 Xuehan Bai1,2†
Xuehan Bai1,2† Jie Tian
Jie Tian Tiewei Lv
Tiewei Lv- 1Department of Cardiology, Children's Hospital of Chongqing Medical University, Chongqing, China
- 2China International Science and Technology Cooperation Base of Child Development and Critical Disorders, Chongqing Key Laboratory of Pediatrics, Ministry of Education Key Laboratory of Child Development and Disorders, Chongqing, China
- 3Chengdu Women's and Children's Central Hospital, Chengdu, China
- 4Department of Biomedical Science, Charlie E. Schmidt College of Medicine, Florida Atlantic University, Boca Raton, FL, United States
Background: The MTUS1 gene encodes a microtubule-associated protein involved in multiple processes including cell polarity and microtubule balance during myocardial development.
Aims: To investigate the association between a de novo c. 2617A->C mutation in MTUS1 (NM_001001924.2) and non-compaction of ventricular myocardium (NVM) and explore the potential mechanisms.
Methods: A de novo mutation in MTUS1 was identified for a familial pedigree with NVM. Lentiviral vectors containing MTUS1 wild type or the mutation MTUS1 were constructed and co-infected into HEK-293 cells. MTUS1, Rac1/Cdc42, α-tubulin, α/β-tubulin, polarity protein (PAR6), and the morphology of daughter cells were measured by real-time PCR, Western blot, and immunofluorescence assays, respectively.
Results: The lentiviral vectors were constructed successfully. Immunofluorescence assays revealed the fluorescence intensity of α-tubulin to be decreased and α/β-tubulin to be increased in the mutation MTUS1 group. The fluorescence intensity of PAR6 was higher and morphology of the daughter cells in the mutation group was different from the wild type group. The phosphorylation of Rac1/Cdc42 in the mutation group was significantly lower than in the wild type group.
Conclusions: A de novo mutation in MTUS1 decreased the stability of microtubules and increased cell polarity via the Rac1/Cdc42 pathway, which may partly elucidate the mechanism underlying cellular protection in NVM.
Introduction
Non-compaction of ventricular myocardium (NVM) is a structural abnormality of the left ventricular myocardium that is accompanied by severe clinical symptoms and a poor prognosis with no currently available effective prevention and therapeutic methods (1–4). NVM is diagnosed based on the ratio of the thickness of the non-compact endocardial layer to the thickness of the compact epicardial layer being >2.0 on echocardiograph (5). In the majority of patients, NVM is associated with genetic disease, particularly neuromuscular disorders and chromosomal defect (6, 7). The polarity of myocardial cells was recently reported to play an important role in the development of NVM (8, 9). However, the underlying molecular mechanisms regulating cell polarity during early cardiac development and trabecular formation remain poorly understood.
Polarity is one of the basic processes that occur in living organisms (10). Cell polarity is the result of asymmetrical organization of cell membrane proteins and cell contents and can influence cell fate and specialized functions, such as migration, development, and proliferation (11). Cell polarity is controlled by Rho GTPase family members, the Par polarity complex, and cytoskeleton (12). Microtubules are a component of the cytoskeleton, found in eukaryotic cells, and formed by the polymerization of a dimer of two globular proteins, α, and β tubulin. These tubular polymers of tubulin are highly dynamic and stabilize the cell structure, transport intracellular substances, and mediate cell movement. The Rho GTPase family consists of six subfamilies: Rho, Rac, Cdc42, Rnd, RhoBTB, and RhoT/Miro (13). The Rho GTPase family mediates the formation of the Par polarity complex, which causes cell polarity. The PAR proteins PAR3, PAR6, and aPKC localize to the anterior cortex, where PAR1 and PAR2 localize to the posterior pole and have essential functions in the first asymmetric division (14). Polar proteins are transported to the cell membrane through microtubule dynamic balance, thus forming polar protein complexes (15). In cardiac development, disruption of the cell polarity complex by targeted gene mutations results in aberrant mitotic spindles, loss of polarized cardiomyocyte division, and loss of normal myocardial trabeculation (9).
