 Giuseppina Rosaria Umano1*
Giuseppina Rosaria Umano1* Carmelo Pistone2
Carmelo Pistone2 Enrico Tondina2
Enrico Tondina2 Alice Moiraghi2
Alice Moiraghi2 Daria Lauretta1
Daria Lauretta1 Emanuele Miraglia del Giudice1
Emanuele Miraglia del Giudice1 Ilaria Brambilla3
Ilaria Brambilla3- 1Department of the Woman, Child, General and Specialized Surgery, University of Campania Luigi Vanvitelli, Naples, Italy
- 2School of Pediatrics, University of Pavia, Pavia, Italy
- 3Department of Pediatrics, Fondazione IRCCS Policlinico San Matteo, University of Pavia, Pavia, Italy
Obesity has reached pandemic proportion and represents a major risk for several comorbidities. In addition to metabolic and cardiovascular obesity-related diseases, recent evidence suggested that obesity might affect immune system function. Adipose tissue is considered an endocrine organ that actively secretes cytokines also referred to as “adipokines.” Adipokines play an important role in the control of human metabolism. The dysfunctional adipose tissue in obese individuals is characterized by an altered cytokine secretion pattern that promotes chronic low-grade inflammation. Epidemiological evidence highlights the association between obesity and allergic and immune-mediated diseases, such as asthma, allergic rhinitis, rheumatic arthritis, and psoriasis. Less is known about underlying pathogenic mechanisms. However, several recent in vivo and in vitro studies have reported that adipokines are involved in inflammatory and autoimmune disorders by influencing both innate and acquired immune responses. In addition, obesity has been associated with reduced immune surveillance and increased risk of cancer. This paper reviews the evidence regarding the role of adipokines in immune system regulation, with particular emphasis on autoimmune, allergic, and inflammatory disorders. Understanding how obesity affects immune system functions may enable researchers to find new potential therapeutic targets in the management of allergic and autoimmune diseases.
Introduction
The obesity epidemic represents a major health issue of the twenty-first century. The prevalence of the disease has significantly increased in the last decades worldwide, including low-income countries (1). In addition, it has been reported that pediatric obesity significantly increases the risk of obesity and its cardiovascular complications during childhood and adulthood (2, 3).
The constant increase in obesity prevalence might be ascribed to changes in lifestyle habits, namely, assumption of junk food and sedentary behavior. Moreover, genetic background plays a role by affecting energy expenditure and food intake (4). The excess of fat present in obese individuals results in a hypertrophic white adipose tissue with a consequent derangement in adipocytes metabolic activity. Adipose tissue in some obese subjects displays a pro-inflammatory activity and has been associated with obesity-related comorbidities in both adults and children (5–8). Adipocytes actively secrete several hormones and cytokines, also referred to as adipokines that exert metabolic and immunological functions, as they could modulate the innate and acquired immune cell activity. Moreover, they are capable of promoting pro-inflammatory signals leading to a chronic systemic low-grade inflammation. In addition, growing evidence reports the association between obesity and immune disorders, such as cancer, autoimmunity, and atopy (9). Therefore, the altered secretion pattern of adipokines in obesity is considered the link between obesity and its metabolic and immunological comorbidities (10).
In this review, we summarize the current knowledge on the basis of endocrine and immunological functions of white adipose tissue. In particular, we describe the interplay between adipokines and both innate and acquired immunity. Moreover, we report the evidences and possible mechanisms of the association between pediatric obesity and immunological disorders.
Obesity and Inflammation
Clinical studies have observed a significant association between serum markers of chronic low-grade inflammation and obesity-related comorbidities in children and adolescents (11–13). Serum levels of C-reactive protein (CRP) directly correlate with insulin resistance and carotid intima-media thickness (cIMT) in children and adolescents with overweight and obesity (11). Moreover, it has been reported that white blood cell count is predictive of increased cIMT and ventricular hypertrophy in obese children and adolescents (12). In addition, weight loss after lifestyle intervention trial was effective in reducing both serum inflammatory markers and insulin resistance in obese children and adolescents (14).
Inflammation is a mechanism that involves a series of non-cellular and cellular mediators involved in the response to infections, tissue damage, cellular death, and cancer. Obesity is accompanied by a sterile chronic low-grade inflammation in the adipose tissue that is not related to infection or tissue damage (15). This process is also referred to as metabolic inflammation or metainflammation. Metainflammation is an active player in the development of obesity-related comorbidities, as obesity-related inflammation might impair the function of other organs (16). Adipose tissue immune response is mediated by adipose tissue-resident immune cells, namely, macrophages, mast cells, neutrophils, and T and B lymphocytes, which are the second more represented cellular type, after adipocytes (17). Mechanisms triggering adipose tissue inflammation are not completely clear. However, adipose tissue hyperplasia and hypertrophy occurring in obese individuals may exert an important role (18). In fact, an increase of adipocyte size leads to hypoxia, intracellular oxidative stress, and release of pro-inflammatory molecules (18). However, some studies have shown that metabolically unhealthy obese adolescents tend to have smaller adipose cells (19), which allows for a smaller storage capacity and favor a more pronounced flux of free fatty acids. Inflammasome activation in subcutaneous adipose tissue (SAT) has been associated with an altered abdominal fat distribution (19). The inflammasome chronic activation impairs the SAT storage capability leading to a lipid spill over into visceral adipose tissue (VAT) (19).
The excess of free fatty acids contributes to an unfavorable metabolic phenotype probably through the macrophage activation in adipose tissue. In fact, free fatty acids directly activate immune cells and induce secretion of pro-inflammatory mediators through interacting with toll-like receptors (TLRs) (20). In VAT, adipocytes develop a dysfunctional phenotype characterized by smaller adipose cells and expression of adhesion molecules for monocytes/macrophages (21).
