 Cristina Oana Mărginean
Cristina Oana Mărginean Lorena Elena Meliţ
Lorena Elena Meliţ Gabriel Grigorescu2
Gabriel Grigorescu2 Iunius Simu
Iunius Simu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 26 August 2020
Sec. Pediatric Pulmonology
Volume 8 - 2020 | https://doi.org/10.3389/fped.2020.00497
Sarcoidosis (SD) is a systemic granulomatous condition that is especially encountered in young adults and rarely in children, affecting predominantly the lungs and lymph nodes. We report the case of a 14-year-old teenage boy admitted to our clinic for nausea, vomiting, and weight loss. Clinical examination at the time of admission revealed malaise, pallor, and abdominal tenderness in the epigastric area at palpation. Laboratory tests revealed an elevated level of hemoglobin, mild thrombocytosis, increased erythrocyte sedimentation rate, and a mild increase in creatinine and urea levels along with hypercalcemia. An abdominal ultrasound revealed a right ectopic kidney, whereas the upper digestive endoscopy showed intense hyperemia and edema of the gastric mucosa. Thoracic computed tomography scan revealed giant hilar and mediastinal lymphadenopathy, along with multiple micronodules within the lung parenchyma and ground-glass aspect. The level of angiotensin-converting enzyme was high, parathormone was normal, and vitamin D level was low. Pathological examination of the bronchial, mediastinal, and lung biopsies established the diagnosis of SD. We administered oral corticosteroids for 2 months with outstandingly favorable outcome and no signs of recurrence 6 months after the cessation of the therapy.
Sarcoidosis (SD) or Besnier–Boeck–Schaumann disease was described for the first time in seventieth century as a systemic granulomatous condition of the lungs and lymph nodes (1). It is mainly a disease of the adults, with a peak onset between 25 and 45 years of age and with an incidence of ~5–45 in 100,000 adults (2), whereas it is almost 10 times less common in children (3). Even though its pathogenesis remains unknown, the current hypothesis involves an association between genetic predisposition and the environmental factors that demonstrate the ability to trigger or enhance the inflammatory and granulomatous processes (3).
The patients with SD express a wide spectrum of clinical manifestations, ranging from a subclinical form affecting only two organs to a life-threatening multiorgan dysfunction (4). The most common symptoms reported in children were related to lung and mediastinal involvement; however, extrarespiratory manifestations were also frequently observed, such as eye impairment, hepatomegaly, splenomegaly, skin anomalies, peripheral lymphadenopathy, or arthralgia (3, 5). Laboratory tests are not usually reliable for the diagnosis because they are not specific. Nevertheless, certain laboratory findings may be associated with SD in children, such as elevated erythrocyte sedimentation rate, anemia, hypergammaglobulinemia, lymphocytosis or lymphopenia, thrombocytosis, increased levels of transaminases and serum angiotensin-converting enzyme (ACE), and hypercalcemia (3), which also suggest other infectious pathologies (6). The assessment of renal function is also essential because hypercalcemia could lead to renal insufficiency or nephrolithiasis (7, 8). Multiple factors contribute to the development of hypercalcemia in SD patients, such as the upregulation of 1-α-hydroxylase activity within the granulomas of SD patients that leads to an increased conversion of 25-(OH) vitamin D3 into 1,25-(OH)2 vitamin D3, parathyroid adenomas, or supplementation of calcium in patients receiving chronic glucocorticoids (9). Nevertheless, normal levels of both forms of vitamin D were observed in hypercalcemic SD patients, which suggested that other mechanisms could also contribute to elevated serum calcium levels in these patients (10).
