 Giulia Solazzo
Giulia Solazzo Giuliana Ferrante
Giuliana Ferrante Stefania La Grutta
Stefania La Grutta- 1Institute for Research and Biomedical Innovation (IRIB), National Research Council (CNR), Palermo, Italy
- 2Department of Health Promotion Sciences, Maternal and Infant Care, Internal Medicine and Medical Specialities, University of Palermo, Palermo, Italy
Asthma is one of the most widespread chronic respiratory conditions. This disease primarily develops in childhood and is influenced by different factors, mainly genetics and environmental factors. DNA methylation is an epigenetic mechanism which may represent a bridge between these two factors, providing a tool to comprehend the interaction between genetics and environment. Most epidemiological studies in this field have been conducted using blood samples, although DNA methylation marks in blood may not be reliable for drawing exhaustive conclusions about DNA methylation in the airways. Because of the role of nasal epithelium in asthma and the tissue specificity of DNA methylation, studying the relationship between DNA methylation and childhood asthma might reveal crucial information about this widespread respiratory disease. The purpose of this review is to describe current findings in this field of research. We will present a viewpoint of selected studies, consider strengths and limitations, and propose future research in this area.
Background
Asthma is a chronic inflammatory condition that affects the airways and is defined by recurrent episodes of wheeze, breathlessness, chest tightness and/or cough associated with variable expiratory airflow limitation (1). Despite environmental exposures explaining about one third of risk factors for asthma, genetic factors are also responsible for vulnerability to the disease (1–3). Epigenetic marks have been suggested as a link between genetic predisposition and environmental exposure, providing a special chance to clarify the relationship between genetics and environment (4, 5).
Much evidence suggest that epigenetic processes, such as DNA methylation, play a role in asthma susceptibility (6). Although most of epidemiological studies have been conducted using blood samples, there is evidence that the study of the nasal methylome allows for making more reliable conclusions about DNA methylation in the lungs. Recently, Brugha et al. provided evidence that the bronchial epithelium and blood are twice as distant as the bronchial and nasal epithelium, emphasizing that DNA methylation in blood samples may not be informative enough to draw conclusions about methylation marks in the airways (7). Thanks to the tissue specificity, the DNA methylation of nasal epithelium may be crucial for understanding the complex molecular patterns involved in asthma. Currently, few studies considered nasal epithelium in pediatric asthma showing that the nose is both a remarkable and less-invasive surrogate for the bronchi in transcriptional profiling studies (8) and discriminates between stable and acute asthma in children (9).
One of the first studies in this field was performed by Baccarelli et al. in 2012 (10). Using pyrosequencing, the authors analyzed the DNA methylation of interleukin-6 (IL-6) and nitric oxide synthase (iNOS) promoters, and DNA methylation of repetitive elements: Alu (Arthrobacter luteus) and LINE-1 (Long Interspersed Nuclear Elements). This revealed that asthmatic children who had higher values of fractional exhaled nitric oxide (FeNO) showed a hypomethylation of the IL-16 and iNOS promoters. More recently, few studies performed epigenome-wide association analysis between the nasal methylome and pediatric asthma (11–14).
In this review, we aim to summarize current evidence about the relationship between DNA methylation in nasal epithelium and childhood asthma. Through this, we will present a viewpoint of selected studies considering the current strengths and limitations and introducing a future scenario in this research field.
Epigenetics Mechanism: The Basics
The term epigenetics refers to DNA modifications that do not change the gene sequence. Modifications in DNA methylation, histone proteins that are responsible for DNA packaging, and microRNAs patterns can change genome function under different environmental stimuli (15). DNA methylation is a well-known epigenetic marker. This modification occurs in the cytosine residue of cytosine-phosphate-guanine (CpG) dinucleotides. Approximately 80% of all CpGs in the mammalian genome are estimated to be methylated. The remaining CpG residues, referred to as CpG islands, are predominantly situated in promoters of genes that are constitutively or inducible active. Usually these promoters are >500 base pairs long and more than 55% of each promoter consist of GC (16, 17). The methylation of these sites is produced by specific enzymes, the DNA methyltransferases (DNMTs). These enzymes are involved in several mechanisms such as transcriptional regulation, genomic stability, chromatin structure modulation, X chromosome inactivation, and the silencing of DNA transposable elements (6).