Macrophages
Macrophages are the most common immune cell type in adipose tissue. The number of macrophages has been associated with size of adipocytes and severity of obesity (21). A small amount of adipose tissue macrophages (ATMs) derives from pre-adipocyte maturation, while the majority comes from systemic circulation (22). Hypertrophic adipose tissue promotes macrophage migration and infiltration via secretion of chemokines. Several chemokines are involved in this pathway. Among them, monocyte-chemoattracting protein 1/chemokine C–C motif receptor 2 (MCP1/CCR2) binds macrophage receptor CCR2, leading to infiltration (23). Moreover, obesity alters the phenotype and distribution of macrophages. Macrophages could be distinguished in two subsets according to membrane antigen and cytokine secretion: classically activated M1 type and alternative activated M2 type. M1 type exerts a pro-inflammatory and antibacterial role; it expresses receptor for inflammatory mediators, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), IFN-γ, and lipopolysaccharides (LPS). Conversely, M2 phenotype is induced by anti-inflammatory molecules, namely, IL-13, IL-10, IL-4, and macrophage colony-stimulating factor (M-CSF), and displays anti-inflammatory and anti-parasitic functions (24, 25). The different polarization of macrophages in human adipose tissue is influenced by nutritional status. In normal weight subjects, the prevalence of eosinophils, T-lymphocytes, and natural killer (NK) cells promote a polarization toward M2 type. These macrophages, in a favorable environment, exert homeostatic actions, regulating cellular proliferation, extracellular matrix deposition, and removing cellular debris in stromal matrix. In obese subjects, oxidative stress, necrotic cellular debris, and overload of free fatty acids lead to a polarization toward an M1 phenotype. In contrast to M2, M1 macrophages do not migrate in stromal matrix; they tend to create aggregates surrounding necrotic adipocytes, in a typical crown-like structure (26). In humans, the number of M1 macrophages directly correlates with systemic inflammation, insulin resistance, type 2 diabetes, and fatty liver disease (15).
Lymphocytes
The second largest immune cell subset in adipose tissue is represented by T-lymphocytes. T-cells could be divided in CD4+ and CD8+ according to surface antigens. The cytokine secretion pattern of CD4+ allows a further categorization in Th1 (INF-γ), Th2 (IL-4, IL-5, and IL-13), Treg (IL-10 and transforming growth factor B), and Th17 (IL-17, IL-21, and IL-22). CD8+ are cytotoxic cells that secrete perforins, granzymes, and a variety of cytokines that mediate activation of other immune cells. In dysfunctional adipose tissue, the T-lymphocyte subpopulation pattern is polarized toward a pro-inflammatory activity. Therefore, Th1, Th17, and CD8+ are more represented compared to anti-inflammatory Th2 and Treg (15).
Other Adipose Tissue-Resident Immune Cells
Neutrophils, mast cells, and dendritic cells constitute a small fraction of immune cells in adipose tissue. Nevertheless, they play an important role in amplifying inflammation process via secretion of pro-inflammatory mediators. The amount of eosinophils, instead, is reduced. They promote M2 macrophage and Th2 differentiation and suppress inflammatory stimuli (15).
Adipokines as Immune Modulators
In the last years, a number of adipose tissue-derived cytokines have been characterized, the so-called adipokines (Figure 1). Besides adipocytes, the vascular-stromal component of adipose tissue concurs with cytokine secretion in an adiposity-dependent manner. Adipokines play a crucial role not only in energy homeostasis but also in inflammatory and immune reaction, most of them promoting inflammation (27).
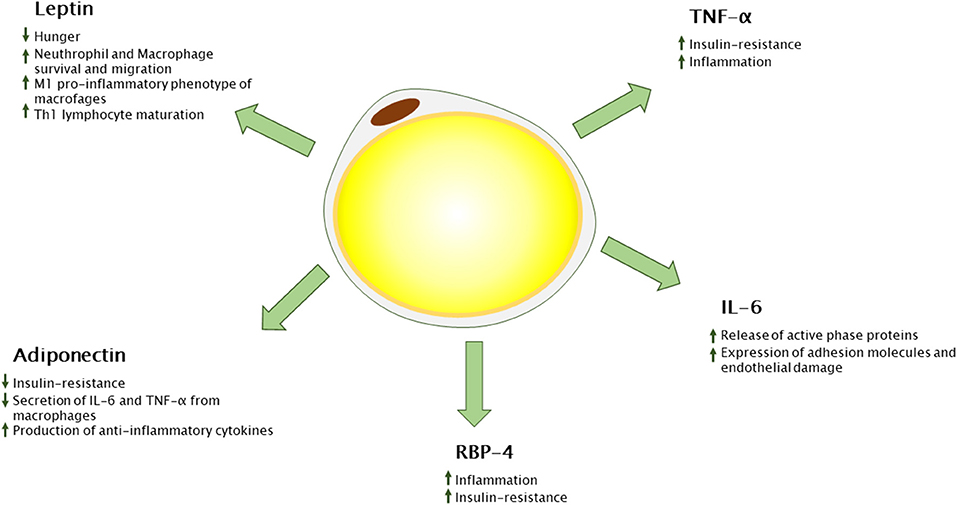
Figure 1. Adipokines secreted from white adipose tissue and their immunological functions. RBP-4, retinol binding protein 4; TNF-α, tumor necrosis factor alpha; Th-1, T-helper 1.
Leptin
Leptin was first described by Zhang et al. (28) in a murine model. It is encoded by murine ob gene and human homologous LEP gene. Animals and human models showed that inhibition of leptin action results in food assumption and insulin resistance (27, 28). Leptin secretion is positively correlated with adipose tissue mass; therefore, obese subjects show high leptin plasmatic levels. Nevertheless, obese subjects develop leptin resistance with a decreased sensitivity to anorexinergic hormone stimulus. Leptin has pleiotropic activities. The main role is suppression of food assumption via inhibiting hypothalamic nuclei that stimulate hunger and stimulation of those which promote satiety (29). Leptin loss of function is associated with hyperphagia, rapid weight gain, and insulin resistance (30). Moreover, leptin influences pubertal development and shows immunomodulatory activity (31). In fact, several immune cell types express leptin receptor (LEPR) (Figure 2). In neutrophils, leptin activates anti-apoptotic signals, resulting in cell survival. In addition, it induces neutrophil activation in terms of chemotaxis, tissue infiltration, and oxygen radicals' release. Similarly, eosinophils and basophils express LEPR on the cellular surface. As for neutrophils, leptin acts as a survival cytokine for these cells and stimulates tissue infiltration and pro-inflammatory molecule release (32). Moreover, leptin is crucial for monocyte/macrophage function. In experimental models of LepR, knock-out mice impaired macrophages killing activity and phagocytosis has been reported (33). Additionally, leptin has been shown to enhance monocyte and macrophage survival, migration, release of pro-inflammatory cytokines (namely, IL-6 and TNF-α), and expression of surface markers (CD39, CD69, CD25, CD71, and IL-1Rα). Leptin also stimulates proliferation and cytotoxicity of NK cell and constitutes a survival cytokine for dendritic cells. With regard to adaptive immune response, hormone inhibits T-lymphocyte apoptosis, promotes Th1 response, suppresses Th2 activity, and enhances IL-2 and INF-γ production. Moreover, it sustains Th17 pro-inflammatory function and B-lymphocyte activity (34). In general, leptin displays a pro-inflammatory activity, interacting with both innate and adaptive immune system. These data highlight the crucial role of leptin in pathogenesis of obesity-related comorbidities, in terms of metabolic derangement and chronic inflammation.
