 Meili Wei
Meili Wei Haibo Fu
Haibo Fu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 17 April 2020
Sec. Pediatric Pulmonology
Volume 8 - 2020 | https://doi.org/10.3389/fped.2020.00138
The mutations in the ABCA3 (ATP-binding cassette transporter subfamily A member 3) gene could result in lethal respiratory distress syndrome (RDS) in neonates and interstitial lung disease (ILD) in infants and children. Here, we describe a full-term newborn who manifested respiratory distress 20 min after birth and then gradually developed hypoxemic respiratory failure and died on 53 days of life. A homozygous missense mutation (c.746C >T) was identified in exon 8 of ABCA3 gene in the neonate by next-generation sequencing, and the mutations were inherited from parents, respectively. This homozygous mutation is the first reported to date.
Neonatal respiratory distress syndrome (NRDS) is a primary or secondary deficiency of pulmonary surfactant and is the primary cause of neonatal death. Survivors may have severe interstitial lung disease (ILD). ILD was a group of clinically, radiologically, and pathologically similar heterogeneous diseases, characterized by progressive oxygenation disorder and respiratory failure. ILD is extremely rare in infants and children, the prevalence is 1 per 100,000, and about 10% of ILD are caused by mutations in genes encoding pulmonary surfactant and related metabolic pathways (1–3), including surfactant proteins (SP)-B, SP-C, SP-D, ATP-binding cassette subfamily A member 3 (ABCA3), NKX2-1, TBX4, etc. There was great heterogeneity in the severity of ILD caused by ABCA3 mutations, ranging from the fetal neonatal respiratory distress after birth to the children's ILD (4–6).
Here, we present clinical characteristics and outcomes of ILD in a neonate that correlated to homozygous missense mutation (c.746C > T) in the ABCA3 gene.
A full-term male Chinese neonate weighting 3,900 g was born (gravida 1, para 1) at 386/7 weeks of gestation via cesarean section due to breech presentation. The Apgar scores at 1, 5, and 10 min were all 10. The parents were both 33-years Han Chinese and denied the family history of similar disease or any other genetic disease.
Respiratory distress occurred at 20 min after birth; the neonate was given humidified oxygen with face mask. Due to the progressively exacerbated respiratory distress, he was admitted to the neonatal intensive care unit. The physical examination was notable for cyanosis, grunting, tachypnea respiratory rate more than 80 breaths per minute, intercostal and subcostal retractions, reduced primitive reflexes. He was supported with nasal continuous positive airway pressure (NCPAP) with FiO2 of 0.25. Arterial blood gas showed a PaO2 of 58 mmHg. Due to the progressively deteriorated respiratory distress, FiO2 was gradually elevated to 0.45 at 12 h of life. Chest radiograph showed decreased transmittance in bilateral lungs (Figure 1A). At 31 h of life, he developed lethal respiratory distress with PaO2 of 36 mmHg. Chest radiograph showed decreased transmittance with blurred cardiac and diaphragmatic margins (Figure 1B). Echocardiography excluded structural heart disease and pulmonary hypertension. He was intubated and given high-frequency oscillatory ventilation with FiO2 of 0.85. After the administration of 360 mg pulmonary surfactant, his respiratory distress moderately ameliorated with FiO2 gradually decreased to 0.6. Blood tests indicated leukocytosis, elevations in C-reactive protein concentration (23 mg/L), procalcitonin concentration (11 ng/ml), and interleukin-6 concentration (228 pg/ml). All the results of culture including blood, urine, and cerebrospinal fluid were negative. Intravenous meropenem and immunoglobulin (1 g/kg/day) were empirically given. At 3 days of life, the patient was given a second dose of pulmonary surfactant due to the persistent respiratory distress. Oxygen demand hereby decreased with FiO2 of 0.5. Chest radiograph showed diffuse decreased transmittance with air bronchograms in the right and left lower lung lobes (Figure 1C). At 4–6 days of life, clinical condition was relatively stable with mild reduction of FiO2 from 0.45 to 0.40. Meropenem was discontinued at 6 days of life.