Nasal Epithelium Sampling
The nasal brushing technique is the most commonly used in children for collecting nasal epithelium samples (10–14). This method has been widely used to study chronic airway diseases including cystic fibrosis, asthma, and other obstructive respiratory diseases. Cells in nasal epithelium have properties resembling bronchial epithelial cells and nasal brushing is much less invasive than bronchial brushing or bronchoalveolar lavage; thus, this technique represents a good surrogate model for lower respiratory tract studies (18). Usually, cells in the nasal epithelium are collected by brushing the inferior turbinate with a cytobrush, but it is also possible to collect nasal cells from the anterior nares by brushing the nasal bone and vigorously scrubbing the nares (19).
Evidence Of The Role Of DNA Methylation In Nasal Epithelium On Children With Asthma
To locate evidence for the review topic, we conducted a keyword search through the PubMed database including the following criteria: (1) biological sample: nasal epithelium; (2) study population age: between 1 month and 20 years; (3) study area: metropolitan; (4) outcomes of interest: atopic and non-atopic asthma; (6) biological modification investigated: DNA methylation; (7) type of analysis: epigenome-wide or pyrosequencing analysis; (8) studies published from 2012 to 2019, as this range includes the first study published on this topic and the most recent studies; (9) language: English. We used the following search strategy: children [ALL] AND DNA methylation [ALL] AND nasal epithelium [ALL]; nasal epithelium. Abstracts (i.e., conference proceedings) and reviews were excluded. Among the 15 studies identified, eleven were excluded due to the following reasons: two studies used nasal cells to replicate results from other tissues, so that nasal epithelium was not the main tissue investigated (19, 20); one study analyzed DNA methylation in relation to single-nucleotide polymorphisms (SNPs) and gene expression in childhood asthma (21); seven studies did not focus the association between DNA methylation and asthma, but other topics such as lung function and corticosteroid treatment response (7, 22–27); one study was conducted on newborns and findings were verified in another cohort of children to characterize early DNA methylation patterns in airways depending on gender (28). Finally, 4 studies were included in this review (Table 1).
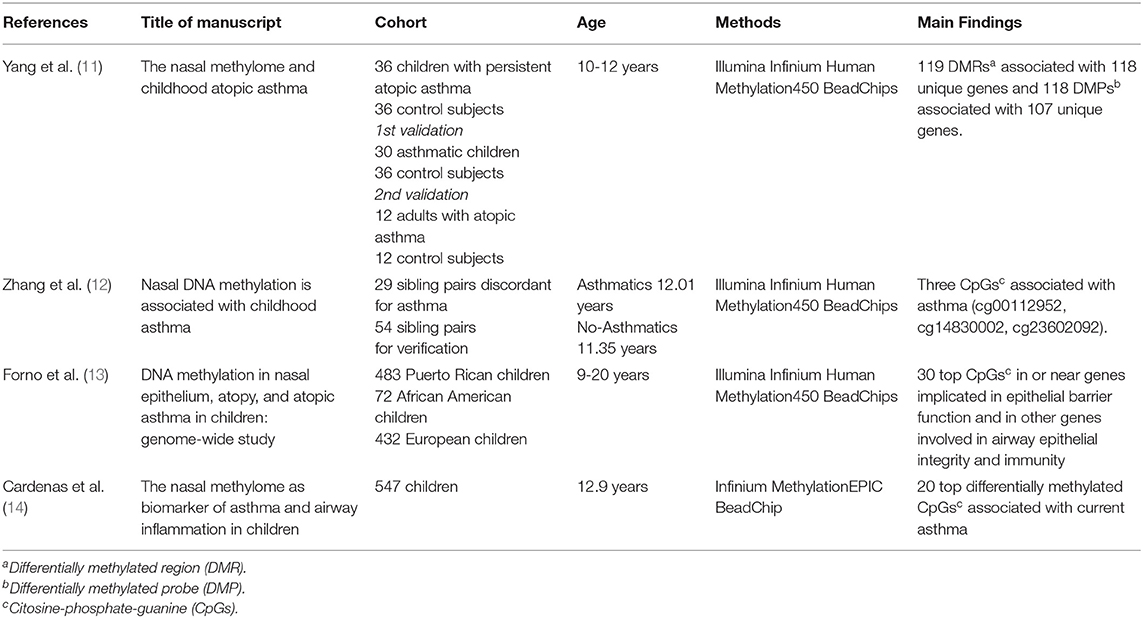
Table 1. Main characteristics of the four studies selected.
