 Eva Pauline Macheroux1
Eva Pauline Macheroux1 Matthias C. Braunisch1,2Stephanie Pucci Pegler1Robin Satanovskij1,2
Matthias C. Braunisch1,2Stephanie Pucci Pegler1Robin Satanovskij1,2 Korbinian M. Riedhammer1,2Roman Günthner1,2
Korbinian M. Riedhammer1,2Roman Günthner1,2 Oliver Gross3Mato Nagel4Lutz Renders2
Oliver Gross3Mato Nagel4Lutz Renders2 Julia Hoefele1*
Julia Hoefele1*- 1Institute of Human Genetics, School of Medicine, Technical University of Munich, Munich, Germany
- 2Department of Nephrology, School of Medicine, Technical University of Munich, Munich, Germany
- 3Clinic of Nephrology and Rheumatology, University Medical Center Goettingen, University of Goettingen, Goettingen, Germany
- 4Center for Nephrology and Metabolic Medicine, Weisswasser, Germany
Background: Alport syndrome (AS) is a progressive kidney disorder leading to end stage renal disease (ESRD). Extrarenal symptoms like hearing loss and ocular changes can be observed. Approximately 85% of the patients carry pathogenic variants in COL4A5 (X-linked inheritance). The variant c.1871G>A, p.(Gly624Asp) in COL4A5 is described in the literature as a hypomorphic variant associated with thin basement membrane nephropathy (TBMN). ESRD was only seen rarely at a median age of 50 years and extrarenal manifestations have only been described in single cases.
Case report and Methods: This is a report on a family with X-linked AS. In the female index patient, microscopic hematuria, and proteinuria were observed beginning at the age of 20 years and 41 years, respectively. Microscopic hematuria was also present in the daughter (from 6th month of life), the son (from 22nd month of life), the mother and the maternal grandniece. Proteinuria was observed in the maternal aunt and paternal grandmother. The father of the index patient, a paternal uncle and a second cousin presented with ESRD at the age of 49, 34, and 70 years of life, respectively. Extrarenal manifestations were absent in the whole family. In the index patient, her children and her mother molecular diagnostics were performed using Sanger and exome sequencing.
Results: In all examined family members the variant c.1871G>A, p.(Gly624Asp) in COL4A5 was identified. With the exception of the index patient, who was homozygous for this variant, all family members carried the variant heterozygously, or hemizygously. A different or additional monogenic hereditary nephropathy could not be detected by exome sequencing of the index patient.
Discussion: This is the first report of a patient with the variant p.(Gly624Asp) in COL4A5 in a homozygous state. The variant was previously reported as a mild variant requiring dialysis in less than 10%. The family presented, however, with a more severe clinical course. We therefore suggest to question the term “hypomorphic” in the context of the variant p.(Gly624Asp) although molecular diagnostics could not be done in all affected family members.
Introduction
Alport syndrome (AS) is a rare hereditary kidney disorder with an estimated prevalence of 1:5,000 (X-linked form). It is characterized by hematuria, proteinuria, and progressive renal failure leading to chronic kidney disease (1–7). AS causes end stage renal disease in about 1–2% of patients in infancy, adolescence, and young adulthood. Frequent extrarenal manifestations are sensorineural hearing loss (60–80% of patients), ocular changes (lenticonus of the anterior lens capsule, retinopathy; 25–40% of patients) and rarely, mental retardation, and leiomyomatosis (8, 9).
AS is caused by pathogenic variants in the genes COL4A3, COL4A4, and COL4A5, encoding the α3, α4, and α5 chain of collagen type IV (2). In ~85% of patients with AS, pathogenic variants in COL4A5 on chromosome Xq22.3 are causative (10, 11). AS can also be inherited in an autosomal recessive pattern (about 15% of the cases; biallelic pathogenic variants in COL4A3 and COL4A4). Other presumed modes of inheritance are autosomal dominant or a digenic inheritance (12, 13).