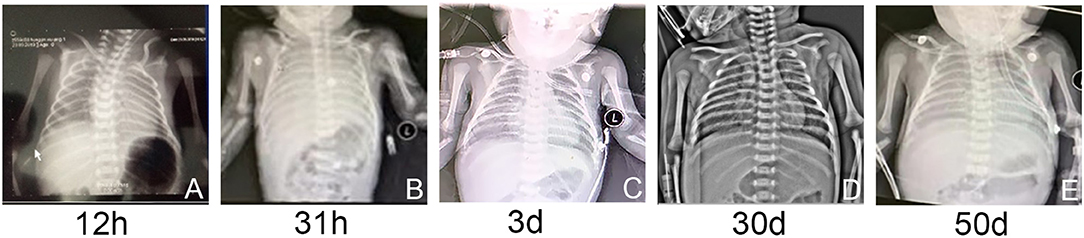
Figure 1. (A–E) The chest radiographs showed varying degrees of decreased transmittance and lung consolidation.
At 7 days of life, due to the improved conditions, the patient was extubated and instead given NCPAP with FiO2 of 0.35. At 10 days of life, a high-resolution computed tomography (HRCT) scan revealed diffuse ground glass opacity in bilateral lungs (Figure 2A). At 13 days of life, he was intermittently given NCPAPC with FiO2 of 0.21. During 14–17 days of life, he was supplied with humidified high-flow oxygen (3 L/min) via nasal cannula. At 18 days of life, dyspnea with hypoxemia reoccurred and NCPAP was hereby reinitiated with FiO2 of 0.25. On 15–19 days of life, dexamethasone was given at a dose of 0.3 mg/kg/day but no improvement was observed. HRCT scan revealed decreased transmittance and ground glass opacity in most of lung lobes (Figure 2B). Several laboratory test results were anormal, including neutrophilic leukocytosis (leukocytes 18.630/mm3; neutrophils 11.790/mm3), mildly elevated C-reactive protein concentration (28.6 mg/L), and procalcitonin concentration (3.6 mg/L). Cefoperazone-sulbactam sodium was administered, showing no effect.
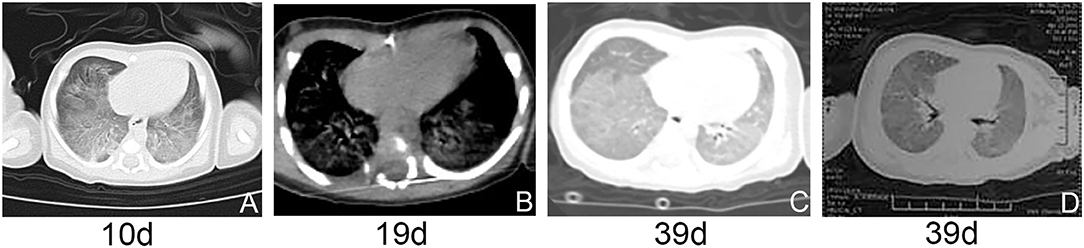
Figure 2. (A–D) HRCT and airway reconstruction showed decreased transmittance and ground glass opacity in bilateral lungs.
Given the recurrent hypoxemic respiratory failure and ground glass opacity in both lungs, we highly suspected ILD. With the informed consent from the parents, 2-ml blood samples were collected from the neonate and the parents for genetic testing.
The genetic analysis revealed a homozygous missense mutation (c.746C>T, p. P249L) in the coding region of exon 8 of ABCA3 gene in the neonate. Sanger sequencing verified that both parents were heterozygous carriers of this mutation (Figure 3A). The mutation occurred in an evolutionarily conserved site by cross-species comparison of ABCA3 protein sequences (Figure 3B).