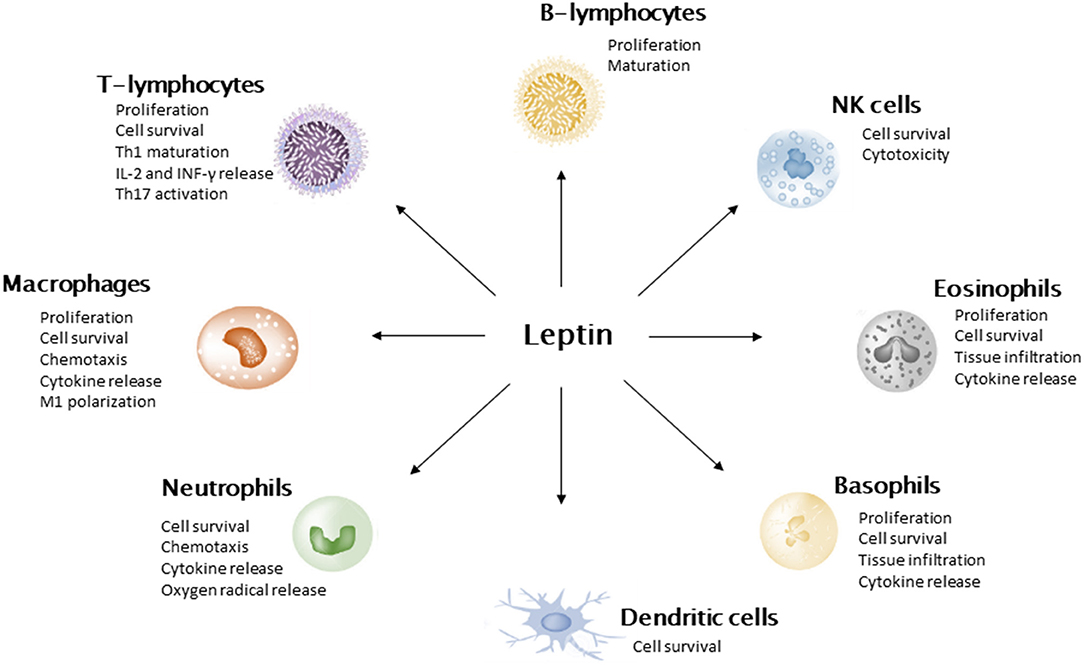
Figure 2. Leptin action on immune cells. Leptin promotes cell survival, stimulates the secretion of pro-inflammatory cytokines, and suppresses cell apoptosis. It induces M1 polarization of macrophages and stimulates Th1 pathway in T-lymphocytes.
Resistin
Resistin is mainly secreted by monocytes and macrophages and a small fraction derives from adipocytes. It is associated with insulin resistance as it inhibits insulin receptor signaling via suppression of cytokine signaling-3 (SOCS-3). Consequently, it can influence plasmatic glucose levels and insulin sensitivity (35). Moreover, it stimulates the release of IL-6 and TNF-α from neutrophils and counteracts adiponectin anti-inflammatory activity on endothelial cells (36).
Interleukin-6 (IL-6)
One of the most important pro-inflammatory cytokines is IL-6. About one-third of total circulating levels of IL-6 derives from adipocytes (37). IL-6 directly stimulates secretion of active phase proteins, such as CRP and fibrinogen. Waist circumference mainly correlates with IL-6 and CRP plasma levels, highlighting the crucial role of visceral adiposity in inflammation (38). In addition, it promotes expression of adhesion molecules on endothelial cells for leucocytes, enhancing vascular damage and inflammation (38, 39).
Tumor Necrosis Factor-Alpha (TNF-α)
TNF-α has a central role in pathogenesis of several inflammatory and autoimmune diseases. Monocyte/macrophage cells are responsible for the major fraction of serum TNF-α. In addition, cytokine levels positively correlate with adiposity measures and insulin resistance. TNF-α inhibits the expression of PPAR-γ (peroxisome proliferator-activated receptor-gamma). PPAR-γ exerts anti-inflammatory activity throughout, reducing pro-inflammatory cytokine secretion in human macrophages (40). Furthermore, it interferes with insulin receptor IRS-1 phosphorylation, leading to insulin resistance (41). Moreover, TNF-α antagonist treatment in subjects affected by autoimmune diseases improves insulin sensitivity (42). However, these findings have not been confirmed in models of obesity-related insulin resistance (43).
Retinol Binding Protein-4 (RBP-4)
Retinol-binding protein 4 (RBP-4) is an adipokine involved in the pathogenesis of insulin resistance. In particular, it reduces insulin sensitivity, decreasing IRS-1 insulin-induced phosphorylation. Hepatocytes are the main source of RBP-4, while visceral adipocytes and macrophages secrete a minor fraction. However, RBP-4 is secreted in a dependent manner to VAT mass, as it has been suggested as a marker of visceral adiposity and chronic low-grade inflammation (43).
A variety of pro-inflammatory adipokines have been described, namely, visfatin, lipocalin, angiopoietin-like protein 2 (ANGPTL2), CC-chemokine ligand 2 (CCL2), and CXC-motif chemokine ligand 5 (CXCL5) (Figure 3). However, their role in metainflammation has not been completely understood (43).
