 Yi-ling Dai
Yi-ling Dai Xue Tang1,2†
Xue Tang1,2† Xia Guo
Xia Guo- 1Department of Pediatrics, West China Second University Hospital, Sichuan University, Sichuan, China
- 2Key Laboratory of Birth Defects and Related Diseases of Women and Children, Sichuan University, Ministry of Education, Sichuan, China
Hereditary thrombotic thrombocytopenic purpura (TTP) is caused by ADAMTS13 mutations with autosomal recessive inheritance. It typically presents during childhood and is frequently misdiagnosed as immune thrombocytopenia. We present a case of hereditary TTP with an undescribed compound heterozygous ADAMTS13 mutation in a Chinese boy. A 12-year-old boy with a history of intermittent thrombocytopenia in the prior 6 years had severe deficiency of plasma ADAMTS13 and harbored a novel compound heterozygous mutation which was also identified in his sister. The c.577C>T was a pathogenic variant reported exclusively in Japanese cases. The undescribed c.2397C>A non-sense mutation was predicted to encode a truncated protein. Identification of the specific novel heterozygous ADAMTS13 mutation in the Chinese family, consisting a variant restricted to Asian individuals and an undescribed c.2397C>A non-sense mutation, demonstrates genetic diversity underlying hereditary TTP, and possibly ethnic skewed mutation profiles.
Background
Hereditary thrombotic thrombocytopenic purpura (TTP), also known as Upshaw–Schulman syndrome (USS), is the congenital form of TTP, with an annual incidence of less than 1 per million (1), caused by mutations of the ADAMTS13 (A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13) gene encoding von Willebrand factor (vWF)-cleaving protease (2, 3). ADAMTS13 mutations with resultant reduced synthesis or increased clearance lead to severe deficiency of ADAMTS13, failure of cleavage of ultralarge VWF multimers, platelet hyperaggregation, and consumptive thrombocytopenia (4, 5). USS and acquired immune-mediated TTP are both characterized by Coombs test-negative microangiopathic hemolytic anemia, consumptive thrombocytopenia, and a variety of organ damages, and both are categorized under the heading of thrombotic microangiopathy (TMA) (6, 7). Over 200 mutations associated with USS have been reported, which differ in different geographical areas and ethnic groups (1). Here, we present a novel compound heterozygous mutation which has not been described, in a Chinese boy and his older sister.
Case Report
The patient, a 12-year-old boy, firstly presented with petechiae over his trunk and extremities for no obvious reasons at age of six. Isolated thrombocytopenia was revealed with platelet counts ranging from 20 to 30 × 109/L in local hospitals. A diagnosis of primary immune thrombocytopenia (ITP) was made, and oral steroid therapy was prescribed for several months. He had remained symptom-free for 4 years, and his platelet counts were within the normal ranges thereafter. Then, he suffered several acute attacks of severe thrombocytopenia (lowest platelet count of 5.0 × 109/L) with concomitant anemia (lowest Hb concentration being 62 g/L), without renal symptoms, neurological symptoms, headaches, abdominal pain, and lethargy, typically triggered by upper respiratory infections. Bone marrow aspiration showed markedly increased megakyrocytes and maturation arrest. Diagnoses of primary ITP and bleeding anemia were made and steroid, intravenous immunoglobulin, blood, and platelet transfusions were given when indicated in other hospitals, but platelet counts were still below normal. The patient presented to our hospital on 31 December 2018, because of thrombocytopenia (platelet count 1.0 × 109/L) and epitaxis. Careful history-taking and relevant laboratory investigations strongly suggested the possibility of thrombotic microangiopathy, because the patient suffered thrombocytopenia, anemia which could not be explained by hemorrhage, conspicuous reticulocytosis (15.2%), and remarkably elevated LDH levels (2,357 IU/L) during acute attacks. Coombs test, DIC screening tests, and liver and renal functions were negative; urinary trace proteins were positive. His old sister suffered thrombocytopenia at 5 years of age but remained disease-free thereafter. Parental consanguinity was denied. Massive plasma infusions (3,600 mL during the 4-day therapy) were given on an emergency basis, which successfully stopped epitaxis, and platelet count was normalized on the third day. Plasma ADAMTS13 enzymatic activity assayed by the SELDI-TOF (surface-enhanced laser resorption/ionization-time of flight) method in a third-party laboratory after the plasma infusion (600 mL) was 32.1%, while the platelet count was 86 × 109/L. A repeated assay 2 weeks after the last plasma infusion returned an enzymatic activity of <5%, and the platelet count was 43 × 109/L. Assay of ADAMTS13 autoantibody was not available at the time of initial diagnosis of USS.
