 Jinzhi Gao
Jinzhi Gao Ling Chen
Ling Chen- Department of Pediatrics, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Primary adrenocortical insufficiency (PAI) is an important cause of morbidity in neonates. The most common cause of PAI in neonates is congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency (21-OHD). Other rarer monogenic cases, for example, adrenal hypoplasia congenita (AHC) or familial glucocorticoid deficiency, also simulate clinical manifestation of 21-OHD, leading to misdiagnosis. The therapies and prognosis of these monogenic cases of PAI are entirely different. This study aimed to compare the differences of clinical data and identify genetic etiologies of PAI cases in the neonatal period. All 7 neonates initially presented with hyperpigmentation, hyponatremia, hyperkalemia, and high serum adrenocorticotropic hormone levels. Only CAH patients showed hyperandrogenism and remarkably elevated serum 17-hydroxyprogesterone levels. All the pathogenic mutations found in CYP21A2 were well known, except c.1069C>T (exon 8). The male patient with AHC had a novel hemizygous deletion of exon 2 in DAX1. The other one with familial glucocorticoid deficiency type 1 had two novel heterozygous mutations in the gene coding melanocortin 2 receptor, c.701C>T (exon 2) and c.119delT (exon 2). Glucocorticoid and/or mineralocorticoid replacement therapy depends on the cause of PAI. Genetic testing can be performed as a alternative diagnostic approach to provide information about therapy, prognosis, and genetic counseling.
Introduction
The clinical feature of primary adrenocortical insufficiency (PAI) depends on the class of deficient hormone including glucocorticoid, mineralocorticoid, and adrenal androgens, and the extent of hormone deficiency. Glucocorticoid deficiency may cause fatigue, nausea, hypoglycemia, recurrent infections, hyperbilirubinemia, and cholestasis. As a consequence of cortisol deficiency, the production of pro-opiomelanocortin is increased, which is the prohormone of adrenocorticotropic hormone (ACTH) and melanocyte-stimulating hormone. The elevated melanocyte-stimulating hormone results in increased melanin synthesis, causing hyperpigmentation. Mineralocorticoid deficiency may cause failure to thrive, anorexia, dehydration, hypotension, hyponatremia, and hyperkalemia. Andadrenal androgens are associated with sexual development. In general, the above clinical findings of neonates with PAI are non-specific, misdiagnosis and delayed diagnosis are likely to result in potentially life-threatening adrenal crisis. The most common cause of PAI in the neonatal period is congenital adrenal hyperplasia (CAH), which occurs in approximately 1 of 14,200 live births (1, 2). 21-hydroxylase deficiency (21-OHD) caused by mutations in the CYP21A2 gene represents 95–99% of cases of CAH (3). 21-OHD results in the defective conversion of 17-hydroxyprogesterone (17-OHP) to 11-deoxycortisol, which leads to impaired synthesis of cortisol and aldosterone, and increased secretion of adrenocorticotropic hormone (ACTH). ACTH can increase the production of androgens by stimulating adrenal glands. Except for clinical findings caused by glucocorticoid and/or mineralocorticoid deficiency, the increasing androgens may cause clitoridauxe and virilization in female neonates, and hyperpigmentation of scrotum and enlarged phallus in male neonate. According to the chinical presentation determined by residual enzyme activity, CAH is classified into classic forms (salt wasting, simple virilizing) and non-classic form (late onset). Even when the detectable enzyme activity is as low as 1–2 percent, the patient could present with the form of simple virilizing (4).
In the neonatal period, except for 21-OHD, the other rarer genetic disorders including 11β-hydroxylase deficiency, adrenal hypoplasia congenita (AHC) and familial glucocorticoid deficiency (FGD), et al. should also be considered (Table 1). The most common cause of AHC is X-linked genetic defects in the differentiation of adrenocortical cells. The prevalence is 1:140000~1:1200000 (5). The clinical feature of AHC caused by glucocorticoid and mineralocorticoid deficiency is similar to CAH in neonates, but without high androgens levels, along with impaired pubertal development and hypogonadism (6, 7). FGD is a rarer autosomal recessive condition of PAI with a prevalence of < 1:1000000 (8), as it has little more than 50 cases reported in the literature (9). FGD is characterized by unresponsiveness of the adrenal gland to ACTH and preserved aldosterone/renin secretion, so that their clinical presentation is mainly caused by glucocorticoid deficiency (10). The clinical feature of these monogenic cases of PAI are similar, but the therapies and prognosis are entirely different. CAH due to 21-OHD is common, other genetic disorders causing PAI need more attention. In the present study, we conducted genetic analyses of 7 non-consanguineous Chinese children with biochemically characterized PAI. We aim to compare the differences in clinical manifestation and auxiliary examinations, and identify genetic etiologies of PAI cases in the neonatal period, which could contribute to further treatment, prognosis prediction, genetic consultation, and prenatal diagnosis.