In SD children with respiratory symptoms, the first imaging investigation is chest radiography, which either can reveal hilar and mediastinal lymphadenopathies or lung fibrosis, or can be normal. Computed tomography (CT) is more sensitive and specific for mediastinal and lung involvement because it can highlight multiple imaging aspects suggestive of SD, such as interstitial lung disease, micronodules involving pleural surfaces, fissures, and peribronchovascular areas, perilymphatic nodules, ground-glass opacities, thickened septa, or alveolar consolidation (3). Flexible bronchoscopy using bronchoalveolar lavage is also reliable in assessing lung involvement. Nevertheless, pathological examination of the biopsied organs that reveal non-caseating granulomas remains the gold standard for SD diagnosis. Lung tissue biopsies could be obtained through wedge transbronchial lung biopsy; however, surgical biopsies are also a potential alternative (3). Other commonly biopsied organs in SD patients are peripheral or mediastinal lymph nodes and skin lesions (11).
Treatment may not be necessary for SD patients who present only hilar lymphadenopathy without parenchymal lung inflammation (12). Moreover, in children with SD, there is a lack of standardized therapeutic protocols, and they are usually treated in a similar manner as adults. Thus, administration of corticosteroids remains the most common therapeutic approach with significant improvement within 6 months. Considering the unproven favorable long-term outcome for lung impairment, other drugs/agents may also be useful, such as methotrexate, cytotoxic agents, or antimalarials (13). The prediction of SD prognosis in children is difficult owing to its rarity at these ages; however, the mortality rates are estimated to vary between 1 and 5% (14).
The aim of this case report was to underline the uncommon onset of SD in a teenage boy due to associated hypercalcemia.
Written informed consent was obtained from the patient's mother prior to the publication of this case report.
A 14-year-old boy who presented with nausea, vomiting, and weight loss (~6 kg within 1 month) was admitted to the Department of Pediatric Gastroenterology with the suspicion of acute gastritis. The patient denied previous vitamin D supplementation or sun exposure.
Clinical examination at the time of admission revealed malaise, pallor, and abdominal tenderness in the epigastric area at palpation. The patient weighed 50 kg.
A complete blood count revealed an elevated level of hemoglobin (15.7 g/dL) and mild thrombocytosis (518,000/μL) along with an elevated erythrocyte sedimentation rate (23 mm/h). Biochemical blood analysis showed high levels of creatinine (156.5 μmol/L) and urea (22.35 mmol/L), along with hypercalcemia (total serum calcium 3.66 mml/L, serum ionized calcium 1.77 mmol/L). A qualitative test performed from a urinary sample proved hypercalciuria, and the routine urine examination revealed microscopic hematuria (10 erythrocytes per field). Renal ultrasound revealed a right ectopic kidney without any associated pathological findings in terms of kidney morphology and structure. We consulted an endocrinologist, who ruled out any associated conditions. An upper digestive endoscopy demonstrated intense hyperemia and edema of the gastric mucosa, and pathological examination of the gastric biopsies established the diagnosis of Helicobacter pylori–induced gastritis. This was treated according to the standard guidelines, using double-antibiotic regimen (amoxicillin and clarithromycin) that influences proton pump inhibitors. The elevated levels of urea and creatinine were initially interpreted as signs of prolonged dehydration; however, despite proper intravenous fluid administration, they remained above the normal range. Thus, we decided to perform thoracic and abdominal CT. Thoracic CT revealed giant hilar and mediastinal lymphadenopathies (Figure 1), multiple micronodules (Figure 2) within the lung parenchyma, and ground-glass opacities (Figure 3), which raised the suspicion of SD, without ruling out tuberculosis or lymphoma, whereas the abdominal scan confirmed the ultrasound findings showing only a right ectopic kidney. We found an elevated level of ACE (244.3 U/L), normal level of parathormone (15.1 pg/mL), and decreased levels of 1,25-(OH)2 vitamin D (14.75 ng/mL). The cardiologic and ophthalmologic examinations were normal. Additional tests, such as peripheral smear, protein electrophoresis, human immunodeficiency virus serology, and serum copper were performed in order to rule out other conditions that evolve with lung and lymph node impairment. In order to confirm our suspicion of SD, a bronchoscopy with bronchial biopsy and open surgery with lung and mediastinal biopsies were performed because an endobronchial ultrasound approach was not possible. The pulmonary function tests performed prior to the bronchoscopy were within normal ranges. The pathological examination revealed multiple non-caseating granulomas in all the biopsy specimens, which established the diagnosis of SD.