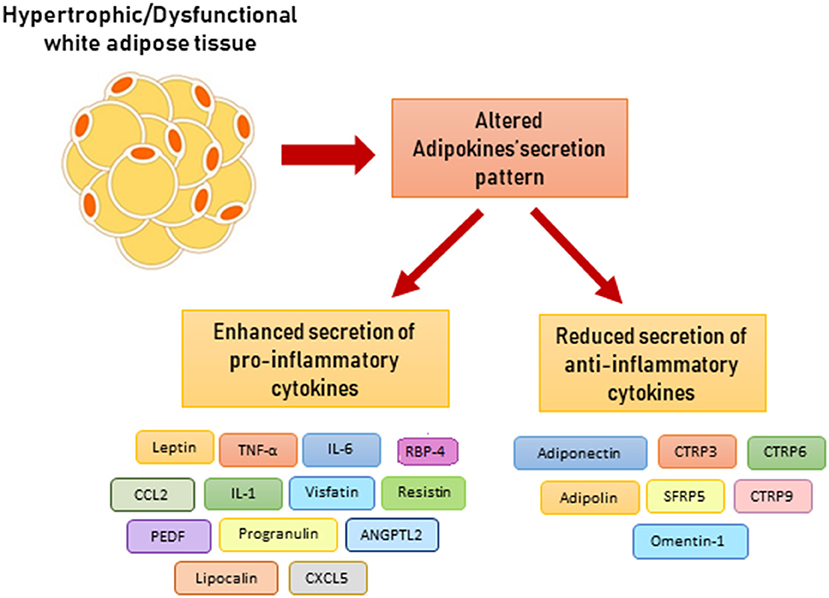
Figure 3. Altered adipokine secretion pattern in dysfunctional adipose tissue. ANGPTL2, angiopoietin-like protein 2; CCL2, CC-chemokine ligand 2; CXCL5, CXC-chemokine ligand 5; CTRP, C1q/TNF-related protein-3; CTRP, C1q/TNF-related protein-6; CTRP, C1q/TNF-related protein-9; PEDF, pigment epithelium-derived factor; RBP-4, retinol binding protein 4; SFRP5, secreted frizzled related protein 5.
Adiponectin
Adipose tissue releases a small amount of adipokines with an anti-inflammatory activity, among them adiponectin is the best characterized (Figure 3). Adiponectin stimulates fatty acid oxidation and glucose uptake in skeletal muscle and liver, thus improving insulin sensitivity (44). In macrophages, adiponectin signal promotes an M2 phenotype polarization, reduction of TNF-α secretion, and enhancement of scavenger activity. Moreover, it stimulates release of anti-inflammatory IL-10. Pro-inflammatory cytokines (IL-6 and TNF-α), oxidative stress, and adipocyte hypoxia decrease adiponectin secretion. In fact, clinical studies have reported that hormone levels are inversely correlated with CRP plasma levels (45). Moreover, obesity is associated with lower adiponectin levels and increased risk of cardiovascular diseases (46, 47). Similarly to adiponectin, the C1q/TNF-related proteins (CTRPs) promote anti-inflammatory pathway. Some CTRPs are expressed in adipose tissue, such as CTRP3, CTRP6, CTRP9, and CTRP12, and inhibit macrophage pro-inflammatory activity (48). In addition, omentin-1 is secreted from VAT and reduces vascular inflammation via inhibition of endothelial adhesion molecules expression (48).
In conclusion, adipokine secretion pattern from adipose tissue exerts a central role in the link between obesity and altered immune response, leading to systemic inflammation and reduced immune tolerance.
Obesity, Atopy, and Asthma
Paralleling with the obesity epidemic, a rise in the prevalence of allergic diseases has been registered. The higher exposure to indoor and outdoor air pollutants has a crucial role in this phenomenon. However, evidence suggests that obesity and obesity-related inflammation might be at least in part responsible of this process. Data from the National Health and Nutrition Examination Study III (NHANES III) have reported a positive association between BMI and atopy rates (49). Nevertheless, obese subjects do not show a significant increase of serum atopy markers, such as IgE plasma levels and eosinophils count (50, 51). In addition, evidence about a possible link between obesity and other allergic disorders, namely, allergic rhinitis and atopic dermatitis, is sparse. Therefore, this field needs to be more deeply investigated. Conversely, several data are available about the relation between asthma and obesity in both adults and children (9). Cross-sectional epidemiological studies have reported that obese children are frequently affected by asthma (49, 52, 53). Moreover, a metanalysis including six perspective studies found that obese children show a twofold higher risk for asthma compared to normal weight children (54). In addition, obese children commonly show a more severe asthma phenotype, which tend to be resistant to pharmacologic therapies with frequent exacerbations. Nevertheless, obese children do not show elevation of expiratory levels of inflammatory markers, such as exhaled nitric oxygen (eNO). Several underlying mechanisms have been proposed for obesity-related asthma (Figure 4). In particular, excess of truncal adiposity leads to a mechanical overload to respiratory muscles. This results in reduced functional residual capacity, decreased residual volume, and expiratory reserve volume. This volume reduction exposes obese children to an impairment of forced expiratory volume in 1 s and forced vital capacity ratio (FEV1/FVC). In addition to mechanical overload, metabolic derangement might also play a role in obesity-related asthma. Dyslipidemia and insulin resistance have been associated with an impaired FEV1/FVC ratio. Insulin is a trophic stimulus for low airway smooth muscle cells. It stimulates laminin production through phospho-inositide-3 kinase/Akt (PI3K/AKT) pathway, leading to muscle hypertrophy. Additionally, it enhances airway hyper-responsiveness via stimulating parasympathetic innervation. These mechanisms promote airway obstruction during physical exercise and a perception of respiratory effort during inspiration (55). Moreover, chronic low-grade inflammation occurring in obesity plays an important role. In obese children, imbalance between Th1/Th2 phenotype toward a Th1 reaction has been related to decrease FEV1/FVC ratio. Similarly, macrophage pro-inflammatory M1 type induces airway obstruction (56). Finally, derangement of adipokines' milieu has a role in adult asthma. In particular, leptin serum levels are inversely correlated with pulmonary volumes and FEV1/FVC ratio, while it increases bronchial hyper-responsiveness (57). However, most of the available evidence is based on cross-sectional studies, and this makes it difficult to completely understand underlying mechanisms of obesity-related asthma.
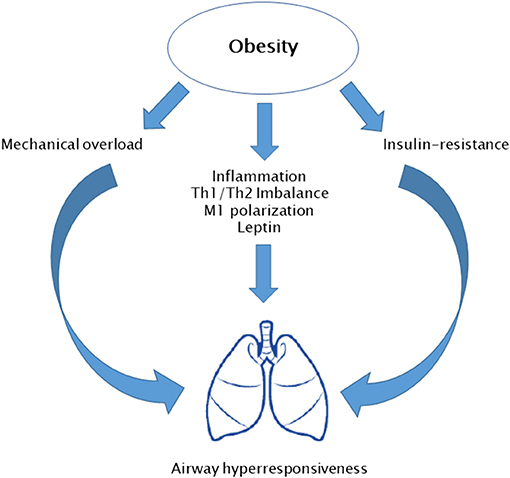
Figure 4. Potential mechanisms involved in obesity-related asthma.