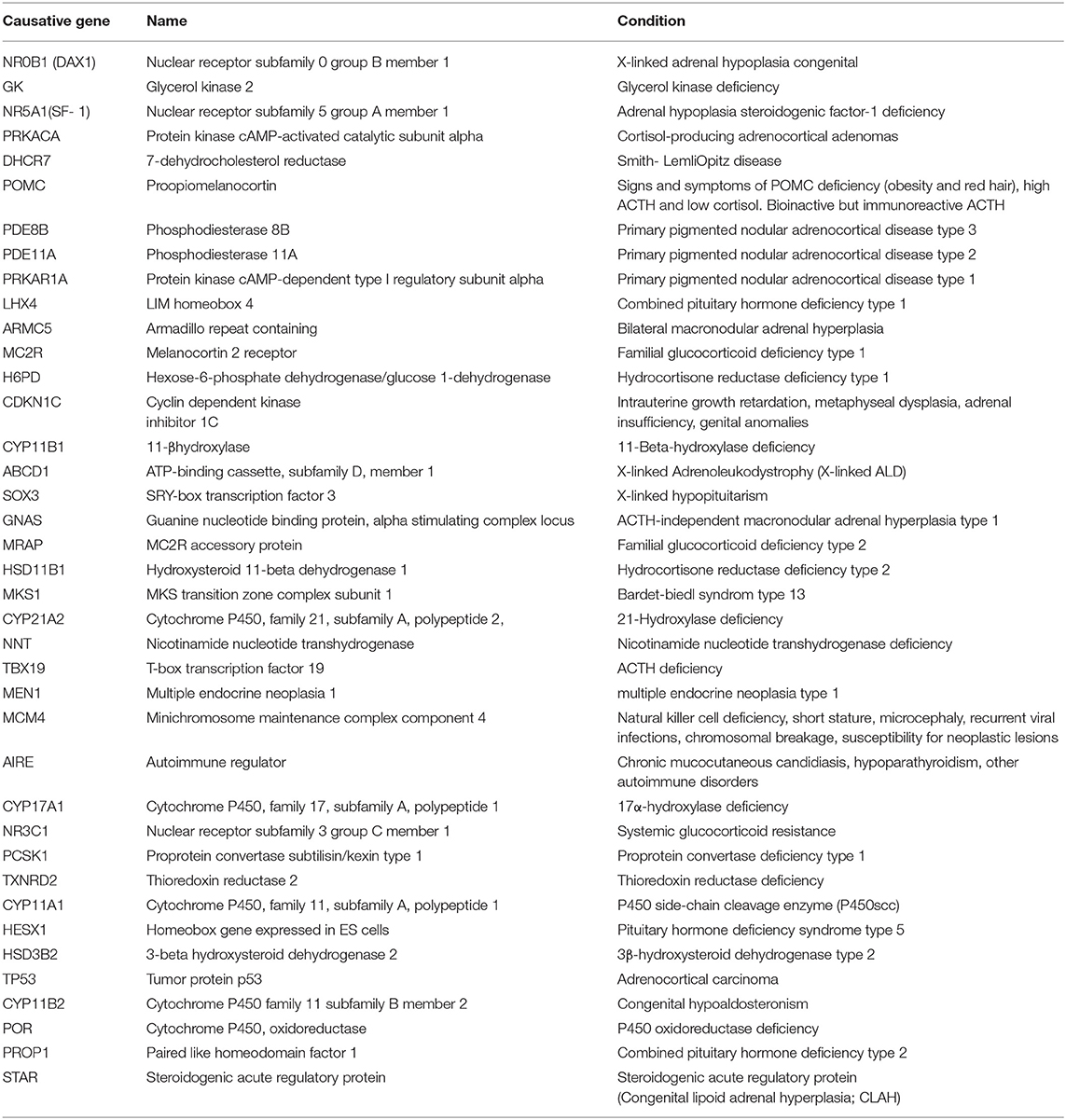
Table 1. Candidate genes were sequenced to identify mutations in patients with adrenocortical insufficiency.
Case Presentation
Subjects
The patients in this study were all from Wuhan, which is located in the center of mainland China. We identified 7 patients who had been diagnosed with PAI in the neonatal period. Patients were not picked up by a newborn screening program but instead were identified by salt-wasting in both males and females. This study was approved by the institutional Human Investigations Committee. Written informed consent was obtained from the legal guardian of each subject.