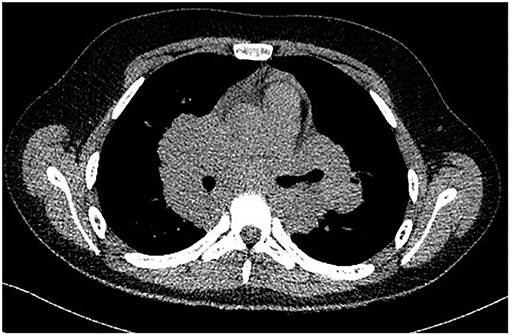
Figure 1. CT aspect of mediastinal lymphadenopathies.
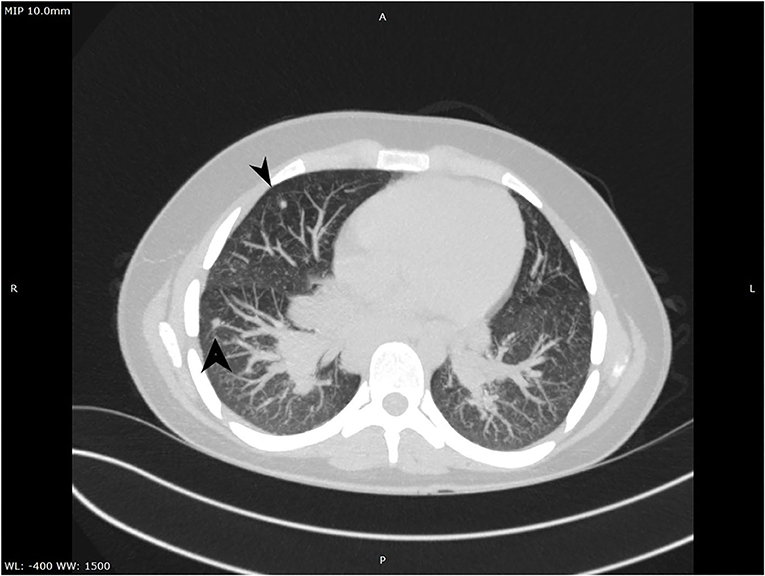
Figure 2. CT aspect of lung micronodules.
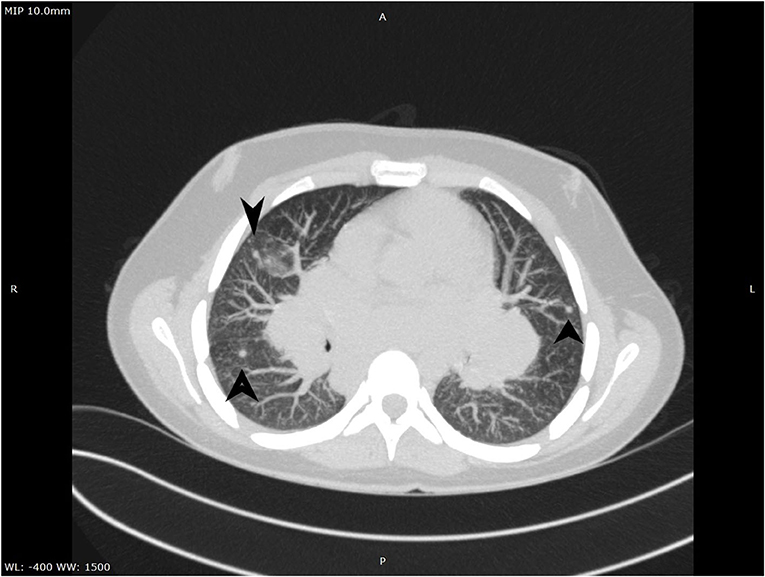
Figure 3. Ground-glass aspect on CT.
We initiated the treatment with oral administration of prednisone at a loading dose of 1 mg/kg per day, that is, 50 mg/d, with tapering doses for 2 months, while considering the improvement in the patients' symptoms after the first week of therapy and findings of the blood tests after ~2 weeks.