Microtubule-associated tumor suppressor 1 (MTUS1) encodes the microtubule-associated protein ATIP3, which cooperates with type-2 angiotensin II receptor (AGTR2) to inhibit extracellular signal-regulated kinase 2 (ERK2) activation and cell proliferation, which are closely related to cell division and migration (16, 17). A recently published study showed that MTUS1 knock-out mice developed spontaneous heart hypertrophy (18), suggesting that MTUS1 may affect cardiovascular system development.
However, the specific mechanisms underlying these processes have not yet been elucidated. Our team discovered a de novo mutation in MTUS1, c. 2617A->C (rs187103704), in a rare NVM family, that is likely associated with the mechanism underlying NVM. Therefore, the aim of this study was to investigate the association between a de novo mutation in MTUS1 with NVM and to explore the potential mechanisms underlying this association. The current findings may help understand the genetic basis of NVM development, provide a theoretical basis for genetic counseling, prenatal diagnosis, and early intervention, and facilitate the development of new strategies for personalized medicine.
Materials and Methods
Subjects
A rare NVM family pedigree was discovered at the Children's Hospital of Chongqing Medical University. Blood samples were collected from the propositus and her family (sister, mother and aunt) for DNA extraction and whole exome sequencing (WES) (Deyi Oriental Translational Medicine Research Center, China). The original WES data were analyzed to confirm the biological relationships between the daughters, their mother and their aunt. First, the mutations identified by WES were selected by bioinformatics analysis and then functional predictions were made by making comparisons using Genebank, including the UCSC Genome Browser (http://genome.ucsc.edu/), GENECARDS (https://www.genecards.org/), the NCBI database (https://www.ncbi.nlm.nih.gov/), UNIPROT (https://www.uniprot.org/), and STRING (https://string-db.org/). Then the well-conserved mutations that caused amino acid polarity changes in important functional domains were screened as possibly pathogenic for NVM. The mutations were then identified by Sanger sequencing, with the Chromas software used for data analysis.
Cell Culture and Transfections
HEK-293 cells, a classic cell line used in cell biology and gene research, were maintained at 37°C in a humidified atmosphere with 5% CO2 in Dulbecco's Modified Eagle Medium (Hyclone) containing 4.5 g/l glucose, 10% fetal bovine serum (Hyclone), and 100 mg/ml penicillin/streptomycin (19). Plasmosin (55 μl; InvivoGen, ant-mpt) was added to 550 ml complete medium to avoid mycoplasma contamination. MTUS1 has multiple different isoforms with distinct functions. ATIP3 isoform (encoded by MTUS1 gene), which expresses most in heart, has been studied most extensively. This isoform is known to play an important role in microtubule functions. We avoided selecting truncations or short isoforms which contain the mutant site of MTUS1, and we chose full length genes to construct lentiviruses, in order to avoid that the mutation site may affect the function of different functional domains through interaction. Lentiviruses containing GFP and FLAG-tags were completed by Genechem (China). To co-transfect with lentiviruses, HEK-293 cells were plated at a density of 2 ×105 cells/5 ml in T25 culture flasks. Media were replaced with fresh media daily. Once the cells reached a density of about 40–50%, they were co-transfected with mutation variant, wild type, and vector lentiviruses at an MOI of 5. A total of 12 h after transfection, lentivirus-containing medium was replaced with fresh medium.
MTUS1 Expression Based on Real-Time PCR
At 24 h post-transfection, total RNA was extracted from HEK-293 cells using a TRIzol Reagent kit (Ambion) per the manufacturer's instructions and quantified by spectrophotometry at 260 nm. The mRNA was reversely transcribed using the Prime Script RT reagent kit containing gDNA Eraser (Takara, NO: RR047A) as described (20). The primers used for amplification were as follows: MTUS1, GAGCTGAGCACTTACAGCAACAA (forward) and TTCAACTGCATTAAGAGCTGTAA (reverse); and β-actin, CTCTTCCAGCCTTCCTTCCT (forward) and AGCACTGTGTTGGCGTACAG (reverse). The mRNA levels were normalized using β-actin as a housekeeping gene. Each experiment was repeated at least three times.