Method
Plasma levels of 17-hydroxyprogesterone (17-OHP), testosterone (T), corticotropin-releasing hormone (ACTH), and cortisol were determined at diagnosis by an electrochemiluminescence immunoassay (Roche, Switzerland). The work was conducted at Tongji Hospital affiliated with Tongji Medical College, Huazhong University of Science and Technology, and a commercial genetic testing company (MyGenostics Research Institute). Clinical, laboratory, and genetic studies were undertaken in the 7 patients as well as in their parents. Targeted next-generation sequencing combined with multiplex ligation-dependent probe amplification (MLPA) was used to identify the genetic etiologies of PAI. Candidate genes are shown in Table 1.
Results
There was no familial relationship among the 7 patients in our report. Each pregnancy was uneventful with term delivery of each patient. All of them presented with hyperpigmentation spreading from head to foot, especially at the genitals and areola, and none of them presented with ambiguous genitalia or intersex conditions. Adrenal imaging, if available, was unremarkable in all cases. Five patients were diagnosed with CAH, one patient was diagnosed with AHC, and the other one was diagnosed with familial glucocorticoid deficiency type 1 (FGD1). Refer to Table 2 for a summary of clinical and biochemical feature.
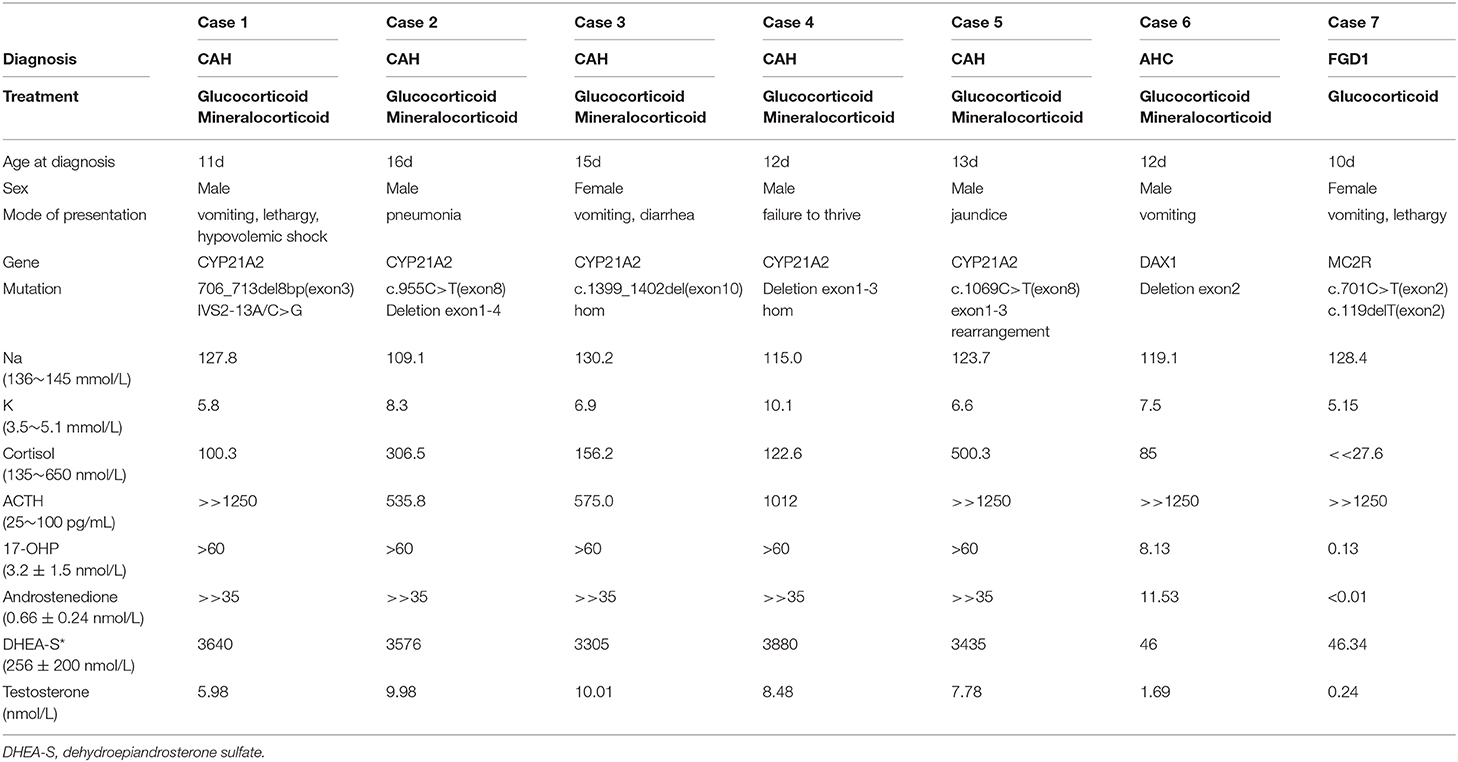
Table 2. The clinical data, mutation spectrum, and laboratory results in the seven consulters diagnosed with PAI.