Mutation analysis revealed that both the patient and his sister harbored the same compound heterozygous ADAMTS13 mutation: c.2397C>A and c.577C>T, inherited from his mother and father, respectively (Figure 1). The pathogenic c.577C>T (p.R193W) variant within exon 6 had been previously reported (8, 9). Molecular modeling analysis using the SWISS-MODEL indicated that tryptophan (a basic amino acid with molecular weight of 204) in substitution for arginine (an aromatic amino acid, with molecular weight of 174) changed the molecular weight, polarization, and folding, probably leading to accelerated protein degradation (Figure 2). While the novel c.2397C>A (p.C799X), a previously undescribed non-sense mutation, created a stop codon and encoded a truncated ADAMTS13 protein. Therefore, a definitive diagnosis of USS caused by a novel compound heterozygous mutation in this Chinese boy was firmly established.
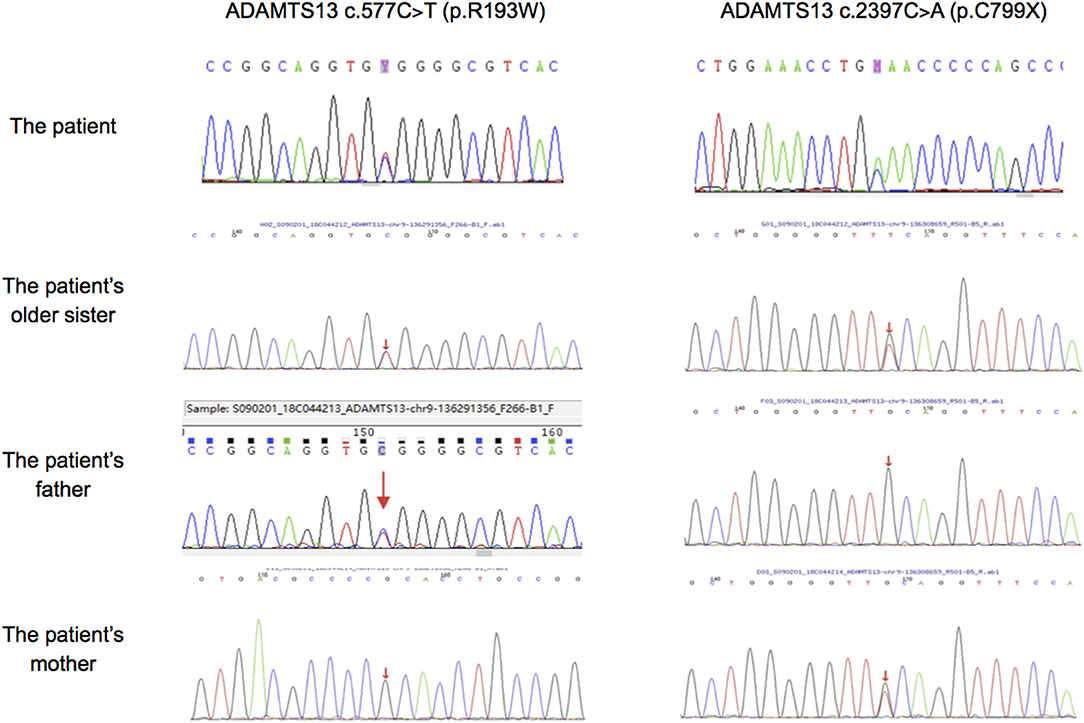
Figure 1. Sanger sequencing results in ADAMTS13 of our patient, his older sister and his parents. The patient, his older sister and his farther had mutation of c.577C>T, while the patient, his older sister and his mother had mutation of c.2397C>A.