Obesity and Autoimmunity
The characterization of immunological function of adipose tissue has suggested a possible link between obesity and autoimmune disorders. Autoimmunity is strongly influenced by genetic background; however, environmental factors are central in the beginning of immune response. Several observational studies have reported the association between obesity and a number of autoimmune diseases, namely, autoimmune thyroiditis, type 1 diabetes, multiple sclerosis, rheumatic arthritis, systemic lupus erythematosus, inflammatory bowel diseases, and psoriasis. However, data for pediatric age are sparse. Longitudinal studies have reported that obese children and adolescents display a twofold higher risk of developing multiple sclerosis in adulthood; this risk is higher in girls compared to boys and increases in subjects with a predisposing genetic background (HLA DRB1*15) (58–60). Several potential underlying mechanisms have been proposed: vitamin D deficiency, imbalance of macrophages M1/M2 phenotypes, increased levels of leptin, and reduced adiponectin levels (60). With regard to rheumatoid arthritis (RA), obese subjects present a 20% higher risk of RA in longitudinal studies (61). In addition, RA severity and treatment responsiveness is negatively influenced by adiposity, as obese subjects are more prone to develop a severe disease with reduced remission and high rate of comorbidities (62). Adipokines are involved in joint damage; they stimulate secretion of pro-inflammatory cytokines and metalloproteinases from synovial chondrocytes and fibroblasts (63). Conversely, no solid evidence is available to confirm the hypothesized relationship between obesity and systemic lupus erythematosus pathogenesis, while it has been reported that obesity increases disease activity (64, 65). More solid evidence confirms that obesity and psoriasis are correlated (66), but the pathogenic underpinnings of this association are still debated. Subjects with psoriasis and psoriatic arthritis tend to gain weight because of a sedentary lifestyle; obesity worsens disease severity and the risk of comorbidities (67). Additionally, weight loss improves disease control and response to treatment (68). Several mechanisms underlie this observation. First of all, altered adipokines' milieu characterized by a prevalence of IL-6, TNF-α, and leptin promotes immune cell activation, proliferation, and migration (30). Moreover, reduction of Treg cells with a concurrent increase of Th17 has been associated with autoimmunity (11). Nutrients also play a role, as a western diet with high-fat and high-sugar content leads to intestinal dysbiosis and Th17/Treg imbalance (69).
Furthermore, adipose tissue is involved in peripheral aromatization of androgens to estrogens, and this may represent a means through which excess adiposity might predispose to autoimmunity. In fact, females are more affected by autoimmune diseases compared to males. This observation might be related to the influence of sexual hormones on immune tolerance. Interestingly, women show higher leptin plasma levels compared to men. It is well-known that estrogens and dehydroepiandrosterone (DHEA) are the major mediators of females' susceptibility to autoimmune disorders. These hormones promote immunoglobulin release, stimulate adaptive immune responses, and induce secretion of pro-inflammatory cytokines (70).
As for RA, adipokines are central players in this scenario. Leptin and resistin plasma levels are elevated in subjects with psoriasis. Moreover, in vitro experiment showed that leptin stimulates cytokine release in human keratinocytes. Contrasting results have been produced about the role of adiponectin in psoriasis pathogenesis and severity (63). In conclusion, more studies are needed to confirm and investigate the pathophysiological mechanisms underlying the potential relationship between autoimmunity and obesity.
Obesity and Cancer
Previous studies have shown that nutritional status affects immunocompetence, as both undernutrition and overweight influence immune system functions. Epidemiological studies reported that subjects with obesity are at higher risk of infectious disease, infectious disease-related complications, and cancer (71, 72). In regard to cancer, it has been estimated that up to 50% of a variety of cancer types, namely, endometrial, breast, colon, liver, and prostate, might be attributed to obesity in adults (73). Additionally, a growing body of evidence suggests that pediatric obesity might increase the risk for cancer occurrence in adulthood. As for atopy and autoimmunity, more complex mechanisms interplay in obesity-related carcinogenesis. The chronic low-grade systemic inflammation has been recognized as the trigger of carcinogenesis and cytokines exert a crucial role in this process. IL-6 and IL-1β promote cell proliferation and survival. Moreover, TNF-α can increase DNA damage and cell proliferation through the NF-KB pathway with upregulation of anti-apoptotic proteins (74). In addition, IL-6 induces transcription of genes involved in angiogenesis, invasiveness, and metastasis via STAT3 activation. Similarly, leptin activates STAT3 pathway, enhancing cell proliferation and angiogenesis. Conversely, adiponectin has an anti-inflammatory and anti-tumorigenic activity through activation of AMPK (adenosine monophosphate-activated protein kinase) and inhibition of mTORC1 and other tumorigenic mediators. Therefore, dysfunctional hypertrophic adipose tissue secretes a relatively higher amount of carcinogenic mediators over anti-tumorigenic molecules. On the other hand, obesity is associated with impaired immune surveillance as suggested by increased risk of infection and lower response to immunization. Obese subjects have a reduced number of NK, dendritic, and CD8+ cells that mediate cytotoxic functions (75). In addition, Michelet et al. observed that NK cells from obese humans secrete lower amounts of INF-γ. The authors hypothesized that free fatty acid overload induces a shift in NK cell metabolism from glycolysis to lipid beta-oxidation, thus deranging cellular homeostasis and function. The direct consequence of reduced immune surveillance is tumor growth and invasiveness (76).
To date, data in pediatric age are sparse, epidemiological evidence comes from adult population, and potential mechanisms have been investigated only in mice. However, it might be hypothesized that these processes might initiate already during childhood.
Conclusions
Obesity is a major risk factor for allergic, autoimmune, and neoplastic diseases, and it is at least in part responsible for the recent burden of these disorders. Dysfunctional adipose tissue is the key pathogenic factor leading to this association. However, to date, a small number of studies have addressed this issue in obese children and adolescents; therefore, more studies are needed to confirm these observations and to understand the complex interplay between adipose tissue and immune system. The gain in knowledge in metainflammation patterns and mediators might allow the development of novel treatments for pediatric obesity. To date, there are no studies evaluating inflammatory targets in the treatment of pediatric obesity. However, PPAR-γ and PPAR-α agonists have shown to modulate monocyte function in activated human adipose tissue in vitro. Moreover, they reduced the release of pro-inflammatory mediators and expression of adhesion molecules in monocytes (77). Therefore, they could represent a promising tool in the treatment of obesity-related inflammation and insulin resistance.