Initial Presentation of the Neonates With PAI
The patients came to our attention at the age of 10–16 days with generalized hyperpigmentation. Their manifestations leading to diagnosis with PAI were non-specific. Five patients presented with digestive tract symptoms such as vomiting, diarrhea, and loss of appetite. Two patients presented with pneumonia and jaundice, and electrolyte disturbances were found occasionally. Steroid hormones levels were measured, because the patients presented with the clinical feature of hyperpigmentation and electrolyte disturbance.
Initial Laboratory Results of the Neonates With PAI
All 7 patients showed hyperkalemia and hyponatremia. Among the 5 patients with CAH, the serum cortisol levels were under normal value in 2 patients and normal in 3 patients, while all the serum ACTH levels were remarkably elevated. Meanwhile, the serum cortisol levels were severely low and ACTH levels were remarkably elevated in patients with AHC and FGD1. Hormonal analyses revealed elevated serum 17-OHP, androstenedione, and dehydroepiandrosterone sulfate (DHEA-S) in patients with CAH, as shown in Table 2. While the male patient with AHC had relatively normal values in serum 17-OHP and androstenedione, lower values in DHEA-S and testosterone. The female patient with FGD1 had an extremely low serum 17-OHP, and low serum androstenedione, DHEA-S, and testosterone values. The mineralocorticoid level in the patient with FGD1 was normal, while her hyponatremia might be due to vomiting, and the extent of hyperkalemia was extremely mild. Hydrocortisone with or without fluorine hydrocortisone therapy with additional saline and glucose infusions were started timely, and the condition was improved markedly.
Genetic Causes of the Neonates With PAI
Genetic testing in 5 unrelated neonates with CAH all showed mutations in CYP21A2 gene (Table 2). Patient 1 had two heterozygous mutations, 706_713del8bp (exon 3) and IVS2-13A/C>G, inherited from each of his parents. Patient 2 had two heterozygous mutations, including a deletion of exon 1-4 inherited from his mother, and c.955C>T (exon 8) inherited from his father. His mother and father were unaffected carriers. We found a novel homozygous mutation, c.1399_1402del4bp (exon 10), in patient 3. This mutation has not been reported before, and no relevant mutation was found in her parents. Patient 4 had homozygous deletion of exon 1–3, and his patents were both carriers of a deletion of exon 1–3. Patient 5 had two heterozygous mutations. One was c.1069C>T (exon 8) inherited from his father, and the other one was exon 1–3, rearranged from CYP21A2 to a CYP21A1P pseudogene, inherited from his mother.
We found a novel hemizygous deletion of exon 2 in DAX1 in Patient 6 diagnosed with AHC. There were two novel heterozygous mutations, c.701C>T (exon 2) and c.119delT (exon 2) in MC2R gene, in Patient 7 diagnosed with FGD1.
Discussion
This is a retrospective study of PAI case series. In early infancy, diagnoses of PAI are very easily confused, as all cases present much like each other. This case series focused exclusively on primary adrenocortical insufficiency in neonatal period. The strength of this study was that we were able to obtain genetic testing on all patients.
Diagnosis and Differential Diagnosis by Clinical Feature and Auxiliary Examinations
The clinical feature of PAI case series is variable but closely similar. As we known, if a patient develops any of the following symptoms: intractable hypoglycemia, hyperpigmentation, hyperkalemia, and hyponatremia with or without circulatory collapse, clitoridauxe and virilization in female neonates, hyperpigmentation of scrotum and enlarged phallus in male neonate, PAI should be considered and steroid hormones should be measured. Isolated hypoaldosteronism could be as first sign of neonates with AHC (7). Patients AHC and CAH often initially present with severe hyperkalemia and hyponatremia, with or without failure to thrive, salt wasting, and vomiting in the neonatal period due to mineralocorticoid deficiency, while patients with FGD do not have salt-wasting. However, transient hyponatremia has been reported in several children with FGD1, sometimes leading to a misdiagnosis of AHC (11). The clinical findings of FGD is very diverse, also including subclinical hypothyroidism, characteristic facial features such as hypertelorism, medial epicanthus and frontal bossing, marginal gross motor developmental delay, gigantism and so on, except for symptoms due to glucocorticoid deficiency (10). Note, though that skin hyperpigmentation is familiar in PAI, absence of obvious hyperpigmentation can not rule out adrenal insufficiency (12). CAH often present with ambiguous or male-appearing external genitalia in girls, while the externalia of patients with AHC or FGD1 often present normal in the neonatal period (6, 13).