Because of the common X-linked inheritance, mostly males are affected and show a distinct genotype-phenotype correlation (14). Missense or in-frame variants lead to a milder progress of disease with a later onset of end-stage renal disease (ESRD) (37 years) compared to splice-site and truncating variants which lead to a more severe phenotype with early onset of ESRD (25–28 years). Additionally, a strong relationship between variant position and age at ESRD has been demonstrated, with younger age at ESRD associated with pathogenic variants at the 5′ end of the gene. Furthermore, patients with splice-site variants and truncating variants had two-fold greater odds of developing ocular changes and/or hearing impairment than those with missense variants (14, 15).
Some variants in the COL4A3-5 genes are described as hypomorphic as they are not fully destructive and therefore obtain a residual protein function. One of these variants is p.(Gly624Asp) in COL4A5 which has been described in the context of thin basement membrane nephropathy (TBMN) in males. The clinical phenotype was comparable to a heterozygous carrier of a variant in COL4A3 or COL4A4 (16, 17).
This report describes a family with several individuals with symptoms ranging from proteinuria up to ESRD requiring dialysis. In some of the family members the variant p.(Gly624Asp) in COL4A5 could be confirmed. Furthermore, this is the first description of a female patient with this hypomorphic variant in a homozygous state. Additionally, this is the first report of a female patient with a homozygous variant in COL4A5 at all.
Materials and Methods
Clinical Case Information
The study was approved by the local Ethics Committee of the Technical University of Munich and performed according to the standards of the 2013 Helsinki Declaration. Written informed consent was obtained from the index patient and their relatives for publication. Clinical and phenotype information was gathered out of clinical reports and medical history.
Genetics
Blood samples were collected after written informed consent. Genomic DNA was extracted from peripheral blood of the index patient (Figure 1, V-3) and her children (Figure 1, VI-3 and VI-4) using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany) according to manufacturer's instructions.
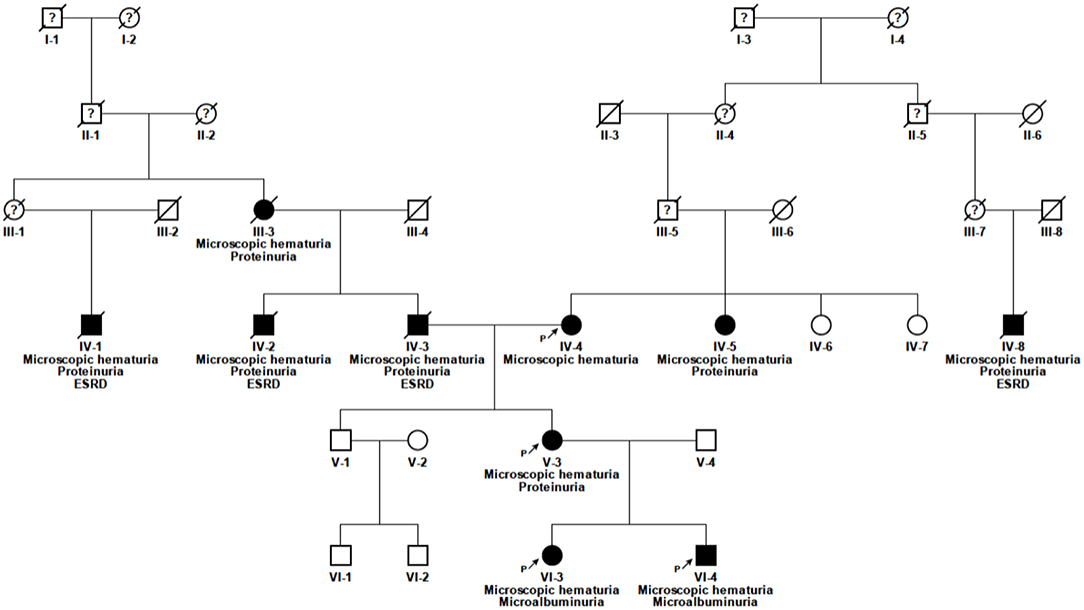
Figure 1. Pedigree of the family. Solid symbols, affected individuals; circles, females; squares, males; n.d., no data available; P, proband; ?, unknown medical history.
