 10.1038/ejhg.2015.175
48. Greenlee R, Hoyme H, Witte M, Crowe P, Witte C. Developmental disorders of the lymphatic system. Lymphology. (1993) 26:156–68.
49. Mansour S, Brice GW, Jeffery S, Mortimer P. Lymphedema-Distichiasis Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. editors. GeneReviews®. Seattle, WA: University of Washington (1993)
50. Alders M, Al-Gazali L, Cordeiro I, Dallapiccola B, Garavelli L, Tuysuz B, et al. Hennekam syndrome can be caused by FAT4 mutations and be allelic to van maldergem syndrome. Hum Genet. (2014) 133:1161–7. doi: 10.1007/s00439-014-1456-y
51. Jeltsch M, Jha SK, Tvorogov D, Anisimov A, Leppänen VM, Holopainen T, et al. CCBE1 enhances lymphangiogenesis via a disintegrin and metalloprotease with thrombospondin motifs-3-mediated vascular endothelial growth factor-c activation. Circulation. (2014) 129:1962–71. doi: 10.1161/CIRCULATIONAHA.113.002779
52. Scheuerle AE, Sweed NT, Timmons CF, Smith ED, Alcaraz WA, Shinde DN. An additional case of Hennekam lymphangiectasia-lymphedema syndrome caused by loss-of-function mutation in ADAMTS3. Am J Med Genet A. (2018) 176:2858–61. doi: 10.1002/ajmg.a.40633
53. McReynolds LJ, Calvo KR, Holland SM. Germline GATA2 mutation and bone marrow failure. Hematol Oncol Clin North Am. (2018) 32:713–28. doi: 10.1016/j.hoc.2018.04.004
54. Couto JA, Huang AY, Konczyk DJ, Goss JA, Fishman SJ, Mulliken JB, et al. Somatic MAP2K1 mutations are associated with extracranial arteriovenous malformation. Am J Hum Genet. (2017) 100:546–54. doi: 10.1016/j.ajhg.2017.01.018
55. Al-Olabi L, Polubothu S, Dowsett K, Andrews KA, Stadnik P, Joseph AP, et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J Clin Invest. (2018) 128:1496–508. doi: 10.1016/j.jid.2018.03.765
56. Goss JA, Huang AY, Smith E, Konczyk DJ, Smits PJ, Sudduth CL, et al. Somatic mutations in intracranial arteriovenous malformations. PLoS ONE. (2019) 14:e0226852. doi: 10.1371/journal.pone.0226852
57. Starke RM, McCarthy D, Komotar RJ, Connolly ES. Somatic KRAS mutation found in sporadic arteriovenous malformations. Neurosurgery. (2018) 83:E14–5. doi: 10.1093/neuros/nyy163
58. Kritharis A, Al-Samkari H, Kuter DJ. Hereditary hemorrhagic telangiectasia: diagnosis and management from the hematologist's perspective. Haematologica. (2018) 103:1433–43. doi: 10.3324/haematol.2018.193003
59. McDonald J, Pyeritz RE. Hereditary hemorrhagic telangiectasia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, and Amemiya A. editors. GeneReviews®. University of Washington, Seattle (1993). Available online at: http://www.ncbi.nlm.nih.gov/books/NBK1351/ (accessed October 25, 2018).