Identifying the main laboratory characteristics is helpful for differential diagnosis. 17-OHP, androstenedione, and DHEA-S are substantial elevated in CAH, while all of them are normal or low-level in AHC and FGD1 in the neonatal period. So 17-OHP level is oftern used for neonatal screening for CAH or differential diagnosis between AHC, FGD1, and 21-OHD. Although high serum ACTH and low serum cortisol values are good predictors for PAI, ACTH and cortisol values cannot be used to diagnose PAI alone. That is probably because ACTH and cortisol levels in normal healthy neonates are variable, without a diurnal rythm. Furthermore, the adrenal ultrasound structure characterized by absence or near absence of the permanent zone of the adrenal cortex in patients with AHC. Theoretically, large glands in patients with CAH may be helpful for differentiation. In our report, adrenal ultrasound examination did not demonstrate any apparent abnormality in any of the patients, which may be associated with diagnosis in neonates. Therefore, even if adrenal imaging examination is normal, PAI can not be completely excluded.
Genetic Causes for PAI Case Series
Although genetic testing for the diagnosis of CAH due to 21-OHD is not essential, it is useful to evaluate borderline cases with equivocal biochemical testing. In order to identify other rarer cases of PAI, such as AHC and FGD (10, 14), and get more information for genetic counseling and prognosis predicting, genetic testing is recommended.
CYP21A2 is located in the human leukocyte antigen class III region on the short arm of chromosome 6p21.3. In this region, CYP21A2 and the CYP21A1P pseudogene are arranged in tandem, and both of them contain ten exons spaced over 3.1 kb. Their nucleotide sequences are 98% identical in their exons and approximately 96% identical in their introns (15). The CYP21A1P pseudogene adds new complexities in detecting mutations in CYP21A2 gene. However, nested PCR sequencing combined with MLPA can detect the actual mutations of CYP21A2 gene effectively. Currently, a general correlation between genotype and phenotype of CAH has been established, but the mutation phenotype does not always correlate precisely the genotype and phenotype (16). The most frequent genetic defect in classic form of CAH is IVS2-13A/C>G (36.1%), followed by deletion of exon 1–3 (19.4%), which both lead to extremely low enzyme activity (17). The combination of mutations IVS2-13A/C>G or c.955C>T (exon 8) with a large deletion has been associated with the most severe salt-wasting phenotype (18–20). The combination of c.955C>T (exon 8) with c.1069C>T (exon 8) has also been associated with salt-wasting phenotype (17). An 8-bp deletion in exon 3, reported as 706_713del8bp in this study, caused a nonsense mutation in exon 3 and was associated with salt wasting (17). The correlation between mutation phenotype and phenotype in our cases was consistent with previous reports. In Chinese patients, the frequencies of IVS2-13A/C>G and c.955C>T are 33.9 and 13.3%, respectively, which are much higher than the frequencies of these mutations in any other population (21).
DAX-1 (dosage-sensitive sex-reversal, on the X-chromosome, gene 1), located on the short arm of chromosome Xp21, also known as NR0B1 gene, consists of two exons with a single 3.4-kb intron. It encodes an orphan member of the nuclear receptor superfamily that lacks the characteristic zinc finger DNA-binding domain. Most studies indicate that DAX-1 acts as a transcriptional repressor of genes involved in the steroidogenic pathway and the development of a functioning hypothalamic-pituitary-gonadal axis (5, 22). Loss of function of DAX1 is associated with adrenal hypoplasia and reproductive dysfunction (22). More than 200 different mutations in the DAX1 gene have been identified to date, but the relationship between mutations and the phenotype of AHC has not been documented (23). The age of onset of adrenal insufficiency, disease severity, and clinical symptoms could vary despite carrying the same mutation (24). It is probable that epigenetic or non-genetic mechanisms could modulate patients' phenotype in X-linked AHC. Frameshift and non-sense mutations may occur within the entire open reading frame, while missense mutations are found mainly in the region encoding the C-terminus of the DAX1 protein (22, 25). Other pathogenic alterations comprise copy-number variations (CNVs) of different sizes (26). In patient 6, AHC was caused by a pathogenic deletion encompassing the entire exon 2 of DAX1, which has not been reported before. This hemizygous deletion was inherited from his mother. His mother is an unaffected carrier. Although we did not confirm the presence of mutant protein, the mutation may result in the most truncated inactive protein so far reported, except for one patient's absence of the entire DAX1 gene sequence region (27).