In 2017, Yang et al. (11) compared DNA methylation markers in genome and gene expression in 36 children with persistent atopic asthma and 36 non-asthmatic children. The results from this analysis were verified using a child' population of 30 subjects (aged 10 to 12 years) with atopic asthma vs. the same control group, as shared control samples [N = 36], and validated in an independent adult population (ranged 27 to 74 years) with atopic asthma [N = 12] and without asthma [N = 12]. The authors identified 119 epigenome-wide significant differentially methylated regions (DMRs) associated with 118 unique genes and 118 differentially methylated probes (DMPs) associated with 107 unique genes. Among 186 allergic asthma-associated differentially methylated genes (DMR and/or DMP), they found genes with an established role in asthma and atopy, immunity, in addition to genes related to extracellular matrix, cell adhesion, epigenetic regulation, and airflow obstruction. Gene expression analysis identified 53 differentially expressed genes. Among those genes, 32 had significant methylation-expression relationships within 5 kb. This was the first epigenome-wide association study on childhood asthma. Advantages of this type of analysis are that it detects DNA methylation signals in different gene regions and eventually allows to study how the methylation status of these gene regions relates to their expression. This study detected new genes with an expression status that can be involved in childhood asthma.
In 2018, Zhang et al. (12) used a cohort of 29 sibling pairs discordant for asthma. This epigenome-wide analysis identified six CpG sites for which methylation was nominally associated with asthma status. Four of them were placed in promoter regions and three of these sites were verified in a cohort of 54 sibling pairs through pyrosequencing. Two CpG sites (cg00112952 and cg14830002) were identified within the promoter region of OR2B11(olfactory receptor family 2 subfamily B member 11), and the other was located in the TET1 (tet methylcytosine dioxygenase 1) promoter (cg23602092), which was already identified in a previous study on children (26). The OR2B11 is a gene located in chromosome 1 which encodes an olfactory receptor that interacts with odorant molecules in the nose to initiate a neuronal response. The protein encoded by TET1 is a demethylase that belongs to the TET (ten-eleven translocation) family. Interestingly, a previous study in humans (26) proposed a role for TET1 in the pathogenesis of pediatric asthma. The finding that cg14830002 is associated with allergies in non-asthmatics and that cg23602092 is associated with asthma symptoms reveals new disease-contributing epigenetic mechanisms and novel CpG sites in nasal epithelial cells which could serve as useful asthma biomarkers.
In 2019, Forno et al. (13) carried out a genome-wide study of DNA methylation in nasal epithelium and atopy or atopic asthma in 483 Puerto Rican children and young adults (9-20 years). The authors identified specific methylation profiles associated with atopy or atopic asthma. In particular, 30 top CpG sites were selected. Many genes reported in this study are involved in epithelial barrier mechanisms and immunity, like CDH26 (cadherin 26), CDHR3 (cadherin related family member 3), GJA4 (gap junction protein alpha 4), and CAPN14 (calpain 14). Among genes of the 30 top CpG sites, there were also genes involved in atopy and inflammation like NTRK1 (neurotrophic receptor tyrosine kinase 1), and lung function like SLC9A3 (solute carrier family 9 member A3). The authors detected a hypomethylation and increased expression of NTRK1 in atopy; this gene is induced by interleukin 13 (IL-13) and is increased in eosinophilic esophagitis. SLC9A3 gene codes for an Na+/H+ exchanger and has been linked to reduced lung function in cystic fibrosis. Furthermore, the authors described two genes, which are expressed in many tissues, including the lung: PCSK6 (proprotein convertase subtilisin/kexin type 6) encodes a protease constitutively secreted in the extracellular matrix, while the product of METTL1 (methyltransferase like 1) gene contains a conserved S-adenosylmethionine-binding motif and is inactivated by phosphorylation. Notably, the 30-CpG panel was able to discriminate atopic subjects in three cohorts regardless of racial and ethnic differences.
In the same year Cardenas et al. (14) performed an epigenome-wide association study (EWAS) in children with asthma (mean age 12.9 years) and healthy controls. Using nasal swabs, the authors reported the top 20 differentially methylated CpGs associated with current asthma compared to non-asthmatic subjects. They also reported the differential DNA methylation of genomic regions of three genes previously associated with asthma: TNIP-1 (TNFAIP3 interacting protein 1), IL-13, and CHI3L1 (chitinase 3 like 1) (29–31), showing that the nasal methylome may serve as a reliable biomarker of asthma in children.