The patient's follow-up for ~6 months after the cessation of corticosteroid administration revealed favorable clinical outcome with normal results of all blood tests, inclusively renal function parameters, and normal ACE levels. Moreover, the chest X-ray showed no enlarged lymph nodes and a normal aspect of the lung parenchyma (Figure 4).
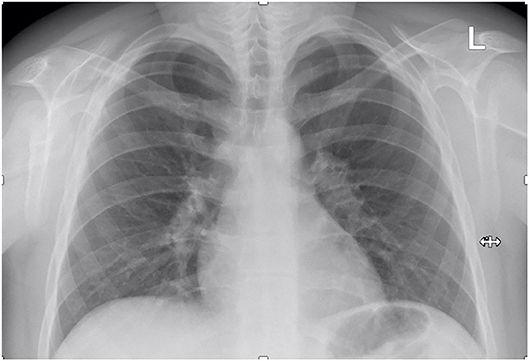
Figure 4. Follow-up chest X-ray.
SD rarely occurs in children; it primarily affects young adults (2). Nevertheless, here, we describe a case of SD in a 14-year-old teenage boy with lung and mediastinal involvement. Respiratory symptoms are most commonly observed at onset in the patients with this type of involvement (3). Nevertheless, our patient presented with nausea, vomiting, and weight loss most likely due to H. pylori–induced gastritis.
SD patients frequently exhibit anemia, thrombocytosis, lymphocytosis, or lymphopenia (3). In our patient, we encountered only thrombocytosis and, contrary to the general observation, an increased level of hemoglobin. As observed for our patient, erythrocyte sedimentation rate may also be elevated in SD patients as a sign of mild inflammation similar to that observed in other conditions (15–17). Among the other altered laboratory parameters previously reported in SD children, including hypergammaglobulinemia, elevated transaminases, increased ACE, and hypercalcemia, our patient exhibited elevated levels of ACE and hypercalcemia. Moreover, he exhibited impairment of renal function as reflected by elevated creatinine and urea. Despite their initial indications as dehydration signs, the increase in renal function parameters was most likely caused by SD-associated hypercalcemia. Other studies stated that ~30% of the patients diagnosed with SD exhibit asymptomatic hypercalcemia, although nephrocalcinosis is rarely observed in these patients (11). The imaging assessment excluded nephrocalcinosis in our patient, but the increase in urea and creatinine levels was strongly related to hypercalcemia. Moreover, Milman et al. (18) stated that hypercalcemia, and cutaneous sarcoid lesions and generalized lymphadenopathy are markers of poor prognosis in patients with SD. Despite the fact that our patient presented hypercalcemia, the outcome was favorable after ~2 weeks from the initiation of corticosteroids when the serum calcium levels became normal. Nevertheless, we did not find any skin lesions, and the lymphadenopathies were localized. SD-associated renal changes have previously been classified into three main types: primary, parenchymatous, or granulomatous SD; glomerulonephritic changes resembling collagen diseases or nephrotic syndrome; and hypercalcemic renal damage, which occurs most frequently (19). Thus, patients with SD-associated hypercalcemia may present a wide spectrum of signs and symptoms, including renal colic (due to recurrent kidney stones), mental disorders, loss of appetite, constipation, diffuse abdominal pain, gastric ulcers, myopathy, or neuromuscular symptoms (19). The authors described two cases of SD-associated hypercalcemia and uremia in two adult patients with favorable outcome after administration of corticosteroids, which demonstrated the high efficacy of steroid therapy in these cases (19). Nevertheless, the reports in children are scarce.
ACE is thought to be produced by both SD granulomas and lung endothelium (20). Although the levels of this enzyme have been shown to be elevated in SD patients, its sensitivity and specificity as a diagnostic marker may vary between 41 and 58% and between 84 and 90%, respectively (21). Regardless of these discordant reports, elevated ACE levels may be associated with disease activity and also represent efficient marker of treatment efficacy (22). A recent study performed on 119 adult SD patients, which aimed to establish the role of ACE, showed that elevated ACE levels are associated with high serum levels of ionized calcium (23). Similarly, in our case, we noticed elevated levels of both ACE and serum levels of ionized calcium that became normal after treatment, suggesting indeed that ACE might be a potential therapeutic marker.