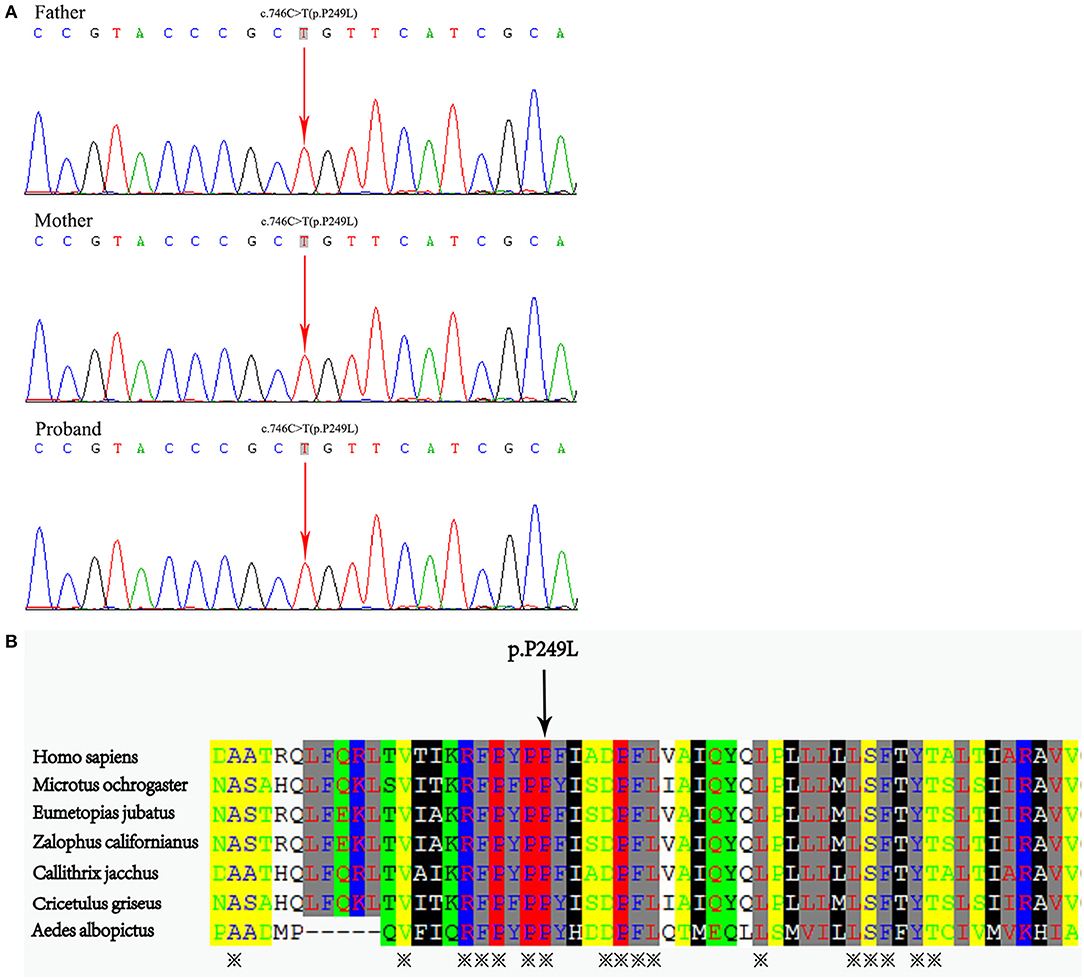
Figure 3. (A) Whole exome sequencing demonstrated a homozygous missense mutation C-to-T transition at position 746 in exon 8 of the ABCA3 gene in the neonate, which resulted in a Pro249Leu mutation. The mutations were detected in his parental DNA, respectively. (B) Protein alignment showed conservation of the P249 residue of ABCA3 across seven species.