Causal mutations of FGD have been identified in many genes, inactivating mutations in the ACTH Receptor gene (melanocortin 2 receptor, MC2R) on chromosome 18p11.2 account for about 25% of FGD cases and cause FGD1 (28). MC2R is a protein consisting of 297 amino acids, which has high affinity for ACTH (10). The genetic mutation in MC2R gene may induce trafficking failure of the receptor from the endoplasmic reticulum to the cell surface, and ineffective binding to ACTH (10). Severe or homozygous truncating mutations in the MC2R gene may disturb the renin-angiotensin-aldosterone axis mildly. Such patients need temporary replacement of mineralocorticoid, but this condition does not cause long-term mineralocorticoid deficiency after the intervention is stopped. A little more than 30 mutations have been reported in literatures (8). One mutation, c.701C>T, in patient 7, which was inherited from her mother, results in an exchange of a proline for a leucine. Another 1-bp deletion, c.119delT, in patient 7, which was inherited from her father, results in a shift of the open reading frame. Her parents were both unaffected carriers. These two novel mutations have not been reported and are low-frequency variants in the normal population database. According to American College of Medical Genetics and Genomics (ACMG) guidelines, the clinical significance of either c.701C>T or c.119delT is uncertain but is likely pathogenic. C.701C>T has been predicted to be deleterious, and c.119delT has been predicted to have uncertain effects, by bioinformatics software (SIFT, PolyPhen_2, MutationTaster, GERP++, REVEL).
The Therapy and Prognosis of PAI Case Series
In all neonates suspected with PAI, glucocorticoid should be initiated to prevent the potentially life-threatening adrenal crisis. If a neonate with PAI presents in adrenal crisis, the confirmatory blood sample is supposed to be obtained first, and then urgent stress doses of hydrocortisone is essential to be supplied. Until the patient is stable and feeding normally, oral glucocorticoid replacement can be chose, and wheather mineralocorticoid replacement is necessary depends on the cause of PAI. Mineralocorticoid replacement is necessary for classic forms of CAH and AHC, not necessary for FGD (29–31). The goal of therapy is to replace deficient steroids and minimize iatrogenic glucocorticoid excess. It is important to use the lowest effective glucocorticoid dose because excessive dosing is associated with poor long-term health outcomes. Patients with CAH should also be monitored for signs of pubertal onset, patients with AHC be monitored for other coexisting endocrine disturbances such as delayed/precocious puberty and infertility (30–33). Because marginal gross motor developmental delay and gigantism may exist in FGD1, growth and development should also be monitored during follow-up in patients with FGD1 (28, 31).
Conclusion
Upon review of our cases, skin hyperpigmentation and serum electrolyte disturbance are important clinical clues to PAI. In the neonatal period, AHC and FGD are rare etiologies, which should be considered in addition to CAH. Serum ACTH, cortisol, 17-OHP, aldosterone, sex hormone levels, and adrenal ultrasound examination are all important potential distinguishing clues. However, PAI is hard to distinguish because of the non-specific and various clinical feature, especially in the neonatal period. Genetic testing should be included as a alternative diagnostic approach to differentiate all these disorders, and provide information about therapy, prognosis and genetic counseling.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
This study was approved by the institutional Human Investigations Committee. Written informed consent was obtained from the legal guardian of each subject.
Author Contributions
JG participated in cases clinical management, data collection, performed the literature search, and drafted the manuscript. LC managed the clinical cases, conceived the work and reviewed the manuscript for important intellectual content.
Funding
This work was financially supported by Independent Innovation Study Fund of Huazhong University of Science and Technology (2017KFYXJJ129).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Perry R, Kecha O, Paquette J, Huot C, Van Vliet G, Deal C. Primary adrenal insufficiency in children: twenty years experience at the Sainte-Justine Hospital, Montreal. J Clin Endocrinol Metab. (2005) 90:3243–50. doi: 10.1210/jc.2004-0016
2. Wijaya M, Huamei M, Jun Z, Du M, Li Y, Chen Q, et al. Etiology of primary adrenal insufficiency in children: a 29-year single-center experience. J Pediatr Endocrinol Metab. (2019) 32:615–22. doi: 10.1515/jpem-2018-0445
3. Raisingani M, Contreras MF, Prasad K, Pappas JG, Kluge ML, Shah B, et al. Unusual phenotype of congenital adrenal hyperplasia (CAH) with a novel mutation of the CYP21A2 gene. J Pediatr Endocrinol Metab. (2016) 29:867–71. doi: 10.1515/jpem-2015-0457
4. Chiou SH, Hu MC, Chung BC. A missense mutation at Ile172—Asn or Arg356—Trp causes steroid 21-hydroxylase deficiency. J Biol Chem. (1990) 265:3549–52.