60. McGarry ME. Long term oncologic surveillance in maffucci syndrome: a case report. J Oncol Sci. (2017) 3:140–4. doi: 10.1016/j.jons.2017.08.003
61. Cetinkaya A, Xiong JR, Vargel I, Kösemehmetoglu K, Canter HI, Gerdan ÖF, et al. Loss-of-function mutations in ELMO2 cause intraosseous vascular malformation by Impeding Rac1 signaling. Am J Hum Genet. (2016) 99:299–317. doi: 10.1016/j.ajhg.2016.06.008
62. Ricci KW. Advances in the medical management of vascular anomalies. Semin Intervent Radiol. (2017) 34:239–49. doi: 10.1055/s-0037-1604297
63. Cioffi A, Reichert S, Antonescu CR, Maki RG. Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am. (2013) 27:975–88. doi: 10.1016/j.hoc.2013.07.005
64. Behjati S, Tarpey PS, Sheldon H, Martincorena I, Van Loo P, Gundem G, et al. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat Genet. (2014) 46:376–9. doi: 10.1038/ng.2921
65. Sardaro A, Bardoscia L, Petruzzelli MF, Portaluri M. Epithelioid hemangioendothelioma: an overview and update on a rare vascular tumor. Oncol Rev. (2014) 8:259. doi: 10.4081/oncol.2014.259
66. Cheung YH, Gayden T, Campeau PM, LeDuc CA, Russo D, Nguyen VH, et al. A recurrent pdgfrb mutation causes familial infantile myofibromatosis. Am J Hum Genet. (2013) 92:996–1000. doi: 10.1016/j.ajhg.2013.04.026
67. Zhao G, Zhu M, Qin C, Liu X, Zhao X. Infantile myofibromatosis: 32 patients and review of the literatures. J Pediatr Hematol Oncol. (2020). doi: 10.1097/MPH.0000000000001603
68. Hammill AM, Wentzel M, Gupta A, Nelson S, Lucky A, Elluru R, et al. Sirolimus for the treatment of complicated vascular anomalies in children. Pediatric Blood Cancer. (2011) 57:1018–24. doi: 10.1002/pbc.23124
69. Reinhardt MA, Nelson SC, Sencer SF, Bostrom BC, Kurachek SC, Nesbit ME. Treatment of Childhood Lymphangiomas With Interferon-α. J Pediatr Hematol Oncol. (1997) 19:232–6. doi: 10.1097/00043426-199705000-00010
70. Dickerhoff R, Bode VU. Cyclophosphamide in non-resectable cystic hygroma. Lancet. (1990) 335:1474–5. doi: 10.1016/0140-6736(90)91512-9
71. Léauté-Labrèze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taïeb A. Propranolol for severe hemangiomas of infancy. N Engl J Med. (2008) 358:2649–51. doi: 10.1056/NEJMc0708819
72. Hagen R, Ghareeb E, Jalali O, Zinn Z. Infantile hemangiomas: what have we learned from propranolol? Curr Opin Pediatr. (2018) 30:499–504. doi: 10.1097/MOP.0000000000000650
73. Zhao F, Yang X, Xu G, Bi J, Lv R, Huo R. Propranolol suppresses HUVEC viability, migration, VEGF expression, and promotes apoptosis by downregulation of miR-4295. J Cell Biochem. (2019) 120:6614–23. doi: 10.1002/jcb.27957
74. Sun B, Dong C, Lei H, Gong Y, Li M, Zhang Y, et al. Propranolol inhibits proliferation and invasion of hemangioma-derived endothelial cells by suppressing the DLL4/Notch1/Akt pathway. Chem Biol Interact. (2018) 294:28–33. doi: 10.1016/j.cbi.2018.08.018
75. Shirazi F, Cohen C, Fried L, Arbiser JL. Mammalian target of rapamycin (mTOR) is activated in cutaneous vascular malformations in vivo. Lymphatic Res Biol. (2007) 5:233–6. doi: 10.1089/lrb.2007.1012
76. Phung TL, Ziv K, Dabydeen D, Eyiah-Mensah G, Riveros M, Perruzzi C, et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell. (2006) 10:159–70. doi: 10.1016/j.ccr.2006.07.003
77. Perry B, Banyard J, McLaughlin ER, Watnick R, Sohn A, Brindley DN, et al. AKT1 overexpression in endothelial cells leads to the development of cutaneous vascular malformations in vivo. Arch Dermatol. (2007) 143:504–6. doi: 10.1001/archderm.143.4.504
78. Arbiser JL, Weiss SW, Arbiser ZK, Bravo F, Govindajaran B, Caceres-Rios H, et al. Differential expression of active mitogen-activated protein kinase in cutaneous endothelial neoplasms: implications for biologic behavior and response to therapy. J Am Acad Dermatol. (2001) 44:193–7. doi: 10.1067/mjd.2000.111632
79. Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard J, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. (2008) 358:140–51. doi: 10.1056/NEJMoa063564
80. Herry I, Neukirch C, Debray MP, Mignon F, Crestani B. Dramatic effect of sirolimus on renal angiomyolipomas in a patient with tuberous sclerosis complex. Eur J Intern Med. (2007) 18:76–7. doi: 10.1016/j.ejim.2006.07.017
81. Stallone G, Schena A, Infante B, Di Paolo S, Loverre A, Maggio G, et al. Sirolimus for kaposi's sarcoma in renal-transplant recipients. N Engl J Med. (2005) 352:1317–23. doi: 10.1056/NEJMoa042831
82. Blatt J, Stavas J, Moats-Staats B, Woosley J, Morrell DS. Treatment of childhood kaposiform hemangioendothelioma with sirolimus. Pediatr Blood Cancer. (2010) 55:1396–8. doi: 10.1002/pbc.22766
83. Adams DM, Trenor CC, Hammill AM, Vinks AA, Patel MN, Chaudry G, et al. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics. (2016) 137:e20153257. doi: 10.1542/peds.2015-3257
84. Keppler-Noreuil KM, Parker VE, Darling TN, Martinez-Agosto JA. Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am J Med Genet Part C. (2016) 172:402–21. doi: 10.1002/ajmg.c.31531
85. Castel P, Carmona FJ, Grego-Bessa J, Berger MF, Viale A, Anderson KV, et al. Somatic PIK3CA mutations as a driver of sporadic venous malformations. Sci Transl Med. (2016) 8:332ra42. doi: 10.1126/scitranslmed.aaf1164
86. Kurek KC, Luks VL, Ayturk UM, Alomari AI, Fishman SJ, Spencer SA, et al. Somatic mosaic activating mutations in PIK3CA Cause CLOVES Syndrome. Am J Hum Genet. (2012) 90:1108–15. doi: 10.1016/j.ajhg.2012.05.006
87. Venot Q, Blanc T, Rabia SH, Berteloot L, Ladraa S, Duong JP, et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature. (2018) 558:540. doi: 10.1038/s41586-018-0217-9
88. di Blasio L, Puliafito A, Gagliardi PA, Comunanza V, Somale D, Chiaverina G, et al. PI3K/mTOR inhibition promotes the regression of experimental vascular malformations driven by PIK3CA -activating mutations. Cell Death Dis. (2018) 9:1–15. doi: 10.1038/s41419-017-0064-x
89. Castillo S, Baselga E, Graupera M. PIK3CA mutations in vascular malformations. Curr Opin Hematol. (2019) 26:170–8. doi: 10.1097/MOH.0000000000000496
90. Lekwuttikarn R, Lim YH, Admani S, Choate KA, Teng JMC. Genotype-guided successful treatment of an arteriovenous malformation in a child. JAMA Dermatol. (2019) 155:256–7. doi: 10.1001/jamadermatol.2018.4653
91. Maynard K, LoPresti M, Iacobas I, Kan P, Lam S. Antiangiogenic agent as a novel treatment for pediatric intracranial arteriovenous malformations: case report. J Neurosurg Pediatr. (2019) 24:673–9. doi: 10.3171/2019.7.PEDS1976
92. Li D, March ME, Gutierrez-Uzquiza A, Kao C, Seiler C, Pinto E, et al. ARAF recurrent mutation causes central conducting lymphatic anomaly treatable with a MEK inhibitor. Nat Med. (2019) 25:1116–22. doi: 10.1038/s41591-019-0479-2
Keywords: vascular malformations, genetic, mutation, anomalies, somatic
Citation: Borst AJ, Nakano TA, Blei F, Adams DM and Duis J (2020) A Primer on a Comprehensive Genetic Approach to Vascular Anomalies. Front. Pediatr. 8:579591. doi: 10.3389/fped.2020.579591
Received: 02 July 2020; Accepted: 27 August 2020;
Published: 19 October 2020.
Edited by:
Andrew Landstrom, Duke University, United StatesReviewed by:
Vinicio De Jesus Perez, Stanford University, United StatesFan Jin, Zhejiang University, China
Copyright © 2020 Borst, Nakano, Blei, Adams and Duis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandra J. Borst, YWxleGFuZHJhLmJvcnN0QHZ1bWMub3Jn; Jessica Duis, amVzc2ljYS5kdWlzQGNoaWxkcmVuc2NvbG9yYWRvLm9yZw==