Immunofluorescence Staining for Tubulin and PAR6
Cells plated on coverslips that were 30–40% confluent were fixed in an ice-cold 4% formaldehyde solution for 20 min prior to incubating for 15 min at room temperature with 0.05% Triton-X100 (21). The slides were then blocked with normal goat serum for 30 min. Slides were washed three times for at least 3 min each time after each incubation and then incubated overnight at 4°C with mouse anti-α-tubulin antibodies (Santa Cruz, USA, sc-5286, 1:50), rabbit anti-α/β-tubulin antibodies (Cell signaling, USA, Cat:2148,1:50), or rabbit anti-PAR6 antibodies (Abcam, USA, ab49776, 1:200). After washing three times the following day, cells were incubated for 60 min at room temperature with either Cy-3-conjugated anti-mouse antibodies or Cy-3-conjugated anti-rabbit antibodies (Seville, wuhan, 1:250) and then washed three times. The cell nuclei were identified by staining with DAPI (Roche, 10236276001, 5 μg/ml) for 15 min at room temperature. Coverslips were mounted on glass slides using DAPI Fluoromount-GTM (YEASEN, 36308ES11, Shanghai) and examined by confocal microscopy. Image analysis was performed using NIH and Image J software. Each experiment was repeated at least three times.
Western Blot Analysis of Rac1/Cdc42
Protein was extracted from cells in exponential growth. Total cellular protein was extracted on ice using 1 × lysis buffer (KeyGEN BioTECH, NO: KGP250) supplemented with protease inhibitor (Roche, Switzerland) and phosphatase inhibitor (Roche, Switzerland). The entire protein extraction was performed strictly on ice. Protein concentrations were measured with the Coomassie (Bradford) Protein Assay (KeyGEN BioTECH, China). Total protein, 150 μg per lane, was separated on 12% SDS-PAGE gels. The SDS-PAGE gels and PVDF membranes were cut into corresponding sizes according to the molecular weights of the target proteins. Proteins were then transferred onto 0.22-μm PVDF membranes. Non-specific bands were blocked with Tris-buffered saline and Tween 20 (TBST) containing 5% bovine serum albumen for 1.5 h at room temperature. Then the membranes were incubated with specific primary antibodies at 4°C overnight. The next day, the PVDF membranes were incubated with the corresponding secondary antibodies at room temperature for 1.5 h (19, 20). Proteins bound to the 0.22-μm PDVF membranes were detected using primary antibodies against β-actin (4A Biotech, China, 1:1000), Rac1/Cdc42 (Cell signaling, 4651, USA, 1:1000), phospho-Rac1/Cdc42 (Ser71) (Cell signaling, 2461, USA, 1:500), or OctA-Probe (FLAG-tagged proteins; Santa Cruz, sc-166384, 1:500). Secondary antibodies were goat anti-mouse IgG (Millipore, GGHL-90P, 1:10000) or goat anti-rabbit IgG (Millipore,GGHL-15P,1:10000). Each experiment was repeated at least five times.
Statistical Data Analysis
Statistical analysis was performed in SPSS version 20. At least three grids were prepared for each experimental condition examined in this study. Results are expressed as mean ± standard deviation. Differences among groups were analyzed by one-way analysis of variance (ANOVA). All P-values were two sided. P < 0.05 was considered statistically significant.
Results
Gene Sequencing Outcomes in Patients in Pedigree With NVM
In a rare patient with an NVM family pedigree, the propositus and her elder sister (Figure 1A) were definitively diagnosed with NVM based on clinical manifestations, echocardiography, and related examinations. Figure 1B shows the echocardiography analysis result of the propositus, which indicates that the ratio of the thickness of the non-compact endocardial layer to the thickness of the compact epicardial layer was 2.076 (0.791/0.381). Interestingly, their parents had normal clinical phenotypes. DNA samples from the two sisters and their mother and aunt were screened by WES and 32 mutation sites in 18 genes were selected for bioinformatics analysis and functional predictions. Other potential functional variants are shown in Supplementary Materials.