According to the findings described in the aforementioned four studies, a few loci do overlap. In the 30 top CpGs found by Forno et al. (13), GJA4 and METTL1 sites overlap with CpGs previously described in the study by Yang et al. (11), which had shown a hypomethylation in these sites in asthma cases when compared to controls. Interestingly, Forno et al. (13) replicated their top results in the Yang and colleague's cohort and 28 of 30 CpGs were significant (p-value < 0.01). Among the results described (11, 13), in both studies PCSK6 was hypomethylated in children with atopy or atopic asthma compared to controls. Cardenas et al. (14) found two CpGs, cg22855021 in TSHR (thyroid stimulating hormone receptor) and cg24707200 in NTRK1, associated with allergic asthma and also overlapping the top results described in the study by Forno et al. (13). The authors replicated their top results in Forno et al. (13) and Yang et al. (11) cohorts, finding some genes which appeared significant in both the cohorts. In particular, they observed significant sites in EPX (eosinophil peroxidase) and EVL (Enah/Vasp-like) genes for asthma.
The top five results of each study selected in the current review are described in Table 2. Overall, all these studies identified two main groups of loci: those in genes related to immunity, like ALOX15 (arachidonate 15-lipoxygenase), and those in genes involved in epithelial cells function and integrity like receptor, extracellular matrix protein and connexin as GJA4, OR2B11, NTRK1. These findings proved the suitability of using the nasal methylome in research settings and pave the way toward developing a nasal methylation panel for future clinical applications in childhood asthma.
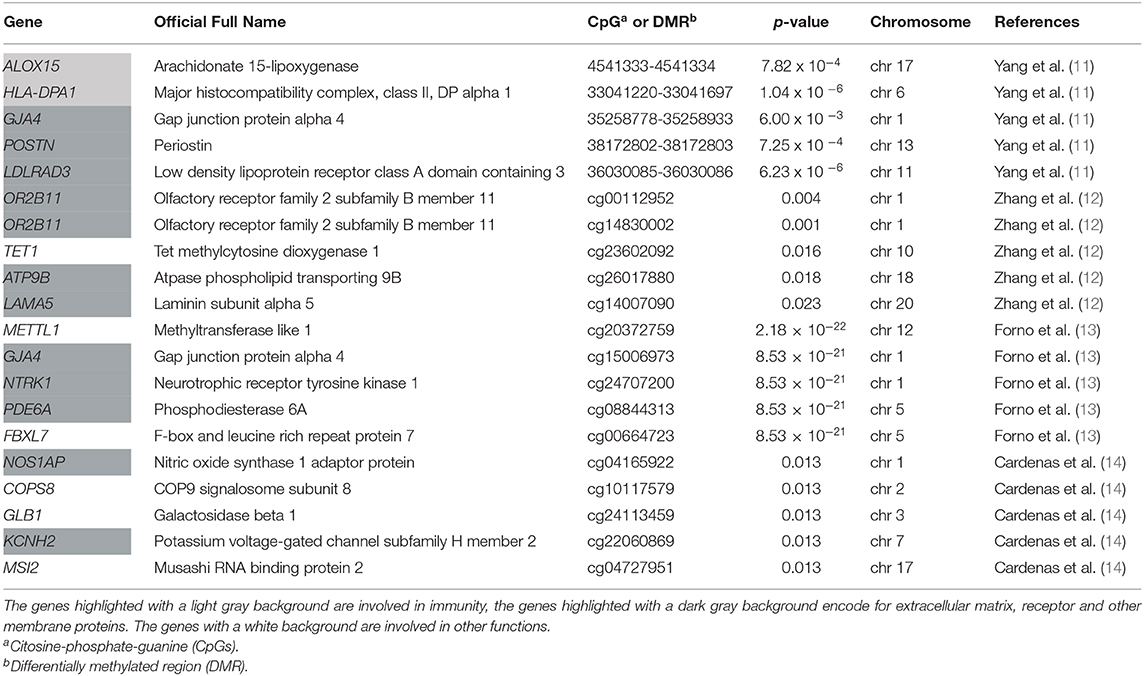
Table 2. Top five results of the four studies selected.
Comparison of Selected Studies
All the studies selected in the current review were focused on asthma and atopic asthma (11–14). In all the studies asthma was defined by a medical diagnosis. In particular, Yang et al. (11) defined asthma following these criteria: a physician diagnosis and positive skin prick-test to at least one indoor aeroallergen. Zhang et al. (12) used a diagnosis of asthma acquired from parental reports and confirmed by medical records. Forno et al. (13) described atopic asthma as a doctor diagnosis plus at least one episode of wheeze in the last 12 months and at least one positive IgE to common aeroallergens. Finally, in the study by Cardenas et al. (14), asthma was obtained from parental report of a medical diagnosis plus report of wheeze or asthma medication in the past year and any positive IgE to common indoor or outdoor aeroallergens. Only Zhang et al. (12) reported the level of asthma severity, defined by symptom frequency using a respiratory symptom score.