5. Nagel SA, Hartmann MF, Riepe FG, Wudy SA, Wabitsch M. Gonadotropin- and adrenocorticotropic hormone independent precocious puberty of gonadal origin in a patient with adrenal hypoplasia congenita due to DAX1 gene mutation – a case report and review of the literature: implications for the pathomechanism. Horm Res Paediatr. (2019) 91:336–45. doi: 10.1159/000495189
6. Pereira BD, Pereira I, Portugal JR, Gonçalves J, Raimundo L. X-linked adrenal hypoplasia congenita: clinical and follow-up findings of two kindreds, one with a novel NR0B1 mutation. Arch Endocrinol Metab. (2015) 59:181–5. doi: 10.1590/2359-3997000000032
7. Iughetti L, Lucaccioni L, Bruzzi P, Ciancia S, Bigi E, Madeo SF, et al. Isolated hypoaldosteronism as first sign of X-linked adrenal hypoplasia congenita caused by a novel mutation in NR0B1/DAX-1 gene: a case report. BMC Med Genet. (2019) 20:98. doi: 10.1186/s12881-019-0834-7
8. Al Jneibi F, Hen T, Rajah J, Nair R. Early diagnosis in familial glucocorticoid deficiency. Dermatoendocrinol. (2017) 9:e1310787. doi: 10.1080/19381980.2017.1310787
9. Sarkar UK, Sarma N, Debbarma S, Mandal AK, Bala AK. A pilot study evaluating therapeutic response of different dosage of oral glucocorticoid in two children with familial glucocorticoid deficiency presenting with diffuse mucocutaneous hyperpigmentation. Indian J Dermatol. (2017) 62:191–4. doi: 10.4103/ijd.IJD_716_16
10. Uyangoda K, Kamalanathan P, Mettananda S. Familial glucocorticoid deficiency presenting with hyperpigmentation, gigantism, and motor development delay: a case report. J Med Case Rep. (2019) 13:280. doi: 10.1186/s13256-019-2206-5
11. Guran T, Buonocore F, Saka N, Ozbek MN, Aycan Z, Bereket A, et al. Rare causes of primary adrenal insufficiency: genetic and clinical characterization of a large nationwide cohort. J Clin Endocrinol Metab. (2016) 101:284–92. doi: 10.1210/jc.2015-3250
12. Khattab A, Nelson-Williams C, Cabreza V, Macdonald A, Loring E, Saland J, et al. A novel de novo frameshift mutation in NR0B1 and low prenatal estriol in adrenal hypoplasia congenita. Ann N Y Acad Sci. (2018) 1433:7–11. doi: 10.1111/nyas.13962
13. Chen C, Zhou R, Fang Y, Jiang L, Liang L, Wang C. Neonatal presentation of familial glucocorticoid deficiency with a MRAP mutation: a case report. Mol Genet Metab Rep. (2016) 9:15–7. doi: 10.1016/j.ymgmr.2016.09.003
14. Al Amer AM, Al Rubaya KM, Alzahrani AS. Adrenal hypoplasia congenita in identical twins. Saudi Med J. (2019) 40:87–92. doi: 10.15537/smj.2019.1.23337
15. Keegan CE, Killeen AA. An overview of molecular diagnosis of steroid 21-hydroxylase deficiency. J Mol Diagn. (2001) 3:49–54. doi: 10.1016/S1525-1578(10)60651-4
16. Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet. (2005) 365:2125–36. doi: 10.1016/S0140-6736(05)66736-0
17. Neocleous V, Fanis P, Phylactou LA, Skordis N. Genotype is associated to the degree of virilization in patients with classic congenital adrenal hyperplasia. Front Endocrinol. (2018) 9:733. doi: 10.3389/fendo.2018.00733
18. Lee HH. Chimeric CYP21P/CYP21 and TNXA/TNXB genes in the RCCX module. Mol Genet Metab. (2005) 84:4–8. doi: 10.1016/j.ymgme.2004.09.009
19. Concolino P, Mello E, Minucci A, Giardina E, Zuppi C, Toscano V, et al. A new CYP21A1P/CYP21A2 chimeric gene identified in an Italian woman suffering from classical congenital adrenal hyperplasia form. BMC Med Genet. (2009) 10:72. doi: 10.1186/1471-2350-10-72
20. L'Allemand D, Tardy V, Gruters A, Schnabel D, Krude H, Morel Y. How a patient homozygous for a 30-kb deletion of the C4-CYP21 genomic region can have a nonclassic form of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2000) 85:4562–7. doi: 10.1210/jcem.85.12.7018