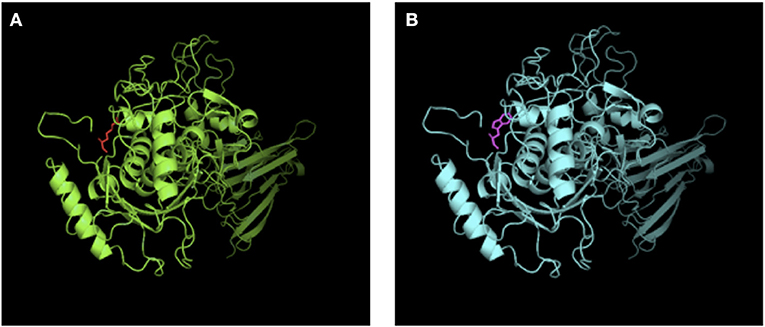
Figure 2. SWISS-MODEL-predicted structures of ADAMST13WT (A) and ADAMST13 p.R193W (B). Tryptophan (a basic amino acid with molecular weight of 204) in substitution for arginine (an aromatic amino acid, with molecular weight of 174) changed the molecular weight, polarization and folding, probably leading to accelerated protein degradation.
Discussion
Hereditary thrombotic thrombocytopenic purpura (TTP), or Upshaw–Schulman syndrome (USS), is an exceedingly rare hereditary disorder, which presents primarily in childhood, constituting up to 5% of TTP (1, 9). Genetically, USS is caused by ADAMTS13 gene mutations in an autosomal recessive mode of inheritance and is more prevalent in families of consanguinity. Biochemically, USS is characterized by severe constitutional deficiency of plasma ADAMTS13, generally less than 10% of normal, which results in accumulation of hyperadhesive ultra-large vWF (ULVWF) multimers and triggers activation of the VWF-platelet hemostatic pathway, culminating in platelet-ULVWF microthrombi formation within microcirculation (3, 6), and currently categorized as ADAMTS13 gene mutation-associated vascular microthrombotic disease (VMTD) (10, 11).
More than 200 ADAMTS13 mutations spreading in all protein domains have been identified in patients with USS, with splicing mutations being increasingly reported (1). Nevertheless, type and frequency of genotypes are highly diverse, with some specific gene variants particularly common in certain ethnic populations. Mutation analysis of a German cohort with 30 USS patients identified c.4143_4144dupA (p.Glu1382Arg fs*6) homozygotes in 4 patients and heterozygotes in another 10 patients (12), suggesting a common mutation origin in individuals of northern European ancestry (13). Similarly, the allelic variant c.577C>T (p.R193W) in our index patient was exclusively identified in Japanese patients enrolled in the International Hereditary Thrombotic Thrombocytopenic Purpura Registry and the most prevalent pathogenic mutation in Asia (14), illustrating genetic-background skewed mutation profiling.
USS has been rarely reported in China, which is not a participating country of the International USS registry. With extensive literature search, we just identified a couple of case reports and a small 5-case study in children with genetically confirmed USS, the largest case report in China (15). Nevertheless, the novel mutation of our index patient has not been described in USS patients.
ADAMTS13 deficiency could result from reduced synthesis, impaired proteolytic activity, or increased degradation. In vitro functional analysis of recombinant ADAMTS13 mutants indicates that mutations involving the metalloprotease catalytic domain generally results in severely decreased enzyme activity, with or without impaired secretion (12). Pathogenic c.577C>T missense mutation in exon 6 inactivates metalloprotease, probably adversely affects protein misfolding, and accelerates proteins degradation (Figure 2) (16). The previously undescribed c.2397C>A (p.C799X) non-sense mutation in exon 19 involving the TSP1-3 domain encodes a truncated ADAMTS13 protein likely degraded rapidly by NMD (non-sense-mediated RNA decay) (12) and judged as pathogenic according to the ACMG guideline.
Clinically, USS is highly heterogeneous in terms disease onset, manifestations, and prognosis. Most patients present in childhood, but some have their first episodes in adulthood, particularly during pregnancies (17), which are somewhat correlated with specific mutation profiles (18). In addition, disease manifestations and severity differ remarkably and there are no definitive correlations between genotypes and phenotypes. Six patients out of 30 in the German cohort had a mild clinical course defined as recurrent episodes of isolated thrombocytopenia without any organ involvement in spite of severe ADAMTS13 deficiency of less 6% in mild cases (12). In addition, patients with the same gene mutations could have quite different clinical pictures (12), including patients from the same family (19), as illustrated by our index case and his old sister who just experienced one single attack of isolated thrombocytopenia, suggesting other disease-modifying environmental or genetic factors.