Author Contributions
GU and IB wrote the manuscript. CP and ET selected the articles. AM and DL realized the figures. EM revised the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. NCD Risk Factor Collaboration. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet. (2017) 390:2627–42. doi: 10.1016/S0140-6736(17)32129-3
2. Ward ZJ, Long MW, Resch SC, Giles CM, Cradock AL, Gortmaker SL. Simulation of growth trajectories of childhood obesity into adulthood. N Engl J Med. (2017) 377:2145–53. doi: 10.1056/NEJMoa1703860
3. Valerio G, Maffeis C, Saggese G, Ambruzzi MA, Balsamo A, Bellone S, et al. Diagnosis, treatment and prevention of pediatric obesity: consensus position statement of the Italian Society for Pediatric Endocrinology and Diabetology and the Italian Society of Pediatrics. Ital J Pediatr. (2018) 44:88. doi: 10.1186/s13052-018-0525-6
4. O'Rahilly S. Human genetics illuminates the paths to metabolic disease. Nature. (2009) 462:307–14. doi: 10.1038/nature08532
5. Preis SR, Massaro JM, Robins SJ, Hoffmann U, Vasan RS, Irlbeck T, et al. Abdominal subcutaneous and visceral adipose tissue and insulin resistance in the Framingham heart study. Obesity. (2010) 18:2191–8. doi: 10.1038/oby.2010.59
6. Kaess BM, Pedley A, Massaro JM, Murabito J, Hoffmann U, Fox CS. The ratio of visceral to subcutaneous fat, a metric of body fat distribution, is a unique correlate of cardiometabolic risk. Diabetologia. (2012) 55:2622–30. doi: 10.1007/s00125-012-2639-5
7. Taksali SE, Caprio S, Dziura J, Dufour S, Cali AM, Goodman TR, et al. High visceral and low abdominal subcutaneous fat stores in the obese adolescent: a determinant of an adverse metabolic phenotype. Diabetes. (2008) 57:367–71. doi: 10.2337/db07-0932
8. Umano GR, Shabanova V, Pierpont B, Mata M, Nouws J, Trico D, et al. A low visceral fat proportion, independent of total body fat mass, protects obese adolescent girls against fatty liver and glucose dysregulation: a longitudinal study. Int J Obes. (2019) 43:673–82. doi: 10.1038/s41366-018-0227-6
9. Kelishadi R, Roufarshbaf M, Soheili S, Payghambarzadeh F, Masjedi M. Association of childhood obesity and the immune system: a systematic review of reviews. Child Obes. (2017) 13:332–46. doi: 10.1089/chi.2016.0176
10. Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. (2004) 89:2548–56. doi: 10.1210/jc.2004-0395
11. Giannini C, de Giorgis T, Scarinci A, Ciampani M, Marcovecchio ML, Chiarelli F, et al. Obese related effects of inflammatory markers and insulin resistance on increased carotid intima media thickness in pre-pubertal children. Atherosclerosis. (2008) 197:448–56. doi: 10.1016/j.atherosclerosis.2007.06.023
12. Di Bonito P, Pacifico L, Chiesa C, Invitti C, Miraglia Del Giudice E, Baroni MG, et al. White blood cell count may identify abnormal cardiometabolic phenotype and preclinical organ damage in overweight/obese children. Nutr Metab Cardiovasc Dis. (2016) 26:502–509. doi: 10.1016/j.numecd.2016.01.013
13. Cook DG, Mendall MA, Whincup PH, Carey IM, Ballam L, Morris JE, et al. C-reactive protein concentration in children: relationship to adiposity and other cardiovascular risk factors. Atherosclerosis. (2000) 149:139–50. doi: 10.1016/S0021-9150(99)00312-3
14. Marti A, Morell-Azanza L, Rendo-Urteaga T, Garcia-Calzon S, Ojeda-Rodriguez A, Martin Calvo N, et al. Serum and gene expression levels of CT-1, IL-6, and TNF-alpha after a lifestyle intervention in obese children. Pediatr Diabetes. (2018) 19:217–22. doi: 10.1111/pedi.12561
15. Mraz M, Haluzik M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J Endocrinol. (2014) 222:R113–27. doi: 10.1530/JOE-14-0283
16. Singer K, Lumeng CN. The initiation of metabolic inflammation in childhood obesity. J Clin Invest. (2017) 127:65–73. doi: 10.1172/JCI88882
17. Curat CA, Miranville A, Sengenes C, Diehl M, Tonus C, Busse R, et al. From blood monocytes to adipose tissue-resident macrophages: induction of diapedesis by human mature adipocytes. Diabetes. (2004) 53:1285–92. doi: 10.2337/diabetes.53.5.1285
18. Ye J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int J Obes. (2009) 33:54–66. doi: 10.1038/ijo.2008.229
19. Kursawe R, Dixit VD, Scherer PE, Santoro N, Narayan D, Gordillo R, et al. A role of the inflammasome in the low storage capacity of the abdominal subcutaneous adipose tissue in obese adolescents. Diabetes. (2016) 65:610–8. doi: 10.2337/db15-1478
20. Lee BC, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta. (2014) 1842:446–62. doi: 10.1016/j.bbadis.2013.05.017
21. Harman-Boehm I, Bluher M, Redel H, Sion-Vardy N, Ovadia S, Avinoach E, et al. Macrophage infiltration into omental versus subcutaneous fat across different populations: effect of regional adiposity and the comorbidities of obesity. J Clin Endocrinol Metab. (2007) 92:2240–7. doi: 10.1210/jc.2006-1811
22. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112:1796–808. doi: 10.1172/JCI19246
23. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. (2012) 18:363–74. doi: 10.1038/nm.2627
24. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. (2003) 3:23–35. doi: 10.1038/nri978
25. Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. (2007) 282:35279–92. doi: 10.1074/jbc.M706762200
26. Prieur X, Mok CY, Velagapudi VR, Nunez V, Fuentes L, Montaner D, et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes. (2011) 60:797–809. doi: 10.2337/db10-0705
27. Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. (2006) 6:772–83. doi: 10.1038/nri1937
28. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. (1994) 372:425–32. doi: 10.1038/372425a0
29. Hegyi K, Fulop K, Kovacs K, Toth S, Falus A. Leptin-induced signal transduction pathways. Cell Biol Int. (2004) 28:159–69. doi: 10.1016/j.cellbi.2003.12.003
30. Fischer-Posovszky P, Funcke JB, Wabitsch M. Biologically inactive leptin and early-onset extreme obesity. N Engl J Med. (2015) 372:1266–7. doi: 10.1056/NEJMc1501146
31. Rasouli N, Kern PA. Adipocytokines and the metabolic complications of obesity. J Clin Endocrinol Metab. (2008) 93:S64–73. doi: 10.1210/jc.2008-1613
32. Francisco V, Pino J, Campos-Cabaleiro V, Ruiz-Fernandez C, Mera A, Gonzalez-Gay MA, et al. Obesity, fat mass and immune system: role for leptin. Front Physiol. (2018) 9:640. doi: 10.3389/fphys.2018.00640
33. Dib LH, Ortega MT, Melgarejo T, Chapes SK. Establishment and characterization of DB-1: a leptin receptor-deficient murine macrophage cell line. Cytotechnology. (2016) 68:921–33. doi: 10.1007/s10616-015-9843-3
34. La Cava A. Leptin in inflammation and autoimmunity. Cytokine. (2017) 98:51–8. doi: 10.1016/j.cyto.2016.10.011
35. Steppan CM, Wang J, Whiteman EL, Birnbaum MJ, Lazar MA. Activation of SOCS-3 by resistin. Mol Cell Biol. (2005) 25:1569–75. doi: 10.1128/MCB.25.4.1569-1575.2005
36. Kawanami D, Maemura K, Takeda N, Harada T, Nojiri T, Imai Y, et al. Direct reciprocal effects of resistin and adiponectin on vascular endothelial cells: a new insight into adipocytokine-endothelial cell interactions. Biochem Biophys Res Commun. (2004) 314:415–9. doi: 10.1016/j.bbrc.2003.12.104
37. Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab. (1998) 83:847–50. doi: 10.1210/jc.83.3.847
38. Hermsdorff HH, Zulet MA, Puchau B, Martinez JA. Central adiposity rather than total adiposity measurements are specifically involved in the inflammatory status from healthy young adults. Inflammation. (2011) 34:161–70. doi: 10.1007/s10753-010-9219-y
39. Lapice E, Maione S, Patti L, Cipriano P, Rivellese AA, Riccardi G, et al. Abdominal adiposity is associated with elevated C-reactive protein independent of BMI in healthy non-obese people. Diabetes Care. (2009) 32:1734–6. doi: 10.2337/dc09-0176
40. Nagy ZS, Czimmerer Z, Szanto A, Nagy L. Pro-inflammatory cytokines negatively regulate PPARγ mediated gene expression in both human and murine macrophages via multiple mechanisms. Immunobiology. (2013) 218:1336–44. doi: 10.1016/j.imbio.2013.06.011
41. Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. J Clin Invest. (1994) 94:1543–9. doi: 10.1172/JCI117495
42. Miranda-Filloy JA, Llorca J, Carnero-López B, González-Juanatey C, Blanco R, González-Gay MA. TNF-alpha antagonist therapy improves insulin sensitivity in non-diabetic ankylosing spondylitis patients. Clin Exp Rheumatol. (2012) 30:850–5.
43. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. (2011) 11:85–97. doi: 10.1038/nri2921
44. Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, et al. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci USA. (2001) 98:2005–10. doi: 10.1073/pnas.98.4.2005
45. Ouchi N, Kihara S, Funahashi T, Nakamura T, Nishida M, Kumada M, et al. Reciprocal 461 association of C-reactive protein with adiponectin in blood stream and adipose tissue. Circulation. (2003) 107:671–4. doi: 10.1161/01.CIR.0000055188.83694.B3
46. Sattar N, Wannamethee G, Sarwar N, Tchernova J, Cherry L, Wallace AM, et al. Adiponectin and coronary heart disease: a prospective study and meta-analysis. Circulation. (2006) 114:623–9. doi: 10.1161/CIRCULATIONAHA.106.618918
47. Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA. (2004) 291:1730–7. doi: 10.1001/jama.291.14.1730
48. Ohashi K, Shibata R, Murohara T, Ouchi N. Role of anti-inflammatory adipokines in obesity-related diseases. Trends Endocrinol Metab. (2014) 25:348–55. doi: 10.1016/j.tem.2014.03.009
49. von Mutius E, Schwartz J, Neas LM, Dockery D, Weiss ST. Relation of body mass index to asthma and atopy in children: the National Health and Nutrition Examination Study III. Thorax. (2001) 56:835–8. doi: 10.1136/thorax.56.11.835
50. Visness CM, London SJ, Daniels JL, Kaufman JS, Yeatts KB, Siega-Riz AM, et al. Association of obesity with IgE levels and allergy symptoms in children and adolescents: results from the National Health and Nutrition Examination Survey 2005–2006. J Allergy Clin Immunol. (2009) 123:1163–69, 69.e1161–4. doi: 10.1016/j.jaci.2008.12.1126
51. Tantisira KG, Litonjua AA, Weiss ST, Fuhlbrigge AL, Childhood Asthma Management Program Research Group. Association of body mass with pulmonary function in the Childhood Asthma Management Program (CAMP). Thorax. (2003) 58:1036–41. doi: 10.1136/thorax.58.12.1036
52. Figueroa-Munoz JI, Chinn S, Rona RJ. Association between obesity and asthma in 4–11 year old children in the UK. Thorax. (2001) 56:133–7. doi: 10.1136/thorax.56.2.133
53. Gold DR, Damokosh AI, Dockery DW, Berkey CS. Body-mass index as a predictor of incident asthma in a prospective cohort of children. Pediatr Pulmonol. (2003) 36:514–21. doi: 10.1002/ppul.10376
54. Chen YC, Dong GH, Lin KC, Lee YL. Gender difference of childhood overweight and obesity in predicting the risk of incident asthma: a systematic review and meta-analysis. Obes Rev. (2013) 14:222–31. doi: 10.1111/j.1467-789X.2012.01055.x