With regard to study population, three studies (11, 12, 14) were performed in cohorts aged around 12 years, while Forno et al. (13) used a cohort of subjects aged between 9 and 20 years. The participants' ethnicities also differ between the studies. In Yang et al. (11), most of the participants were African Americans and the rest were Hispanic or Latino; the Zhang et al. (12) cohort included 96% African Americans; Forno et al. (13) recruited Puerto Rican children (all Hispanic or Latino); in the study by Cardenas et al. (14), 67.1% were white and only 16.1% were black. Since DNA methylation patterns can be influenced by age and ethnicity, using cohorts with a small age range and with a homogenous race may limit the generalizability of findings. To overcome this issue, the results by Yang et al. (11) were validated in two different cohorts, one of which was a group of adult Caucasians. To confirm the association between asthma and the six CpGs found, Zhang and colleagues (12) replicated their results using data from the Genomic of Secondhand smoke Exposure in Pediatric (GSEP) study and the Public Inner-City Asthma Consortium (ICAC) study. The top 30 results by Forno et al. (13) were replicated in two cohorts: the Yang et al. (11) cohort and the Prevention and Incidence of Asthma and Mite Allergy (PIAMA) cohort (32). Finally, also Cardenas et al. replicated their results using data from Yang et al. (11) and Forno et al. (13).
With regard to nasal sampling, three of the selected studies used cells from the inferior turbinate (11–13), while Cardenas et al. (14) performed their analysis on nasal cells from the anterior nares. The latter technique is easier to perform, and it does not necessitate a speculum to visualize nasal anatomy or specialized training. One possible limitation could be the lower average of respiratory epithelial cells collected (65%) compared to the inferior turbinate (99%). Nonetheless, the patients' discomfort is lower for anterior nares compared to inferior turbinate sampling (33). This is particularly important in pediatric settings where collecting samples by brushing the anterior nares would be an easier and less invasive method for nasal sampling.
The epigenome-wide DNA methylation was measured on Illumina's Infinium Human Methylation 450k BeadChip in three studies (11–13), while Cardenas et al. (14) performed the measurements with the Infinium MethylationEPIC BeadChip. The difference between these two arrays is that the Infinium Human Methylation 450 k BeadChip measures methylation at 450,000 CpG sites in the genome, while Infinium MethylationEPIC BeadChip measures methylation at more than 850,000 CpG sites. However, they both use the same technology.
The statistical analysis process was similar among the selected studies. The authors identified DMRs and they adjusted for false discovery rates (FDR). However, Yang et al. (11) also analyzed the gene expression and investigated the relationship between DMRs and gene expression changes. Zhang et al. (12) validated the association analysis between DNA methylation and asthma in candidate sites using pyrosequencing. Forno et al. (13) determined if significant methylation signals were associated with gene expression with a transcriptome-wide analysis. Cardenas et al. (14) also calculated the DNA methylation age using an online calculator (https://dnamage.genetics.ucla.edu/). A further strength in the studies performed by Forno et al. (13) and Cardenas et al. (14) is the large sample size, which increased the statistical power. However, to consider the effect size, Zhang and his colleagues (12) performed a sensitivity analysis. Finally, whereas three of the selected studies evaluated the impact of environmental exposures using questionnaires (11, 13, 14), the study by Zhang et al. (12) used a cohort of sibling-pairs, which allowed a better control of environmental confounding factors that may modify DNA methylation and contribute to asthma. Notably, in all the studies, analyses were adjusted for smoke exposure, which is known to affect DNA methylation (33).