At 20 days of age, the treatment strategy was adjusted as follows: intravenous methylprednisolone pulse therapy at a dose of 10 mg/kg/day (3-days courses on a monthly basis), together with azithromycin at a dose of 10 mg/kg every other day, and hydroxychloroquine at a dose of 5 mg/kg/day. At 30 days, chest radiograph showed patchy increased density shadows in bilateral lungs (Figure 1D). At 39 days of age, airway reconstruction and HRCT scan revealed decreased transmittance, ground glass opacity, and mildly reduced lesion extent (Figures 2C,D). During the aforementioned medications, the patient consistently depended on supplemental oxygen.
At 50 days of life, the patient was transferred to a lung transplantation center. Due to the extremely severe hypoxemia with PaO2 of 24 mmHg, mechanical ventilation with FiO2 of 0.6 was given. Chest radiograph showed diffuse decreased transmittance, air bronchogram, and vesicular shadows in the right and left lower lung lobes (Figure 1E). At 52 days of life, he had frequent episodes of cyanosis with oxygen saturation decreased to <50%, and meanwhile, the FiO2 gradually elevated to 0.8. At 53 days of life, further rescue was stopped due to the parental declination and the patient was discharged from the hospital. The autopsy request was declined by the parents as well. The main treatment course is demonstrated in Figure 4.
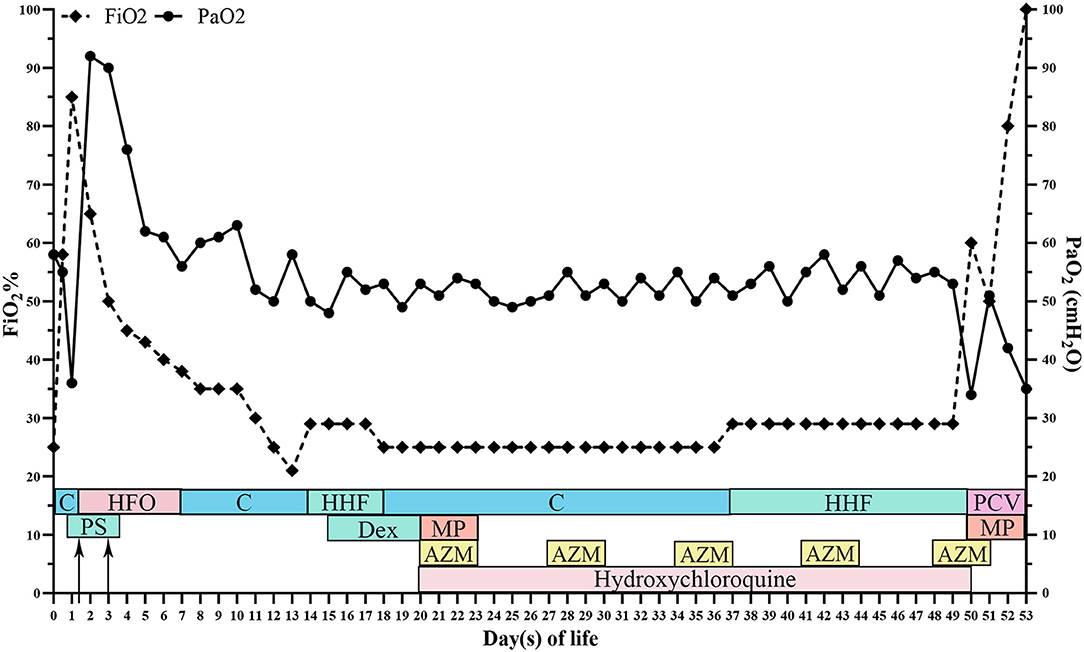
Figure 4. The overall time curve of medication, respiratory support, arterial oxygen partial pressure, and fraction of inspire oxygen. FiO2, fraction of inspire oxygen; PaO2, arterial oxygen partial pressure; C, continuous positive pressure ventilation; HFO, high-frequency oscillatory ventilation; HHF, humidified high-flow oxygen; PCV, pressure controlled ventilation; Dex, dexamethasone; MP, methylprednisolone; AZM, azithromycin; PS, pulmonary surfactant.