55. Vijayakanthi N, Greally JM, Rastogi D. Pediatric obesity-related asthma: the role of metabolic dysregulation. Pediatrics. (2016) 137:e20150812. doi: 10.1542/peds.2015-0812
56. Rastogi D, Holguin F. Metabolic dysregulation, systemic inflammation, and pediatric obesity related asthma. Ann Am Thorac Soc. (2017) 14:S363–7. doi: 10.1513/AnnalsATS.201703-231AW
57. Peters U, Dixon AE, Forno E. Obesity and asthma. J Allergy Clin Immunol. (2018) 141:1169–79. doi: 10.1016/j.jaci.2018.02.004
58. Munger KL, Bentzen J, Laursen B, Stenager E, Koch-Henriksen N, Sorensen TI, et al. Childhood body mass index and multiple sclerosis risk: a long-term cohort study. Mult Scler. (2013) 19:1323–9. doi: 10.1177/1352458513483889
59. Pakpoor J, Pakpoor J. Childhood obesity and risk of pediatric multiple sclerosis and clinically isolated syndrome. Neurology. (2013) 81:1366. doi: 10.1212/WNL.0b013e3182a7af5f
60. Hedstrom AK, Lima Bomfim I, Barcellos L, Gianfrancesco M, Schaefer C, Kockum I, et al. Interaction between adolescent obesity and HLA risk genes in the etiology of multiple sclerosis. Neurology. (2014) 82:865–72. doi: 10.1212/WNL.0000000000000203
61. Crowson CS, Matteson EL, Davis JM III, Gabriel SE. Contribution of obesity to the rise in incidence of rheumatoid arthritis. Arthritis Care Res. (2013) 65:71–7. doi: 10.1002/acr.21660
62. Ajeganova S, Andersson ML, Hafstrom I, BARFOT Study Group. Association of obesity with worse disease severity in rheumatoid arthritis as well as with comorbidities: a long-term follow-up from disease onset. Arthritis Care Res. (2013) 65:78–87. doi: 10.1002/acr.21710
63. Versini M, Jeandel PY, Rosenthal E, Shoenfeld Y. Obesity in autoimmune diseases: not a passive bystander. Autoimmun Rev. (2014) 13:981–1000. doi: 10.1016/j.autrev.2014.07.001
64. Sinicato NA, Postal M, Peres FA, Pelicari Kde O, Marini R, dos Santos Ade O, et al. Obesity and cytokines in childhood-onset systemic lupus erythematosus. J Immunol Res. (2014) 2014:162047. doi: 10.1155/2014/162047
65. Chaiamnuay S, Bertoli AM, Fernandez M, Apte M, Vila LM, Reveille JD, et al. The impact of increased body mass index on systemic lupus erythematosus: data from LUMINA, a multiethnic cohort (LUMINA XLVI) [corrected]. J Clin Rheumatol. (2007) 13:128–33. doi: 10.1097/RHU.0b013e3180645865
66. Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and obesity: a systematic review and meta-analysis of observational studies. Nutr Diabetes. (2012) 2:e54. doi: 10.1038/nutd.2012.26
67. Carrascosa JM, Rocamora V, Fernandez-Torres RM, Jimenez-Puya R, Moreno JC, Coll Puigserver N, et al. Obesity and psoriasis: inflammatory nature of obesity, relationship between psoriasis and obesity, and therapeutic implications. Actas Dermosifiliogr. (2014) 105:31–44. doi: 10.1016/j.ad.2012.08.003
68. Debbaneh M, Millsop JW, Bhatia BK, Koo J, Liao W. Diet and psoriasis, part I: Impact of weight loss interventions. J Am Acad Dermatol. (2014) 71:133–40. doi: 10.1016/j.jaad.2014.02.012
69. Manzel A, Muller DN, Hafler DA, Erdman SE, Linker RA, Kleinewietfeld M. Role of “Western diet” in inflammatory autoimmune diseases. Curr Allergy Asthma Rep. (2014) 14:404. doi: 10.1007/s11882-013-0404-6
70. Verthelyi D. Sex hormones as immunomodulators in health and disease. Int Immunopharmacol. (2001) 1:983–93. doi: 10.1016/S1567-5769(01)00044-3
71. Schaible UE, Kaufmann SH. Malnutrition and infection: complex mechanisms and global impacts. PLoS Med. (2007) 4:e115. doi: 10.1371/journal.pmed.0040115
72. Huttunen R, Syrjanen J. Obesity and the risk and outcome of infection. Int J Obes. (2013) 37:333–40. doi: 10.1038/ijo.2012.62
73. Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. (2008) 371:569–78. doi: 10.1016/S0140-6736(08)60269-X
74. Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. (2002) 3:221–7. doi: 10.1038/ni0302-221
75. Lamas O, Marti A, Martinez JA. Obesity and immunocompetence. Eur J Clin Nutr. (2002) 56 (Suppl 3):S42–5. doi: 10.1038/sj.ejcn.1601484
76. Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol. (2018) 19:1330–40. doi: 10.1038/s41590-018-0251-7
77. Massaro M, Scoditti E, Pellegrino M, Carluccio MA, Calabriso N, Wabitsch M, et al. Therapeutic potential of the dual peroxisome proliferator activated receptor (PPAR)alpha/gamma agonist aleglitazar in attenuating TNF-alpha-mediated inflammation and insulin resistance in human adipocytes. Pharmacol Res. (2016) 107:125–36. doi: 10.1016/j.phrs.2016.02.027
Keywords: obesity, immune system, inflammation, adipokines, atopy, autoimmune diseases
Citation: Umano GR, Pistone C, Tondina E, Moiraghi A, Lauretta D, Miraglia del Giudice E and Brambilla I (2019) Pediatric Obesity and the Immune System. Front. Pediatr. 7:487. doi: 10.3389/fped.2019.00487
Received: 28 May 2019; Accepted: 06 November 2019;
Published: 22 November 2019.
Edited by:
Luigi Daniele Notarangelo, National Institute of Allergy and Infectious Diseases (NIAID), United StatesReviewed by:
Sigrun Ruth Hofmann, Universitätsklinikum Carl Gustav Carus, GermanyEugênia Terra Granado, Brazilian National Cancer Institute (INCA), Brazil
Copyright © 2019 Umano, Pistone, Tondina, Moiraghi, Lauretta, Miraglia del Giudice and Brambilla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppina Rosaria Umano, Z2l1c2VwcGluYXJvc2FyaWEudW1hbm9AdW5pY2FtcGFuaWEuaXQ=