USS can remain symptom-free for years between episodes and is most frequently triggered by infections and other inciting events (1, 14). Therefore, USS are easily misdiagnosed as primary immune thrombocytopenia, especially during childhood. In fact, 12 patients were initially misdiagnosed before 7 years as idiopathic immune thrombocytopenia, and 13 cases as TTP or other disease entities, out of an early Japanese cohort with 43 USS patients (20). The International USS Registry study clearly identified a significant delay of USS diagnosis, with median ages of overt presentation and clinical diagnosis being 4.5 and 16.7 years, respectively (14).
The differential diagnosis of USS from immune thrombocytopenia is of great clinical implication. The correct diagnosis of USS should be based on relevant family history, clinical presentations, plasma ADAMTS13 determination, and possibly mutation analysis. This helps to promptly institute plasma therapy to control acute attacks and prevent organ damages, to plan genetic counseling for family members, and to avoid unnecessary immunosuppressive therapy (1, 5, 21).
Prophylactic plasma given at 1- to 2-week intervals is a substantial measure to prevent recurrent episodes. However, prophylactic plasma infusion requires clinic visits and establishment of venous access. In addition, recombinant ADAMTS13 has been documented to be safe and could be self-administered at home (22).
In summary, we present an undescribed compound heterozygous ADMATS13 mutation, adding new information to the USS mutation database. Further studies are needed to explore ethnic and functional aspects of this specific variant.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
YD and XT: writing—original draft. JG: writing—review & editing. HC: processing pictures. QP: data curation. XG and JG: project administration. All authors have read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81600122); Fund of Science and Technology of Sichuan Province (No. 19ZDYF1202); Fund of Science and Technology of Sichuan Province (No. 2020YFS0253); Fund of Key R&D Program of Chengdu, Ministry of Science and Technology of Chengdu (No. 2019-YF05-01140-SN).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the patient and his parents.
References
1. Kremer Hovinga JA, George JN. Hereditary thrombotic thrombocytopenic purpura. N Engl J Med. (2019) 381:1653–62. doi: 10.1056/NEJMra1813013
2. Zheng XL. ADAMTS13 and von Willebrand factor in thrombotic thrombocytopenic purpura. Annu Rev Med. (2015) 66:211–25. doi: 10.1146/annurev-med-061813-013241
3. Tersteeg C, Verhenne S, Roose E, Schelpe AS, Deckmyn H, De Meyer SF, et al. ADAMTS13 and anti-ADAMTS13 autoantibodies in thrombotic thrombocytopenic purpura-current perspectives and new treatment strategies. Exp Rev Hematol. (2016) 9:209–21. doi: 10.1586/17474086.2016.1122515
4. Sadler JE. Pathophysiology of thrombotic thrombocytopenic purpura. Blood. (2017) 130:1181. doi: 10.1182/blood-2017-04-636431
5. Kremer Hovinga JA, Coppo P, Lämmle B, Moake JL, Miyata T, Vanhoorelbeke K, et al. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primer. (2017) 3:17020. doi: 10.1038/nrdp.2017.20
6. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. (2014) 371:654–66. doi: 10.1056/NEJMra1312353
7. Masias C, Vasu S, Cataland SR. None of the above: thrombotic microangiopathy beyond TTP and HUS. Blood. (2017) 129:2857–63. doi: 10.1182/blood-2016-11-743104
8. Matsumoto M, Kokame K, Soejima K, Miura M, Hayashi S, Fujii Y, et al. Molecular characterization of ADAMTS13 gene mutations in Japanese patients with Upshaw-Schulman syndrome. Blood. (2004) 3:1305–10. doi: 10.1182/blood-2003-06-1796