The ABCA3 gene maps to chromosome 16p13.3 and is highly expressed in the limiting membrane of lamellar bodies of type II alveolar epithelial cells. ABCA3 transports the phospholipids into lamellar bodies, where these lipids, together with the surfactant proteins, assemble into pulmonary surfactants. The newly synthesized surfactants are then released into the alveoli. The ABCA3 mutation could influence a set of physiological processes (7, 8): (1) phospholipid transportation, (2) lamellar body formation, (3) SP-B and SP-C transcription and translation, (4) pulmonary surfactant assembly and structural transformation in type II alveolar epithelial cells, (5) pulmonary surfactant production, and (6) lung epithelial cell apoptosis.
Numerous studies have indicated that ABCA3 mutation is one of the major causes of neonatal fatal RDS and ILD in children (9, 10). ABCA3 mutation is inherited as an autosomal recessive. More than 200 cases of ABCA3 mutations have been reported, most of which are compound heterozygotes (11). Kröner et al. conducted a retrospective study on 1,153 patients with ILD. Of these patients, 242 were sequenced for ABCA3 gene, and at least one variation was identified to be shared in 69 patients. In addition, two pathogenic mutations were identified in 40 patients (12). Shulenin et al. reported that 16 in 21 infants with RDS at birth carried ABCA3 gene mutations (13). El Boustany et al. reported a premature newborn with severe RDS in a few hours after birth, and sequencing analysis revealed two frame shift mutations p.pro1301argfs *45 and p.eu1695argfs*103 in ABCA3 gene (14). López Castillo et al. reported a full-term female newborn with RDS immediately after birth, and the homozygous nonsense mutation c. 4681C>T in ABCA3 gene was detected (15).
A classification scheme of childhood interstitial lung disease (chILD) was elaborated by the chILD Research Network (16). ILD in neonate has a high early mortality. Due to ILD's non-specific clinical manifestations and its progressive deterioration and early lethality, earlier identification of ILD in neonate is extremely difficult but meaningful.
Diagnostic tests for chILD include echocardiography, imaging studies (chest X-ray, thin-section CT) pulmonary function test, bronchoscopy with BAL, genetic test, and lung biopsy. Considering many ILD cases at infant and children stages have genetic pathogenesis, genetic test is hereby recommended for rapid diagnosis. According to the guidelines formulated by the American Thoracic Society, gene diagnosis was listed as one of the main diagnostic tests in infant with ILD, and it is recommended to be performed prior to lung biopsy. In particular, cases with clinical and radiological changes are suspected with inborn problems in surfactant metabolism, which can also be confirmed by genetic tests. If the sequencing results are negative, lung biopsy with electron microscope will be performed (17). In this case, bronchoscopy with BAL was recommended, but it was not performed due to clinical instability and rapid disease progression. In addition, lung biopsy was canceled due to the parental declination. Despite the aggressive treatment, the patient was still unable to wean from supplemental oxygen. The characteristic radiological findings made us highly suspect ILD and led to the immediate decision of genetic testing.
In this study, a homozygous mutation (c.746C>T) in ABCA3 gene was detected in the neonate's genome. Family sequencing analysis concluded that the two mutant alleles were inherited respectively from the phenotypically healthy parents. Pathogenicity analysis was conducted according to the “interpretation standards and guidelines for gene sequence variation” formulated by ACMG and AMP in 2015 (6): (1) The mutation (c.746C>T, Pro249Leu) is located in a mutation hotspot (PM1) where mutations such as Asp253His, Asp253Tyr, Pro248Leu, and Pro248Ser have been reported. (2) The frequency of this variation in the normal population database is 0.00010, a low-frequency variation (PM2). (3) SIFT, Mutation Taster, GERP++, REVEL all predicted that this mutation was harmful. (4) The neonate's clinical manifestations and radiological changes were highly consistent with the phenotype of ILD caused by ABCA3 gene mutations. Therefore, the evidence intensity of c.746C>T was “PM1+PM2+PP3+PP4,” which was judged as a possible pathogenic variation. Having reviewed the HGMD, 1,000 Genomes and EXAC gene databases, we confirmed that the mutation is a novel ABCA3 homozygous mutation.