Future Perspectives
Previous studies investigating DNA methylation in blood samples have detected loci which have been found in the nasal epithelium. In the study by Forno et al. (13), PCSK6 and METTL1 were top results, and they had been previously reported in blood samples in a case-control study performed in asthmatic children and healthy subjects (34). Also Cardenas et al. (14) reported genes that were previously discovered in the same study (34), among which a specific DNA methylation mark, the RUNX3 (RUNX family transcription factor 3), which is involved in T cell maturation. Findings from epigenome-wide meta-analysis (20) also suggested that CpG sites involved in inflammation are associated with asthma in both blood and nasal epithelium. Accordingly, methylome marks in asthma appear to be stable in both blood and nasal cells. The relationship between these two biosamples is still not clear. However, the finding of new genes in the studies included in the current review suggests that DNA methylation in the nasal epithelium may provide more information about airway epithelium pathways involved in childhood asthma. For instance, a study in nasal epithelium found for the first time a different methylation status in one gene, SLC9A3 (solute carrier family 9 member A3), encoding for an Na+/H+ exchanger which was associated with decreased in lung function in cystic fibrosis (13). According to the available evidence, it seems that the methylome changes in blood samples often involved inflammatory mediators, while the methylome changes in nasal cells also included many proteins of extracellular matrix and membrane proteins. In particular, the pathway in nasal cells of asthmatic subjects of genes involved in extracellular matrix organization and disassembly, collagen catabolic and metabolic processes, suggests an important role of dysfunctional airway epithelium in asthma. However, it is necessary to add more epigenome-wide association analysis and to compare the results from studies in both the biosamples, in order to clarify how the information from blood and nasal epithelium differ, and then optimize their use as biomarkers.
With respect to study design, further studies in longitudinal cohorts are required to develop predictive nasal methylation panels suitable for early prediction of asthma, as well as gene candidate studies to analyze genes found to be significantly related with asthma. Since sensitization to aeroallergens has been found to drive an overlap between nasal methylome correlated with asthma and rhinitis (35), future studies should carefully consider the coexistence of rhinitis when investigating the association of DNA methylation with asthma in nasal epithelium. Furthermore, it would be fascinating to study the influence of aging on DNA methylation on this tissue, which is particularly exposed to environmental factors. Indeed, exploring how the estimated epigenetic age differs across a population of the same age may contribute to understand the effect of endogenous or exogenous stress factors on aging (36). Epigenetics biomarkers of aging, otherwise known as “the epigenetic clock,” have already been studied widely in aging-associated diseases, but evidence about the epigenetics clock's role in pediatric allergy and asthma is scant (36). To our knowledge, only one study used the nasal methylome to estimate epigenetic age acceleration in children (14), reporting a significant epigenetic age acceleration in subjects with current asthma and a greater age acceleration for those with allergic asthma. Since several age-related changes occur in lung physiology and morphology and likely have an impact on asthma, introducing aging information will allow the use of DNA methylation as a reliable biomarker for asthma in children, and might contribute to the development of antiaging therapies.
Conclusions
Studies in children suggest that differences in DNA methylation are associated with asthma. Emerging findings in this field of research suggest that the nasal epithelium can be used to reliably infer the presence of asthma. In particular, recent epigenome-wide association studies detected new differentially methylated genes involved in solute carriers and membrane transport proteins and confirmed the association with genes involved in the immune system for asthma. However, there is limited overlap between the studies which prohibits firm conclusions from being draw on the data. This could be due to different study design and heterogeneous techniques as well as to the very limited number of studies in this field. Therefore, extending this analysis using longitudinal cohorts and analyzing gene candidates selected from relevant genes could be a promising evolution of this research area.