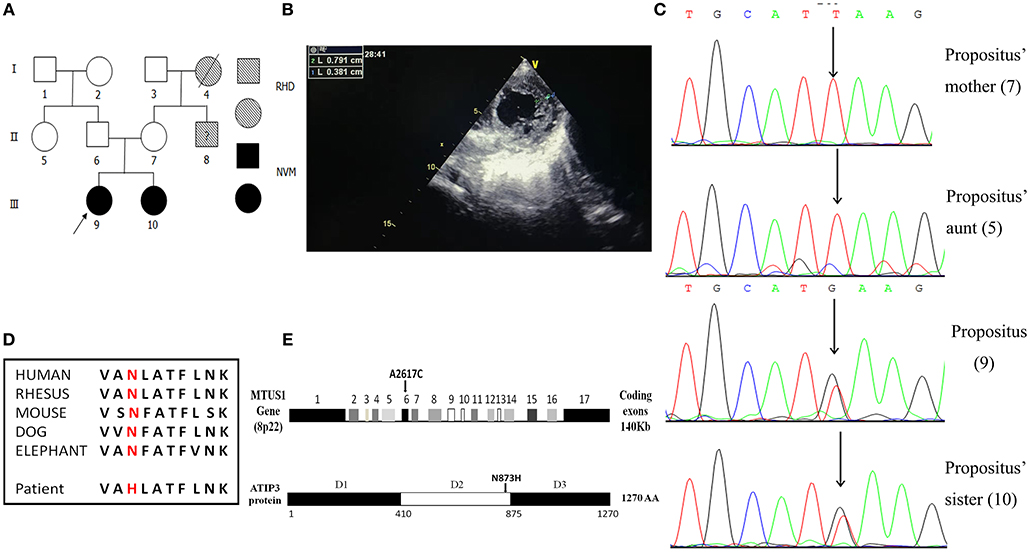
Figure 1. Familial pedigrees of NVM and information of the de novo mutation of MTUS1 c. 2617A->C. (A) Pedigree showing individuals with non-compaction of ventricular myocardium (NVM) and Rheumatic Heart Disease (RHD). The arrow shows the index patient with NVM carrying the MTUS1 mutation (c. 2617A->C). Squares, men; circles, women; black figures, individuals with NVM; oblique line figures, individuals with RHD; oblique line figures with a black question mark in the middle, suspicious RHD patient. (B) The echocardiographic image showing that the ratio of the thickness of the non-compact endocardial layer to the thickness of the compact epicardial layer was >2.0 (0.791/0.381). (C) Backward sequencing chromatogram of propositus and her family (sister, mother, and aunt). Parts of the nucleotide sequence are given below. The mutation site is pointed by an arrow (the position means c. 2617A->C). (D) Evolutionary conservation of c. 2617A->C mutation in all species. (E) Schematic representation of the structural organization shows human MTUS1 gene and ATIP3 (encoded by MTUS1 gene) protein regions. The de novo mutation of MTUS1 c. 2617A->C located in exon 6. Position of the mutant amino-acid sequence was in D2. D1: domain 1, D2: domain 2, D3: domain 3.
By Sanger sequencing analysis of the MTUS1 gene in the two sisters, a heterozygous single nucleotide exchange at the position c. 2617A->C was identified (Figure 1C). In addition, we found that the c. 2617A->C mutation site was highly conserved among different species. The mutation changed the amino acid polarity from the hydrophobic Asn to the hydrophilic His at position 873 (N873H), leading to replacement of neutral amino-acids (amide side chains) by basic residues (Figure 1D). In schematic representation of the human MTUS1 gene, the site of c. 2617A->C mutation located in exon 6, and in structural organization of ATIP3 protein (encoded by MTUS1 gene), the position of the mutant amino-acid sequence was in D2 (Figure 1E), which decorates and stabilizes microtubules (22). It indicated that the mutant localized to an important functional domain. This mutation was present in the two sisters affected by NVM but absent from their mother and aunt. It means that a correlation between the NVM phenotype and the c. 2617A->C mutation was discovered in this NVM family. In a nutshell, the c. 2617A->C mutation in MTUS1 is a de novo mutation and may be pathogenic for NVM.