21. Xu C, Jia W, Cheng X, Ying H, Chen J, Xu J, et al. Genotype-phenotype correlation study and mutational and hormonal analysis in a Chinese cohort with 21-hydroxylase deficiency. Mol Genet Genomic Med. (2019) 9:e671. doi: 10.1002/mgg3.671
22. Suthiworachai C, Tammachote R, Srichomthong C, Ittiwut R, Suphapeetiporn K, Sahakitrungruang T, et al. Identification and functional analysis of six DAX1 mutations in patients with X-linked adrenal hypoplasia congenita. J Endocr Soc. (2018) 3:171–80. doi: 10.1210/js.2018-00270
23. Ali JM, Jalaludin MY, Harun F. Late onset X-linked adrenal hypoplasia congenita with hypogonadotropic hypgonadism due to a novel 4-bp deletion in exon 2 of NR0B1. J Pediatr Endocrinol Metab. (2014) 27:1189–92. doi: 10.1515/jpem-2014-0161
24. Rojek A, Flader M, Malecka E, Niedziela M. A novel mutation in the NR0B1 (DAX1) gene in a large family with two boys affected by congenital adrenal hypoplasia. Hormones. (2014) 13:413–9. doi: 10.14310/horm.2002.1495
25. Yang J, Lv Y, Zhou Y, Xiao X. Identification of a novel mutation of NR0B1 in a patient with X-linked adrenal hypoplasia and symptomatic treatment. J Pediatr Endocrinol Metab. (2017) 30:1299–304. doi: 10.1515/jpem-2017-0237
26. Rodríguez Estévez A, Pérez-Nanclares G, Fernández-Toral J, Rivas-Crespo F, López-Siguero JP, Díez I, et al. Clinical and molecular characterization of five spanish kindreds with X-linked adrenal hypoplasia congenita: atypical findings and a novel mutation in NR0B1. J Pediatr Endocrinol Metab. (2015) 28:1129–37. doi: 10.1515/jpem-2014-0472
27. Khadilkar VV, Mangtani HR, Jahagirdar RR, Khatod KA, Phadke ND, Deepa PS, et al. Entire DAX1 gene deletion in an Indian boy with adrenal hypoplasia congenita. Indian J Pediatr. (2013) 80:631–5. doi: 10.1007/s12098-012-0946-y
28. Abuduxikuer K, Li ZD, Xie XB, Li YC, Zhao J, Wang JS. Novel melanocortin 2 receptor variant in a Chinese infant with familial glucocorticoid deficiency type 1, case report and review of literature. Front Endocrinol. (2019) 10:359. doi: 10.3389/fendo.2019.00359
29. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metabol. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
30. Loureiro M, Reis F, Robalo B, Pereira C, Sampaio L. Adrenal hypoplasia congenita: a rare cause of primary adrenal insufficiency and hypogonadotropic hypogonadism. Pediatr Rep. (2015) 7:5936. doi: 10.4081/pr.2015.5936
31. Mazur A, Ostanski M, Kalina M. Familial glucocorticoid deficiency. Pediatr Endocrinol Diabetes Metab. (2007) 13:91–4.
32. Galeotti C, Lahlou Z, Goullon D, Sarda-Thibault H, Cahen-Varsaux J, Bignon-Topalovic J, et al. Longitudinal evaluation of the hypothalamic-pituitary testicular function in 8 boys with adrenal hypoplasia congenita (AHC) due to NR0B1 mutations. PLoS ONE. (2012) 7:e39828. doi: 10.1371/journal.pone.0039828
Keywords: primary adrenocortical insufficiency, congenital adrenal hyperplasia, adrenal hypoplasia congenita, familial glucocorticoid deficiency, hyperpigmentation, hyponatremia, hyperkalemia, gene mutation
Citation: Gao J and Chen L (2020) Primary Adrenocortical Insufficiency Case Series in the Neonatal Period: Genetic Etiologies Are More Common Than Expected. Front. Pediatr. 8:464. doi: 10.3389/fped.2020.00464
Received: 05 March 2020; Accepted: 03 July 2020;
Published: 12 August 2020.
Edited by:
Henry J. Rozycki, Virginia Commonwealth University, United StatesReviewed by:
Anna Nordenstrom, Karolinska Institutet (KI), SwedenSarantis Livadas, Metropolitan Hospital, Greece
Copyright © 2020 Gao and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ling Chen, NzkwMzU2NzYwQHFxLmNvbQ==