9. Pérez-Rodríguez A, Lourés E, Rodríguez-Trillo Á, Costa-Pinto J, García-Rivero A, Batlle-López A, et al. Inherited ADAMTS13 deficiency (Upshaw-Schulman syndrome): a short review. Thromb Res. (2014) 134:1171–5. doi: 10.1016/j.thromres.2014.09.004
10. Chang JC. Hemostasis based on a novel ‘two-path unifying theory’ and classification of hemostatic disorders. Blood Coagul Fibrinolysis. (2018) 29:573–84. doi: 10.1097/MBC.0000000000000765
11. Chang JC. Disseminated intravascular coagulation: is it fact or fancy? Blood Coagul Fibrinolysis. (2018) 29:330–7. doi: 10.1097/MBC.0000000000000727
12. Hassenpflug WA, Obser T, Bode J, Oyen F, Budde U, Schneppenheim S, et al. Genetic and functional characterization of ADAMTS13 variants in a patient cohort with Upshaw-Schulman syndrome investigated in Germany. Thromb Haemost. (2018) 118:709–22. doi: 10.1055/s-0038-1637749
13. Schneppenheim R, Kremer Hovinga JA, Becker T, Budde U, Karpman D, Brockhaus W, et al. A common origin of the 4143insA ADAMTS13 mutation. Thromb Haemost. (2006) 96:3–6. doi: 10.1160/TH05-12-0817
14. van Dorland HA, Taleghani MM, Sakai K, Friedman KD, George JN, Hrachovinova I, et al. The International Hereditary Thrombotic Thrombocytopenic Purpura Registry: key findings at enrollment until 2017. Haematologica. (2019) 104:2107–15. doi: 10.3324/haematol.2019.216796
15. Fu LL, Ma J, Ma JY, Zhang R, Gu H, Chen ZP, et al. Analysis of 5 children with congenital thrombotic thrombocytopenic purpura. Zhonghua Er Ke Za Zhi. (2019) 57:50–4. doi: 10.3760/cma.j.issn.0578-1310.2019.01.012
16. Lotta LA, Garagiola I, Palla R, Cairo A, Peyvandi F. ADAMTS13 mutation and polymorphisms in congenital thrombotic thrombocytopenic purpura. Hum Mutat. (2010) 31:11–9. doi: 10.1002/humu.21143
17. Scully M, Thomas M, Underwood M, Watson H, Langley K, Camilleri RS, et al. Thrombotic thrombocytopenic purpura and pregnancy: presentation, management, and subsequent pregnancy outcomes. Blood. (2014) 124:211–9. doi: 10.1182/blood-2014-02-553131
18. Joly BS, Boisseau P, Roose E, Stepanian A, Biebuyck N, Hogan J, et al. ADAMTS13 gene mutations influence ADAMTS13 conformation and disease age-onset in the French cohort of Upshaw-Schulman syndrome. Thromb Haemost. (2018) 118:1902–17. doi: 10.1055/s-0038-1673686
19. Noris M, Bucchioni S, Galbusera M, Donadelli R, Bresin E, Castelletti F, et al. Complement factor H mutation in familial thrombotic thrombocytopenic purpura with ADAMTS13 deficiency and renal involvement. J Am Soc Nephrol. (2005) 16:1177–83. doi: 10.1681/ASN.2005010086
20. Fujimura Y, Matsumoto M, Isonishi A, Yagi H, Kokame K, Soejima K, et al. Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan. J Thromb Haemost. (2011) 9(Suppl. 1):283–301. doi: 10.1111/j.1538-7836.2011.04341.x
21. Masias C, Cataland SR. The role of ADAMTS13 testing in the diagnosis and management of thrombotic microaniopathies and thrombosis. Blood. (2018) 132:90. doi: 10.1182/blood-2018-02-791533
Keywords: hereditary thrombotic thrombocytopenic purpura, Upshaw-Schulman syndrome, ADAMTS13, novel mutation, Chinese
Citation: Dai Y, Tang X, Chen H, Peng Q, Guo X and Gao J (2020) Hereditary Thrombotic Thrombocytopenic Purpura in a Chinese Boy With a Novel Compound Heterozygous Mutation of the ADAMTS13 Gene. Front. Pediatr. 8:554. doi: 10.3389/fped.2020.00554
Received: 25 April 2020; Accepted: 30 July 2020;
Published: 10 September 2020.
Edited by:
John Joseph Strouse, Duke University, United StatesReviewed by:
Jeffrey R. Andolina, University of Rochester, United StatesTaizo Nakano, Children's Hospital Colorado, United States
Copyright © 2020 Dai, Tang, Chen, Peng, Guo and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xia Guo, Z3VveGtsQDE2My5jb20=; Ju Gao, Z2FvanU2NTEyMjBAMTI2LmNvbQ==
†These authors share first authorship