Sanger Sequencing
In the index patient exon 1–51 of COL4A5 were examined followed by exon 1–52 of COL4A3 and exon 1–48 of COL4A4 using direct DNA sequencing on both strands applying the dideoxy chain termination method on an ABI capillary sequencer 3730 (Applied Biosystems, Foster City, USA). Primers were designed by Primer3 program (http://frodo.wi.mit.edu/primer3/input.htm). For segregation analysis, subsequent targeted sequencing was performed in mother and children of the index patient in exon 25 of the COL4A5 gene. DNA alignment and sequence variant analysis were carried out using the Sequence PilotCE software (JSI Medical Systems GmbH, Kippenheim, Germany) and compared to EMBL (European Molecular Biology Laboratory) and GenBank databases as well as our in-house database. All variants were validated in a second independent DNA sample.
Exome Sequencing
Exome sequencing was additionally performed in the index patient to examine pathogenic variants and genetic modifier in other genes responsible for the renal phenotype and its severity using a Sure Select Human All Exon 60Mb V6 Kit (Agilent) and a HiSeq4000 (Illumina) as previously described (18, 19). Reads were aligned to the UCSC human reference assembly (hg19) with BWA v.0.5.8. More than 98% of the exome was covered at least 20x. Single-nucleotide variants (SNVs) and small insertions and deletions were detected with SAMtools v.0.1.7. For the analysis of autosomal dominant and mitochondrial variants, only variants with a minor allele frequency (MAF) of less than 0.1% in the in-house database of the Helmholtz center Munich containing over 16,000 exomes were considered. For the analysis of autosomal recessive and X-linked variants (homozygous, hemizygous or putative compound heterozygous), variants with a MAF of <1.0% were considered. As there are pathogenic alleles in hereditary nephropathies with a MAF of more than 1.0% like the NPHS2 p.Arg229Gln or the NEPH3 p.Val353Met allele (20–22), an additional search for recessive and X-linked variants with a MAF up to 3% was used. Variants were classified in accordance to the classification of the American College of Human Genetics (AMCG) (23, 24). Exome depth was used as a CNV calling algorithm (25).
Results
Clinical Phenotype
Index Patient
The 52 year old female index patient is of Caucasian origin (Figure 1, V-3). She is of non-consanguineous German ancestry and presented to our department with microscopic hematuria and proteinuria. The further clinical presentation was unremarkable, besides a nasal septum operation with a tonsillectomy at the age of 10 years, varicose veins at the right lower leg and an atrial septal aneurysm with patent foramen ovale. Notably, there was no report of any extrarenal involvement. At the age of 20 years the index patient was first diagnosed with microhematuria, followed by microalbuminuria (132 mg/24 h) at the age of 34 years and by proteinuria (822 mg/g creatinine) first seen at the age of 41 years. An ultrasound examination performed at the age of 34 years showed morphologically normal kidneys. Therapy with angiotensin-converting enzyme (ACE) inhibitor was initiated at the age of 39 years which was switched to an AT1 antagonist because of ACE inhibitor-induced cough.
Relatives
The daughter of the index patient (VI-3) first presented with microhematuria at the age of 6 months and microalbuminuria (182 mg/g creatinine) at the age of 4 ½ years (see Table 1). She is currently 22 years of age and without medication. The son of the index patient (VI-4) showed microscopic hematuria and microalbuminuria (134 mg/g creatinine) since the age of 22 months. He is currently 20 years of age and is treated with AT1 antagonist since the age of 19 years (microalbuminuria <300 mg/g creatinine). The father of the index patient (IV-3) was on dialysis since the age of 49 years, as well as a paternal uncle (IV-2) and a paternal grand cousin (IV-1) (34 and 70 years of age, respectively). The father of the index patient died from stroke which occurred after kidney transplantation. The mother of the index patient (IV-4) is known to have microscopic hematuria and an eGFR of 65 ml/min/1.73 m2 at the current age of 78 years. The treating physician denied to perform urine analysis in this relative so far. The paternal grandmother (III-3) was known having proteinuria. The maternal aunt (IV-5) developed proteinuria after giving birth to twins at the age of 23 years. Extrarenal manifestations could not be detected in any of the family member.