The neonate's clinical phenotypes, imaging findings, and genetic results strongly support the diagnosis of ILD caused by ABCA3 mutation. The homozygosity of the mutation may explain the severity of clinical status. However, given the absence of lung histopathology, the pathogenicity of the mutations remains unelucidated.
There are no standardized treatment guidelines for chILD. In current practice, methylprednisolone, hydroxychloroquine, and azithromycin were widely used, though their pharmacological mechanisms and clinical effects remain inconclusive. In the study conducted by Kroner et al., of all the 35 ABCA3-mutated patients treated with systemic steroids, eight displayed moderate improvement, and two received complete remission. On the other hand, the administration of hydroxychloroquine resulted in good responses in three cases, and moderate improvement in six cases, of all the 18 patients (14). El Boustany et al. reported that a premature newborn carrying ABCA3 mutation was given high doses of methylprednisolone intravenously plus oral administration of hydroxychloroquine and azithromycin but received no improvement (17). A similar futile case was also reported by López Castillo et al. on a full-term newborn carrying ABCA3 mutation (15). So far, only isolated or a small number of cases have been reported, with varying prognosis. Based on the current inadequate data, we think that systemic steroid could be a double-edged sword, due to the fact that details such as recommended dosages and doses remain unclear. Hence, clinical trials with larger number of such patients are urgently needed to establish more effective therapeutic intervention guidelines. Currently, lung transplantation, an option for end-stage lung disease, may be the only fundamental solution for ILD. However, the rapid development of gene editing technologies may cast light on ILD treatment in the future.
All datasets generated for this study are included in the article/supplementary material.
This study was carried out in accordance with the Ethical Review Committee of the Zibo Central Hospital. Written and informed parental consent was obtained for publication of this case report.
MW designed the study, collected data, drafted the initial manuscript, and carried out the initial analyses. LM reviewed and revised the manuscript. HF and AH collected and collated clinical data of the patient.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are very grateful to Dr. Ran Wei (University of California) for his great support and assistance.
ABCA3, ATP-binding cassette transporter subfamily A member 3; NRDS, neonatal respiratory distress syndrome; PS, pulmonary surfactant; ILD, interstitial lung disease; FiO2, fraction of inspired oxygen; PaO2, arterial oxygen partial pressure; HRCT, high-resolution computed tomography; chILD, childhood interstitial lung disease.