Lentiviral Vector Validation and Transfection
For HEK-293 cells, 48 h post-transfection, GFP and FLAG tag expressions by the lentiviral vector were measured based on fluorescence staining (Figure 2A) and western blot (Figure 2C). Meanwhile, MTUS1 mRNA levels were increased based on real-time PCR (Figure 2B) in the mutation and wild type groups compared to the vector and blank groups. These results indicated transfection of the c. 2617A->C mutation MTUS1 gene was successful.
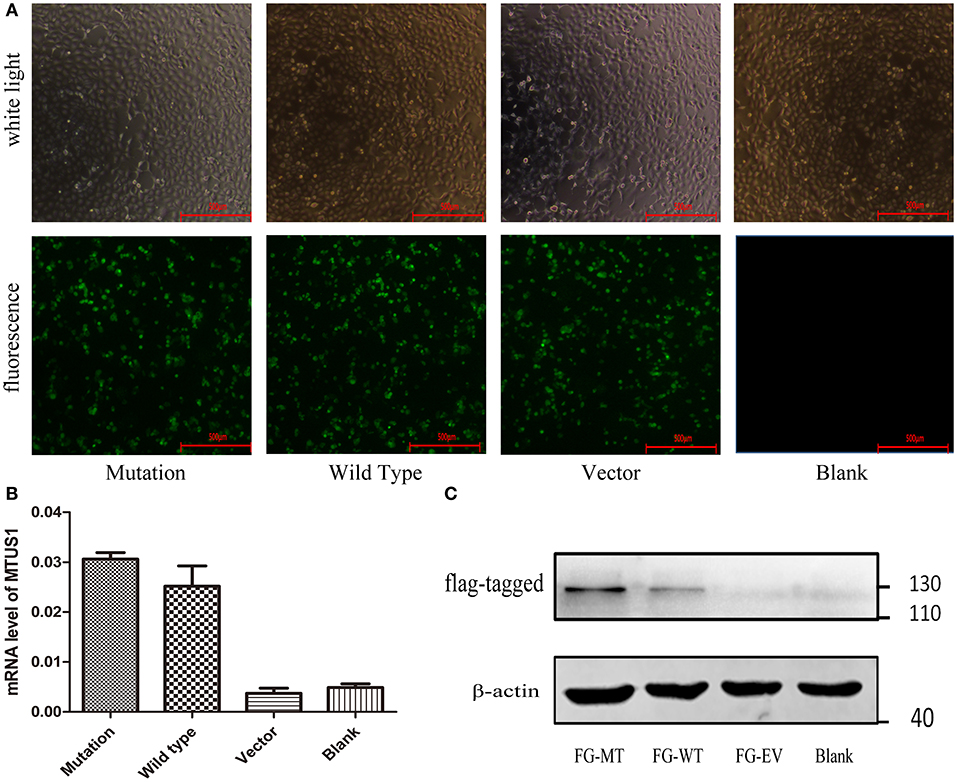
Figure 2. Lentiviral vector validation and transfection. (A) Co-transfect with lentiviruses containing GFP (c. 2617A->C mutation, wild type, vector, respectively), 48 h after transfection, cell state and transfection efficiency of each group were observed under ordinary microscope and fluorescence microscope. (B) After 48 h transfection, the MTUS1 mRNA level expression was increased in the mutation and wild type group. (C) When GFP fluorescence occurs in mutation, wild type and vector group, the expression of flag-MTUS1 protein was detected to verify successful overexpression in HEK293 cells. FG-MT: flag-c. 2617A->C mutation, FG-WT: flag-wild type, FG-EV: flag-vector, Scale bar = 500 μm.
The c. 2617A->C Mutation Decreased α-tubulin Expression and Increased α/β-tubulin Expression
The protein α-tubulin is a globular tubulin that serves as a subunit of microtubules to assess microtubule stability in this study. α/β-tubulin, the heterodimers have roles in the transportation functions of microtubule (21), which were examined to assess PAR protein transportation when MTUS1 contained the c.2617A->C mutation. The fluorescence intensity of α-tubulin was found to be decreased in the c.2617A->C mutation group compared to the wild type group (P < 0.001, Figures 3A,C). Conversely, the fluorescence intensity of α/β-tubulin in the c. 2617A->C mutation group was significantly higher than in the wild type group (P < 0.001, Figures 3B,D).