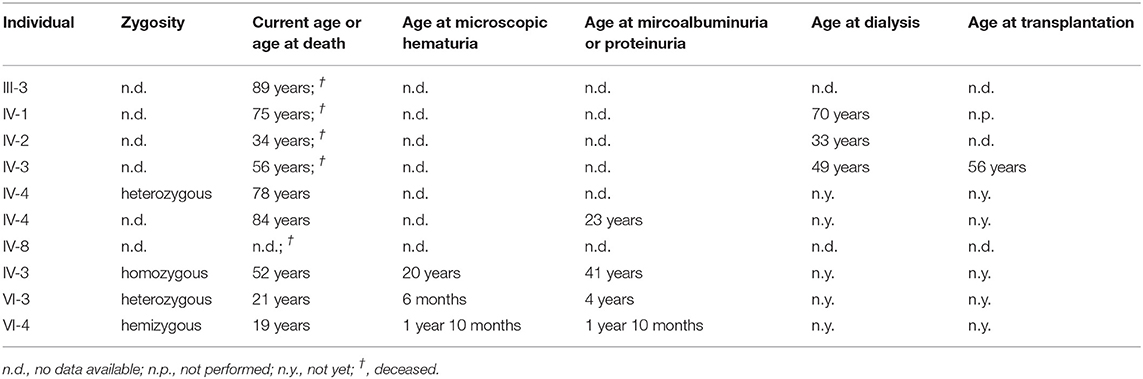
Table 1. Overview about the p.(Gly624Asp) in COL4A5-associated clinical phenotype and the genetic zygosity of the affected family members.
Genetic Analysis
Pathogenic variants in COL4A3 and COL4A4 could not be identified. Sanger sequencing of COL4A5 (NM_033380.2) in the index patient revealed the homozygous missense variant c.1871G>A, p.(Gly624Asp) (Figure 1, V-3). The variant is located in exon 25. The variant leads to a change of an evolutionarily highly conserved nucleotide at the second position of codon 624. The son and the daughter of the index patient carry the same variant in a hemizygous and a heterozygous state, respectively (Figure 1, VI-3 and VI-4). This variant was also present in the mother of the index patient (Figure 1, IV-4). Exome sequencing of the index patient did not reveal any other pathogenic variant responsible for the renal phenotype in this family. Modifier variants in other podocyte genes like NPHS2 or NEPH3 were also not identified.
Discussion
In all available affected family members the already known variant p.(Gly624Asp) in COL4A5 most likely causative for their renal phenotype was identified. The presence of a glycine residue as every third amino acid in the collagenous domain of COL4A3/4/5 is essential. Therefore, most of the pathogenic variants identified in patients with AS affect a glycine residue. However, the variant p.(Gly624Asp) is described in the literature as a hypomorphic variant leading to the mild clinical phenotype of TBMN in males and females caused by a residual function of the remaining protein. In the patients carrying this variant ESRD is very rare and only seen in single patients at a median age of 50 years (16, 17). The variant is also reported in the genome Aggregation Database (gnomAD) with an overall allele frequency of 1 in 11,438 individuals. In Europeans (non-Finnish), the allele frequency is 1 in 5105, including four hemizygotes and no homozygotes. In this family all affected and molecular confirmed individuals developed microalbuminuria or proteinuria later in life. Additionally, all affected adult male individuals presumably carrying this variant presented with ESRD. In contrast to the available literature, this observation suggests that this variant is not as mild as assumed.