Author Contributions
GS, GF, and SL provided substantial contributions to the conception or design of the work, revised the manuscript for important intellectual content, approved the final version, and agreed to be accountable for all aspects of the work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ferrante G, La Grutta S. The burden of pediatric asthma. Front Pediatr. (2018) 6:186. doi: 10.3389/fped.2018.00186
2. GBD 2015 Chronic Respiratory Disease Collaborators. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med. (2017) 5:691–706. doi: 10.1016/S2213-2600(17)30293-X
3. La Grutta S, Indinnimeo L, di Coste A, Ferrante G, Landi M, Pelosi U, et al. Environmental risk factors and lung diseases in children: from guidelines to health effects. Early Hum Dev. (2013) 89:S59–62. doi: 10.1016/j.earlhumdev.2013.07.025
4. Davidson EJ, Yang IV. Role of epigenetics in the development of childhood asthma. Curr Opin Allergy Clin Immunol. (2018) 18:132–8. doi: 10.1097/ACI.0000000000000429
5. DeVries A, Vercelli D. Epigenetic Mechanisms in Asthma. Ann Am Thorac Soc. (2016) 13:S48–S50. doi: 10.1513/AnnalsATS.201507-420MG
6. Adcock IM, Ford P, Ito K, Barnes PJ. Epigenetics and airways disease. Respir Res. (2006) 7:21. doi: 10.1186/1465-9921-7-21
7. Brugha R, Lowe R, Henderson AJ, Holloway JW, Rakyan V, Wozniak E, et al. DNA methylation profiles between airway epithelium and proxy tissues in children. Acta Paediatr. (2017) 106:2011–6. doi: 10.1111/apa.14027
8. Poole A, Urbanek C, Eng C, Schageman J, Jacobson S, O'Connor BP, et al. Dissecting childhood asthma with nasal transcriptomics distinguishes subphenotypes of disease. J Allergy Clin Immunol. (2014) 133:670–8.e12. doi: 10.1016/j.jaci.2013.11.025
9. Guajardo JR, Schleifer KW, Daines MO, Ruddy RM, Aronow BJ, Wills-Karp M, et al. Altered gene expression profiles in nasal respiratory epithelium reflect stable vs. acute childhood asthma. J Allergy Clin Immunol. (2005) 115:243–51. doi: 10.1016/j.jaci.2004.10.032
10. Baccarelli A, Rusconi F, Bollati V, Catelan D, Accetta G, Hou L, et al. Nasal cell DNA methylation, inflammation, lung function and wheezing in children with asthma. Epigenomics. (2012) 4:91–100. doi: 10.2217/epi.11.106
11. Yang IV, Pedersen BS, Liu AH, O'Connor GT, Pillai D, Kattan M, et al. The nasal methylome and childhood atopic asthma. J Allergy Clin Immunol. (2017) 139:1478–88. doi: 10.1016/j.jaci.2016.07.036
12. Zhang X, Biagini Myers JM, Burleson J, Ulm A, Bryan KS, Chen X, et al. Nasal DNA methylation is associated with childhood asthma. Epigenomics. (2018) 10:629–41. doi: 10.2217/epi-2017-0127
13. Forno E, Wang T, Qi C, Yan Q, Xu C-J, Boutaoui N, et al. DNA methylation in nasal epithelium, atopy, and atopic asthma in children: a genome-wide study. Lancet Respir Med. (2019) 7:336–46. doi: 10.1016/S2213-2600(18)30466-1
14. Cardenas A, Sordillo JE, Rifas-Shiman SL, Chung W, Liang L, Coull BA, et al. The nasal methylome as a biomarker of asthma and airway inflammation in children. Nat Commun. (2019) 10:3095. doi: 10.1038/s41467-019-11058-3
15. Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr. (2009) 21:243–51. doi: 10.1097/mop.0b013e32832925cc
16. Robertson KD. DNA methylation and human disease. Nat Rev Genet. (2005) 6:597–610. doi: 10.1038/nrg1655
17. Takai D, Jones PA. The CpG island searcher: a new WWW resource. In Silico Biol (Gedrukt). (2003) 3:235–40.
18. Stokes AB, Kieninger E, Schögler A, Kopf BS, Casaulta C, Geiser T, et al. Comparison of three different brushing techniques to isolate and culture primary nasal epithelial cells from human subjects. Exp Lung Res. (2014) 40:327–32. doi: 10.3109/01902148.2014.925987
19. Reese SE, Xu C-J, den Dekker HT, Lee MK, Sikdar S, Ruiz-Arenas C, et al. Epigenome-wide meta-analysis of DNA methylation and childhood asthma. J Allergy Clin Immunol. (2019) 143:2062–74. doi: 10.1016/j.jaci.2018.11.04320