The communications skills of a physician are essential for a proper anamnesis in order to rule out other potential risk factors or associated conditions (24). Chest X-ray and pulmonary function tests are important for the follow-up of SD patients; however, initial small lesions may not be observed using these diagnostic tools (14). Thus, CT represents the gold standard in diagnosing SD in pediatric patients that require further investigations (25). In our case, thoracic CT revealed giant hilar and mediastinal lymphadenopathies, multiple micronodules within the lung parenchyma, and ground-glass opacities, all suggestive of SD. Despite all the information provided by anamnesis, clinical examination, and imaging assessment, only a pathological examination could confirm SD diagnosis (26). In our patient, non-caseating granulomas were identified in all biopsy specimens. Because the transbronchial approach was unsuccessful, we were forced to perform an open surgery for mediastinal and lung biopsies in order to confirm the diagnosis and to exclude the possibility of other pathologies (27). Thus, other similar granulomatous disorders include Blau syndrome, autoimmune lymphoproliferative syndrome, or Crohn disease. Blau syndrome is defined by a triad consisting in granulomatous dermatitis, arthritis, and uveitis, occurring commonly during early childhood (28). Contrariwise, our patient presented no signs of dermatitis, arthritis, and uveitis. Wegener granulomatosis, an idiopathic inflammatory disorder, is characterized by necrotizing granulomatous inflammation and pauci-immune small-vessel vasculitis affecting the upper and lower respiratory tracts and kidneys, and the diagnosis is usually established based on histopathological examination (29). Nevertheless, our patient presented no signs of vasculitis involving the upper and lower respiratory tracts or kidney, and the histopathological examination confirmed the diagnosis of SD. An uncommon condition that must be taken into account in patients with lymphadenopathy is the autoimmune lymphoproliferative syndrome defined also by splenomegaly and autoimmune cytopenia (30), these were absent in our case. Chron's disease is another granulomatous condition affecting the gastrointestinal tract (31), which was ruled out in our case based on the upper digestive endoscopy and pathological examination of the gastric mucosa biopsy. Except all these rare conditions, all the remaining common infectious or inflammatory disorders, including tuberculosis, were ruled out based on the pathological examination.
Despite possible spontaneous remission, visual observation is not an efficient management strategy in case of pediatric patients (3). Oral or intravenous administration of corticosteroids is primarily efficient in the management of SD children, especially in those with SD-associated hypercalcemia (3). In previously reported cases, the duration of treatment varied from 1 month to 23 years (5, 11, 32, 33). We decided to cease corticosteroids after 2 months owing to the favorable outcome of our patient after 2 weeks of treatment. The prognosis of SD in children depends on the onset age, disease extension, and response to corticosteroids (3). We hope that the patient we presented would have a favorable outcome because of his older onset age, disease limitation to lung and mediastinum, and rapid favorable response to corticosteroids. Nevertheless, prognostic factors still remain unclear in children (3).
SD occurs rarely in children. Even though spontaneous remission is possible in patients with SD, corticosteroids result in an outstandingly favorable evolution in SD children with primarily mediastinal and lung involvement associated with hypercalcemia. SD in children remains an “exclusive diagnosis;” however, awareness of the physician is vital for its early diagnosis.
Written informed consent was obtained from the legal guardian, for the publication of any potentially identifiable images or data included in this article.
CM and LM conceptualized and designed the study, drafted the initial manuscript, and reviewed and revised the manuscript. CP, GG, and IS designed the data collection instruments, collected data, carried out the initial analyses, and reviewed and revised the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We express our sincere thanks to Dr. Popoviciu Bogdan for performing the surgical intervention and lung biopsies and to Dr. Kovecsi Attila for performing the pathological exam.
ACE, angiotensin-converting enzyme; CT, computed tomography; SD, sarcoidosis.
1. Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet P-Y, Müller-Quernheim J. Sarcoidosis. Lancet. (2014) 383:1155–67. doi: 10.1016/S0140-6736(13)60680-7
2. Byg K-E, Milman N, Hansen S. Sarcoidosis in Denmark 1980-1994. A registry-based incidence study comprising 5536 patients. Sarcoidosis Vasc Diffuse Lung Dis. (2003) 20:46–52.
3. Nathan N, Sileo C, Calender A, Pacheco Y, Rosental P-A, Cavalin C, et al. Paediatric sarcoidosis. Paediatr Respir Rev. (2019) 29:53–9. doi: 10.1016/j.prrv.2018.05.003
4. Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. (2008) 6:16. doi: 10.1186/1546-0096-6-16
5. Nathan N, Marcelo P, Houdouin V, Epaud R, de Blic J, Valeyre D, et al. Lung sarcoidosis in children: update on disease expression and management. Thorax. (2015) 70:537–42. doi: 10.1136/thoraxjnl-2015-206825
6. Meliţ LE, Mărginean CO, Georgescu A, Duicu C. Complications of sepsis in infant. A case report. J Crit Care Med. (2016) 2:96–9. doi: 10.1515/jccm-2016-0012
7. Ponce C, Gujral JS. Renal failure and hypercalcemia as initial manifestations of extrapulmonary sarcoidosis. South Med J. (2004) 97:590–2. doi: 10.1097/00007611-200406000-00016
8. Rizzato G, Fraioli P, Montemurro L. Nephrolithiasis as a presenting feature of chronic sarcoidosis. Thorax. (1995) 50:555–9. doi: 10.1136/thx.50.5.555
9. Baughman RP, Janovcik J, Ray M, Sweiss N, Lower EE. Calcium and vitamin D metabolism in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. (2013) 30:113–20.
10. Falk S, Kratzsch J, Paschke R, Koch CA. Hypercalcemia as a result of sarcoidosis with normal serum concentrations of vitamin D. Med Sci Monit. (2007) 13:CS133–6.
11. Hoffmann AL, Milman N, Byg KE. Childhood sarcoidosis in Denmark 1979-1994: incidence, clinical features and laboratory results at presentation in 48 children. Acta Paediatr. (2004) 93:30–6. doi: 10.1111/j.1651-2227.2004.tb00670.x
12. White ES, Lynch JP. Current and emerging strategies for the management of sarcoidosis. Expert Opin Pharmacother. (2007) 8:1293–311. doi: 10.1517/14656566.8.9.1293
13. Fretzayas A, Moustaki M, Vougiouka O. The puzzling clinical spectrum and course of juvenile sarcoidosis. World J Pediatr. (2011) 7:103–10. doi: 10.1007/s12519-011-0261-0
14. Statement on Sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med. (1999) 160:736–55.
15. Mărginean C, Meliţ L, Ghiga D, Mărginean M. Early inflammatory status related to pediatric obesity (STROBE compliant article). Front Pediatr. (2019) 7:241. doi: 10.3389/fped.2019.00241
16. Melit LE, Mărginean MO, Mocan S, Mărginean CO. The usefulness of inflammatory biomarkers in diagnosing child and adolescent's gastritis: STROBE compliant article. Medicine. (2019) 98:e16188. doi: 10.1097/MD.0000000000016188
17. Mǎrginean CO, Mǎrginean C, Melit LE. New insights regarding genetic aspects of childhood obesity: a minireview. Front Pediatr. (2018) 6:271. doi: 10.3389/fped.2018.00271
18. Milman N, Hoffmann AL, Byg KE. Sarcoidosis in children. Epidemiology in Danes, clinical features, diagnosis, treatment and prognosis. Acta Paediatr. (1998) 87:871–8. doi: 10.1111/j.1651-2227.1998.tb01554.x
19. Dawidson I, Jameson S. Sarcoidosis with hypercalcemia, reversible uremia and hyperparathyroidism. Scand J Urol Nephrol. (1972) 6:308–11. doi: 10.3109/00365597209132108
20. Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. (2007) 357:2153–65. doi: 10.1056/NEJMra071714
21. Ungprasert P, Carmona EM, Crowson CS, Matteson EL. Diagnostic utility of angiotensin-converting enzyme in sarcoidosis: a population-based study. Lung. (2016) 194:91–5. doi: 10.1007/s00408-015-9826-3
22. Baughman RP, Ploysongsang Y, Roberts RD, Srivastava L. Effects of sarcoid and steroids on angiotensin-converting enzyme. Am Rev Respir Dis. (1983) 128:631–3.