As variants in other genes can mimic a similar clinical and histological phenotype of AS and albeit the pedigree seems compatible with an X-linked inheritance pattern, a second monogenic disease was not identified by exome sequencing. And even if exome sequencing cannot absolutely exclude other pathogenic variants and not in all affected relatives molecular analysis could be performed, another genetic cause seems unlikely in this family.
Extrarenal manifestations are a common feature in AS patients observed in 25% (ocular changes) and 80% (sensorineural hearing loss) of the cases, respectively. In the literature only two patients carrying the hemizygous variant p.(Gly624Asp) developed late onset hearing impairment so far and one patient ocular lesions (16). As the affected individuals in our family did also not develop any extrarenal malformations, this observation can support the rare occurrence of extrarenal manifestations.
To our knowledge, this is the first report on a patient carrying the variant p.(Gly624Asp) in homozygous state and with a homozygous variant in COL4A5 at all. Female patients with a heterozygous variant in COL4A5 (X-linked AS) have been traditionally described as carrier female subjects even though with variable intra- and interfamilial penetrance, showing a broad spectrum of clinical symptoms ranging from mild microscopic hematuria to severe AS (26–29). In contrast to male patients, genotype-phenotype correlation is less-well described in females so far but loss-of-function variants appear to result in a more severe phenotype (27, 30). As possible causes for phenotypic variability in X-linked AS, the type of variant, degree of mosaicism following lyonization of the X chromosome, and genetic modifiers are discussed, among other things (13, 26, 31, 32).
As the female index patient of the reported family is genetically comparable with a male patient, and even if other genetic causes (e.g., digenic inheritance, intronic variants, genetic modifiers) cannot be excluded for sure in her affected relatives and non-genetic factors might be different, she might develop ESRD according to her affected male relatives.
As this variant seems to lead to a different clinical phenotype and some of the carriers seem to have a more severe phenotype than known in the literature so far, a registry study solely collecting patients with the variant p.(Gly624Asp) should be initiated to answer the open question concerning the severity of this variant. Genetic modifiers in other genes like NPHS2 or NEPH3 could be one possible cause for an aggravation of the symptoms but were not seen in the present case (20–22). However, according to this report the term “hypomorphic” in the context of this variant should be questioned.
Conclusion
This report presents a severely affected family with the so far called “hypomorphic” variant p.(Gly624Asp) in COL4A5. It also underlines that patients with a variant classified as hypomorphic can present with AS and therefore have to be monitored closely as well as treated accordingly.
Data Availability Statement
The datasets analyzed in this manuscript are not publicly available. Requests to access the datasets should be directed to JH, anVsaWEuaG9lZmVsZUB0dW0uZGU=.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee, Technical University of Munich, Munich, Germany. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
MB, KR, and JH analyzed and interpreted the patient data regarding the genetic and clinical findings. EM, SP, RG, and JH wrote the manuscript. KR, RS, and MN performed the molecular diagnostics. OG conducted in-patient treatment. EM, MB, LR, KR, and RG contributed important intellectual content during manuscript drafting and revision. All authors accept accountability for the overall work by ensuring that questions pertaining to the accuracy or integrity of any portion of the work are appropriately investigated and resolved. Text revision was performed by all authors. All authors read and approved the final manuscript.
Funding
This work was supported by the German Research Foundation (DFG) and the Technical University of Munich (TUM) in the framework of the Open Access Publishing Program. JH was supported by the German Pediatric Nephrology Association.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the index patient and her family for their participation in the study.
Abbreviations
ACE, angiotensin-converting enzyme; AS, Alport syndrome; CNV, copy number variant; DNA, deoxyribonucleic acid; ESRD, end-stage renal disease; gnomAD, genome Aggregation Database; SNP, single nucleotide polymorphism; TBMN, thin basement membrane nephropathy.