20. Xu C-J, Söderhäll C, Bustamante M, Baïz N, Gruzieva O, Gehring U, et al. DNA methylation in childhood asthma: an epigenome-wide meta-analysis. Lancet Respir Med. (2018) 6:379–88. doi: 10.1016/S2213-2600(18)30052-3
21. Kim S, Forno E, Yan Q, Jiang Y, Zhang R, Boutaoui N, et al. SNPs identified by GWAS affect asthma risk through DNA methylation and expression of cis-genes in airway epithelium. Eur Respir J. (2019) 55:1902079. doi: 10.1183/13993003.02079-2019
22. Xiao C, Biagini Myers JM, Ji H, Metz K, Martin LJ, Lindsey M, et al. Vanin-1 expression and methylation discriminate pediatric asthma corticosteroid treatment response. J Allergy Clin Immunol. (2015) 136:923–31.e3. doi: 10.1016/j.jaci.2015.01.045
23. Lee AG, Le Grand B, Hsu H-HL, Chiu Y-HM, Brennan KJ, Bose S, et al. Prenatal fine particulate exposure associated with reduced childhood lung function and nasal epithelia GSTP1 hypermethylation: Sex-specific effects. Respir Res. (2018) 19:76. doi: 10.1186/s12931-018-0774-3
24. Zhang X, Biagini Myers JM, Yadagiri VK, Ulm A, Chen X, Weirauch MT, et al. Nasal DNA methylation differentiates corticosteroid treatment response in pediatric asthma: A pilot study. PLoS ONE. (2017) 12:e0186150. doi: 10.1371/journal.pone.0186150
25. Somineni HK, Zhang X, Biagini Myers JM, Kovacic MB, Ulm A, Jurcak N, et al. Ten-eleven translocation 1 (TET1) methylation is associated with childhood asthma and traffic-related air pollution. J Allergy Clin Immunol. (2016) 137:797–805.e5. doi: 10.1016/j.jaci.2015.10.021
26. Pech M, Weckmann M, König IR, Franke A, Heinsen F-A, Oliver B, et al. Rhinovirus infections change DNA methylation and mRNA expression in children with asthma. PLoS ONE. (2018) 13:e0205275. doi: 10.1371/journal.pone.0205275
27. Ji H, Zhang X, Oh S, Mayhew CN, Ulm A, Somineni HK, et al. Dynamic transcriptional and epigenomic reprogramming from pediatric nasal epithelial cells to induced pluripotent stem cells. J Allergy Clin Immunol. (2015) 135:236–44. doi: 10.1016/j.jaci.2014.08.038
28. Nino CL, Perez GF, Isaza N, Gutierrez MJ, Gomez JL, Nino G. Characterization of sex-based Dna methylation signatures in the airways during early life. Sci Rep. (2018) 8:5. doi: 10.1038/s41598-018-23063-5
29. Li X, Ampleford EJ, Howard TD, Moore WC, Torgerson DG, Li H, et al. Genome-wide association studies of asthma indicate opposite immunopathogenesis direction from autoimmune diseases. J Allergy Clin Immunol. (2012) 130:861–8.e7. doi: 10.1016/j.jaci.2012.04.041
30. Demenais F, Margaritte-Jeannin P, Barnes KC, Cookson WOC, Altmüller J, Ang W, et al. Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks. Nat Genet. (2018) 50:42–53. doi: 10.1038/s41588-017-0014-7
31. Ober C, Tan Z, Sun Y, Possick JD, Pan L, Nicolae R, et al. Effect of variation in CHI3L1 on serum YKL-40 level, risk of asthma, and lung function. N Engl J Med. (2008) 358:1682–91. doi: 10.1056/NEJMoa0708801
32. Wijga AH, Kerkhof M, Gehring U, de Jongste JC, Postma DS, Aalberse RC, et al. Cohort profile: the prevention and incidence of asthma and mite allergy (PIAMA) birth cohort. Int J Epidemiol. (2014) 43:527–35. doi: 10.1093/ije/dys231
33. Lai PS, Liang L, Cibas ES, Liu AH, Gold DR, Baccarelli A, et al. Alternate methods of nasal epithelial cell sampling for airway genomic studies. J Allergy Clin Immunol. (2015) 136:1120–3.e4. doi: 10.1016/j.jaci.2015.04.032
34. Yang IV, Pedersen BS, Liu A, O'Connor GT, Teach SJ, Kattan M, et al. DNA methylation and childhood asthma in the inner city. J Allergy Clin Immunol. (2015) 136:69–80. doi: 10.1016/j.jaci.2015.01.025
35. Qi C, Jiang Y, Yang IV, Forno E, Wang T, Vonk JM, et al. Nasal DNA methylation profiling of asthma and rhinitis. J Allergy Clin Immunol. (2020) S0091-6749 30034–8. doi: 10.1016/j.jaci.2019.12.911
Keywords: DNA methylation, nasal epithelium, asthma, biomarker, children
Citation: Solazzo G, Ferrante G and La Grutta S (2020) DNA Methylation in Nasal Epithelium: Strengths and Limitations of an Emergent Biomarker for Childhood Asthma. Front. Pediatr. 8:256. doi: 10.3389/fped.2020.00256
Received: 21 February 2020; Accepted: 23 April 2020;
Published: 15 May 2020.
Edited by:
Mario Barreto, Sapienza University of Rome, ItalyReviewed by:
Jorg Tost, Commissariat à l'Energie Atomique et aux Energies Alternatives, FranceGiuseppe Pingitore, ASL Roma, Italy
Copyright © 2020 Solazzo, Ferrante and La Grutta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuliana Ferrante, Z2l1bGlhbmEuZmVycmFudGVAdW5pcGEuaXQ=