23. Sejdic A, Graudal N, Baslund B. Clinical and biochemical presentation of sarcoidosis with high and normal serum angiotensin-converting enzyme. Scand J Rheumatol. (2018) 47:487–90. doi: 10.1080/03009742.2017.1420818
24. Mărginean CO, Melit LE, Chinceşan M, Mureşan S, Georgescu AM, Suciu N, et al. Communication skills in pediatrics - the relationship between pediatrician and child. Medicine. (2017) 96:e8399. doi: 10.1097/MD.0000000000008399
25. Fauroux B, Clément A. Paediatric sarcoidosis. Paediatr Respir Rev. (2005) 6:128–33. doi: 10.1016/j.prrv.2005.03.007
26. Heinle R, Chang C. Diagnostic criteria for sarcoidosis. Autoimmun Rev. (2014) 13:383–7. doi: 10.1016/j.autrev.2014.01.035
27. Mărginean CO, Melit LE, Horvath E, Gozar H, Chinceşan MI. Non-Hodgkin lymphoma, diagnostic, and prognostic particularities in children - a series of case reports and a review of the literature (CARE compliant). Medicine. (2018) 97:e9802. doi: 10.1097/MD.0000000000009802
28. Wouters CH, Maes A, Foley KP, Bertin J, Rose CD. Blau Syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatr Rheumatol. (2014) 12:33. doi: 10.1186/1546-0096-12-33
29. Almouhawis HA, Leao JC, Fedele S, Porter SR. Wegener's granulomatosis: a review of clinical features and an update in diagnosis and treatment. J Oral Pathol Med. (2013) 42:507–16. doi: 10.1111/jop.12030
30. Lau C-Y, Mihalek AD, Wang J, Dodd LE, Perkins K, Price S, et al. Pulmonary manifestations of the autoimmune lymphoproliferative syndrome. A retrospective study of a unique patient cohort. Ann Am Thorac Soc. (2016) 13:1279–88. doi: 10.1513/AnnalsATS.201601-079OC
31. Mărginean CO, Melit LE, Mocanu S, Mărginean MO. Inflammatory bowel diseases: a burden in pediatrics: case series and a review of the literature. Medicine. (2017) 96:e6329. doi: 10.1097/MD.0000000000006329
32. Gedalia A, Khan TA, Shetty AK, Dimitriades VR, Espinoza LR. Childhood sarcoidosis: Louisiana experience. Clin Rheumatol. (2016) 35:1879–84. doi: 10.1007/s10067-015-2870-9
Keywords: sarcoidosis, children, hypercalcemia, renal failure, uncommon onset
Citation: Mărginean CO, Meliţ LE, Grigorescu G, Puiac C and Simu I (2020) Hypercalcemia, an Important Puzzle Piece in Uncommon Onset Pediatric Sarcoidosis—A Case Report and a Review of the Literature. Front. Pediatr. 8:497. doi: 10.3389/fped.2020.00497
Received: 18 February 2020; Accepted: 15 July 2020;
Published: 26 August 2020.
Edited by:
Bülent Taner Karadaǧ, Marmara University, TurkeyReviewed by:
Aleksandar Sovtic, The Institute for Health Protection of Mother and Child Serbia, SerbiaCopyright © 2020 Mărginean, Meliţ, Grigorescu, Puiac and Simu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorena Elena Meliţ, bG9yeV9jaGltaXN0YTg5QHlhaG9vLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.