References
1. Nagel M, Nagorka S, Gross O. Novel COL4A5, COL4A4, and COL4A3 mutations in Alport syndrome. Hum Mutat. (2005) 26:60. doi: 10.1002/humu.9349
2. Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med. (2003) 348:2543–56. doi: 10.1056/NEJMra022296
3. Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M. Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: impact on clinical counselling. Nephrol Dial Transplant. (2002) 17:1218–27. doi: 10.1093/ndt/17.7.1218
4. Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M. Novel COL4A4 splice defect and in-frame deletion in a large consanguine family as a genetic link between benign familial haematuria and autosomal Alport syndrome. Nephrol Dial Transplant. (2003) 18:1122–7. doi: 10.1093/ndt/gfg157
5. Flinter FA, Cameron JS, Chantler C, Houston I, Bobrow M. Genetics of classic Alport's syndrome. Lancet. (1988) 2:1005–7. doi: 10.1016/S0140-6736(88)90753-2
6. Alport AC. Hereditary familial congenital haemorrhagic nephritis. Br Med J. (1927) 1:504–6. doi: 10.1136/bmj.1.3454.504
7. Williamson DA. Alport's syndrome of hereditary nephritis with deafness. Lancet. (1961) 2:1321–3. doi: 10.1016/S0140-6736(61)90899-6
8. Grunfeld JP. Contemporary diagnostic approach in Alport's syndrome. Renal Fail. (2000) 22:759–63. doi: 10.1081/JDI-100101961
9. Kashtan CE. Familial hematuric syndromes–Alport syndrome, thin glomerular basement membrane disease and Fechtner/Epstein syndromes. Contribut Nephrol. (2001) 136:79–99. doi: 10.1159/000060181
10. Barker DF, Hostikka SL, Zhou J, Chow LT, Oliphant AR, Gerken SC, et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science. (1990) 248:1224–7. doi: 10.1126/science.2349482
11. Martin P, Heiskari N, Zhou J, Leinonen A, Tumelius T, Hertz JM, et al. High mutation detection rate in the COL4A5 collagen gene in suspected Alport syndrome using PCR and direct DNA sequencing. J Am Soc Nephrol. (1998) 9:2291–301.
12. Weber S, Strasser K, Rath S, Kittke A, Beicht S, Alberer M, et al. Identification of 47 novel mutations in patients with Alport syndrome and thin basement membrane nephropathy. Pediatr Nephrol. (2016) 31:941–55. doi: 10.1007/s00467-015-3302-4
13. Mencarelli MA, Heidet L, Storey H, van Geel M, Knebelmann B, Fallerini C, et al. Evidence of digenic inheritance in Alport syndrome. J Med Genet. (2015) 52:163–74. doi: 10.1136/jmedgenet-2014-102822
14. Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. (2000) 11:649–57.
15. Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, Masoumi A, et al. Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol. (2010) 21:876–83. doi: 10.1681/ASN.2009070784
16. Pierides A, Voskarides K, Kkolou M, Hadjigavriel M, Deltas C. X-linked, COL4A5 hypomorphic Alport mutations such as G624D and P628L may only exhibit thin basement membrane nephropathy with microhematuria and late onset kidney failure. Hippokratia. (2013) 17:207–13.