1. Nathan N, Taam RA, Epaud R, Delacourt C, Deschildre A, Reix P, et al. A national internet-linked based database for pediatric interstitial lung diseases: the French network. Orphanet J Rare Dis. (2012) 7:40. doi: 10.1186/1750-1172-7-40
2. Turcu S, Ashton E, Jenkins L, Gupta A, Mok Q. Genetic testing in children with surfactant dysfunction. Arch Dis Child. (2013) 98:490–5. doi: 10.1136/archdischild-2012-303166
3. Suhrie K, Pajor NM, Ahlfeld SK, Dawson DB, Dufendach KR, Kitzmiller JA, et al. Neonatal lung disease associated with TBX4 mutations. J Pediatr. (2019) 206:286–92.e1. doi: 10.1016/j.jpeds.2018.10.018
4. Doan ML, Guillerman RP, Dishop MK, Nogee LM, Langston C, Mallory GB, et al. Clinical, radiological and pathological features of ABCA3 mutations in children. Thorax. (2008) 63:366–73. doi: 10.1136/thx.2007.083766
5. Campo I, Zorzetto M, Mariani F, Kadija Z, Morbini P, Dore R, et al. A large kindred of pulmonary fibrosis associated with a novel ABCA3 gene variant. Respir Res. (2014) 15:43. doi: 10.1186/1465-9921-15-43
6. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
7. Zarbock R, Kaltenborn E, Frixel S, Wittmann T, Liebisch G, Schmitz G, et al. ABCA3 protects alveolar epithelial cells against free cholesterol induced cell death. Biochim Biophys Acta. (2015) 1851:987–95. doi: 10.1016/j.bbalip.2015.03.004
8. Weichert N, Kaltenborn E, Hector A, Woischnik M, Schams A, Holzinger A, et al. Some ABCA3 mutations elevate ER stress and initiate apoptosis of lung epithelial cells. Respir Res. (2011) 12:4. doi: 10.1186/1465-9921-12-4
9. Winter J, Essmann S, Kidszun A, Aslanidis C, Griese M, Poplawska K, et al. Neonatal respiratory insufficiency caused by an (homozygous) ABCA3-stop mutation: a systematic evaluation of therapeutic options. Klin Padiatr. (2014) 226:53–8. doi: 10.1055/s-0033-1363687
10. Naderi HM, Murray JC, Dagle JM. Single mutations in ABCA3 increase the risk for neonatal respiratory distress syndrome in late preterm infants (gestational age 34–36 weeks). Am J Med Genet A. (2014) 164A:2676–8. doi: 10.1002/ajmg.a.36660
11. Beers MF, Mulugeta S. The biology of the ABCA3 lipid transporter in lung health and disease. Cell Tissue Res. (2017) 367:481–93. doi: 10.1007/s00441-016-2554-z
12. Kröner C, Wittmann T, Reu S, Teusch V, Klemme M, Rauch D, et al. Lung disease caused by ABCA3 mutations. Thorax. (2017) 72:213–20. doi: 10.1136/thoraxjnl-2016-208649
13. Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. (2004) 350:1296–303. doi: 10.1056/NEJMoa032178
14. El Boustany P, Epaud R, Grosse C, Barriere F, Grimont-Rolland E, Carsin A, et al. Unusual long survival despite severe lung disease of a child with biallelic loss of function mutations in ABCA3. Respir Med Case Rep. (2018) 23:173–5. doi: 10.1016/j.rmcr.2018.03.004
15. López Castillo MC, Pérez Ruiz E, Caro Aguilera P, Rodríguez Vives MA. ABCA3 deficiency in a newborn with respiratory failure. Arch Bronconeumol. (2018) 54:634–5. doi: 10.1016/j.arbres.2018.03.009
16. Deutsch GH, Young LR, Deterding RR, Fan LL, Dell SD, Bean JA, et al. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med. (2007) 76:1120–8. doi: 10.1164/rccm.200703-393OC
17. Kurland G, Deterding RR, Hagood JS, Young LR, Brody AS, Castile RG, et al. An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med. (2013) 188:376–94. doi: 10.1164/rccm.201305-0923ST
Keywords: full-term neonate, ABCA3 gene, pulmonary surfactant, interstitial lung diseases, lethal respiratory distress syndrome
Citation: Wei M, Fu H, Han A and Ma L (2020) A Term Neonatal Case With Lethal Respiratory Failure Associated With a Novel Homozygous Mutation in ABCA3 Gene. Front. Pediatr. 8:138. doi: 10.3389/fped.2020.00138
Received: 29 December 2019; Accepted: 11 March 2020;
Published: 17 April 2020.
Edited by:
Renato Cutrera, Bambino Gesù Children Hospital (IRCCS), ItalyReviewed by:
Enrico Lombardi, University of Florence, ItalyCopyright © 2020 Wei, Fu, Han and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liji Ma, bWFsajEyNUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.