17. Slajpah M, Gorinsek B, Berginc G, Vizjak A, Ferluga D, Hvala A, et al. Sixteen novel mutations identified in COL4A3, COL4A4, and COL4A5 genes in Slovenian families with Alport syndrome and benign familial hematuria. Kidney Int. (2007) 71:1287–95. doi: 10.1038/sj.ki.5002221
18. Haack TB, Gorza M, Danhauser K, Mayr JA, Haberberger B, Wieland T, et al. Phenotypic spectrum of eleven patients and five novel MTFMT mutations identified by exome sequencing and candidate gene screening. Mol Genet Metab. (2014) 111:342–52. doi: 10.1016/j.ymgme.2013.12.010
19. Wortmann SB, Zietkiewicz S, Kousi M, Szklarczyk R, Haack TB, Gersting SW, et al. CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am J Hum Genet. (2015) 96:245–57. doi: 10.1016/j.ajhg.2014.12.013
20. Tonna S, Wang YY, Wilson D, Rigby L, Tabone T, Cotton R, et al. The R229Q mutation in NPHS2 may predispose to proteinuria in thin-basement-membrane nephropathy. Pediatr Nephrol. (2008) 23:2201–7. doi: 10.1007/s00467-008-0934-7
21. Stefanou C, Pieri M, Savva I, Georgiou G, Pierides A, Voskarides K, et al. Co-inheritance of functional podocin variants with heterozygous collagen IV mutations predisposes to renal failure. Nephron. (2015) 130:200–12. doi: 10.1159/000432406
22. Voskarides K, Stefanou C, Pieri M, Demosthenous P, Felekkis K, Arsali M, et al. A functional variant in NEPH3 gene confers high risk of renal failure in primary hematuric glomerulopathies. Evidence for predisposition to microalbuminuria in the general population. PLoS ONE. 12:e0174274. doi: 10.1371/journal.pone.0174274
23. Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, Working Group of the American College of Medical Genetics Laboratory Quality Assurance C. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. (2011) 13:680–5. doi: 10.1097/GIM.0b013e3182217a3a
24. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
25. Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. (2012) 28:2747–54. doi: 10.1093/bioinformatics/bts526
26. Rheault MN. Women and Alport syndrome. Pediatr Nephrol. (2012) 27:41–6. doi: 10.1007/s00467-011-1836-7
27. Savige J, Colville D, Rheault M, Gear S, Lennon R, Lagas S, et al. Alport syndrome in women and girls. Clin J Am Soc Nephrol. (2016) 11:1713–20. doi: 10.2215/CJN.00580116
28. Yamamura T, Nozu K, Fu XJ, Nozu Y, Ye MJ, Shono A, et al. Natural history and genotype-phenotype correlation in female X-linked alport syndrome. Kidney Int Rep. (2017) 2:850–5. doi: 10.1016/j.ekir.2017.04.011
29. Kashtan CE, Ding J, Garosi G, Heidet L, Massella L, Nakanishi K, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV α345: a position paper of the Alport Syndrome Classification Working Group. Kidney Int. (2018) 93:1045–51. doi: 10.1016/j.kint.2017.12.018
30. Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study. J Am Soc Nephrol. (2003) 14:2603–10. doi: 10.1097/01.ASN.0000090034.71205.74
31. Fallerini C, Baldassarri M, Trevisson E, Morbidoni V, La Manna A, Lazzarin R, et al. Alport syndrome: impact of digenic inheritance in patients management. Clin Genet. (2017) 92:34–44. doi: 10.1111/cge.12919
Keywords: Alport syndrome, COL4A5, p.Gly624Asp, ESRD, hearing impairment
Citation: Macheroux EP, Braunisch MC, Pucci Pegler S, Satanovskij R, Riedhammer KM, Günthner R, Gross O, Nagel M, Renders L and Hoefele J (2019) The Hypomorphic Variant p.(Gly624Asp) in COL4A5 as a Possible Cause for an Unexpected Severe Phenotype in a Family With X-Linked Alport Syndrome. Front. Pediatr. 7:485. doi: 10.3389/fped.2019.00485
Received: 03 September 2019; Accepted: 05 November 2019;
Published: 26 November 2019.
Edited by:
Max Christoph Liebau, Universitätsklinikum Köln, GermanyReviewed by:
Konstantinos Voskarides, University of Cyprus, CyprusHamidreza Badeli, Gilan University of Medical Sciences, Iran
Copyright © 2019 Macheroux, Braunisch, Pucci Pegler, Satanovskij, Riedhammer, Günthner, Gross, Nagel, Renders and Hoefele. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julia Hoefele, anVsaWEuaG9lZmVsZUB0dW0uZGU=