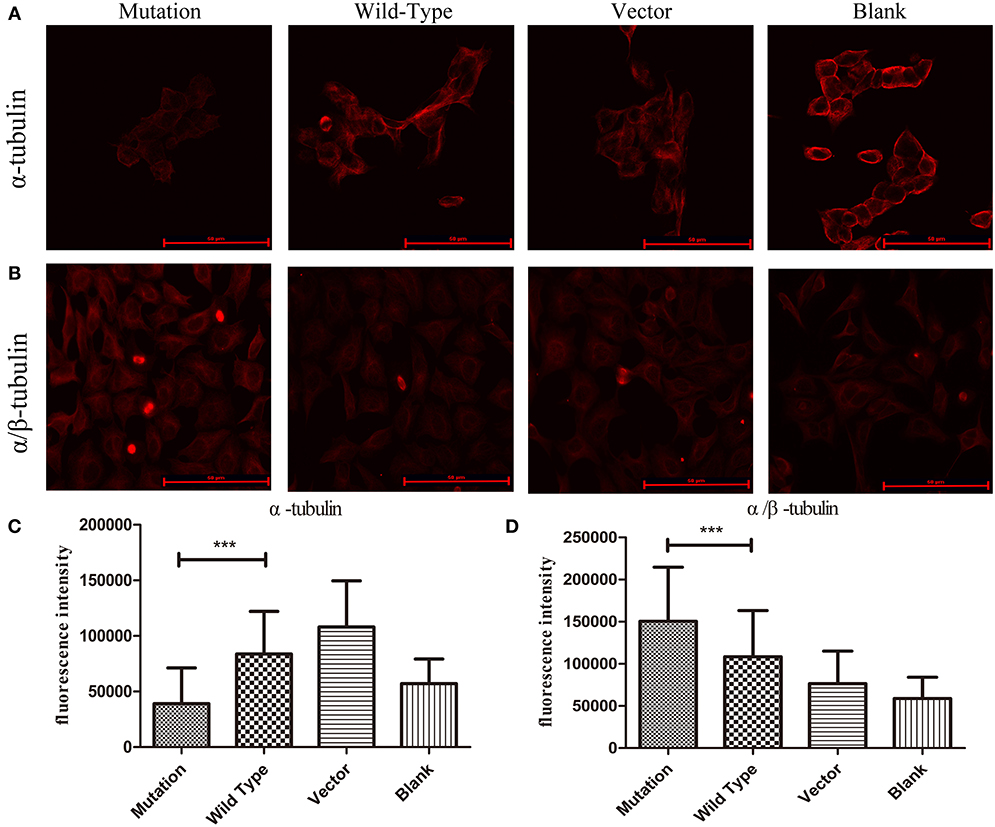
Figure 3. The stability of microtubules in HEK293 cells by immunofluorescence. (A) Images showing α-tubulin expression in four groups. The expression of α-tubulin in c. 2617A->C mutation group was significantly decreased than wild type group. (B) Images showing α/β-tubulin expression in four groups. Increased expression levels of α/β-tubulin in c.2617A->C mutation group were observed compared with wild type group. (C) Quantification of fluorescence intensity measurements of α-tubulin in four groups under the conditions described in (A). (D) The quantification of fluorescence intensity of α/β-tubulin in four groups under the conditions described in (B). ***P < 0.001; scale bar = 50 μm. Error bars show mean ± standard deviation in (C,D).
The c. 2617A->C Mutation Increased PAR6 Protein Expression
PAR6, which acts as a polarity protein, is transported by microtubule heterodimers to one side of the cell membrane and forms polar PAR6-PAR3-aPKC complexes that affect cell polarity (23). The expression and location of PAR6 were analyzed in cells transfected with different recombinant lentiviruses to determine whether the c. 2617A->C mutation influences PAR6 protein. Our study showed that the fluorescence intensity of PAR6 protein was significantly increased in the c. 2617A->C mutation group compared to the wild type group (P < 0.001, Figure 4B), Interestingly, we found that the location of PAR6 protein in the mutation group was abnormal, where it was more partial to the side of daughter cells, compared to the wild type and blank groups (Figure 4A).
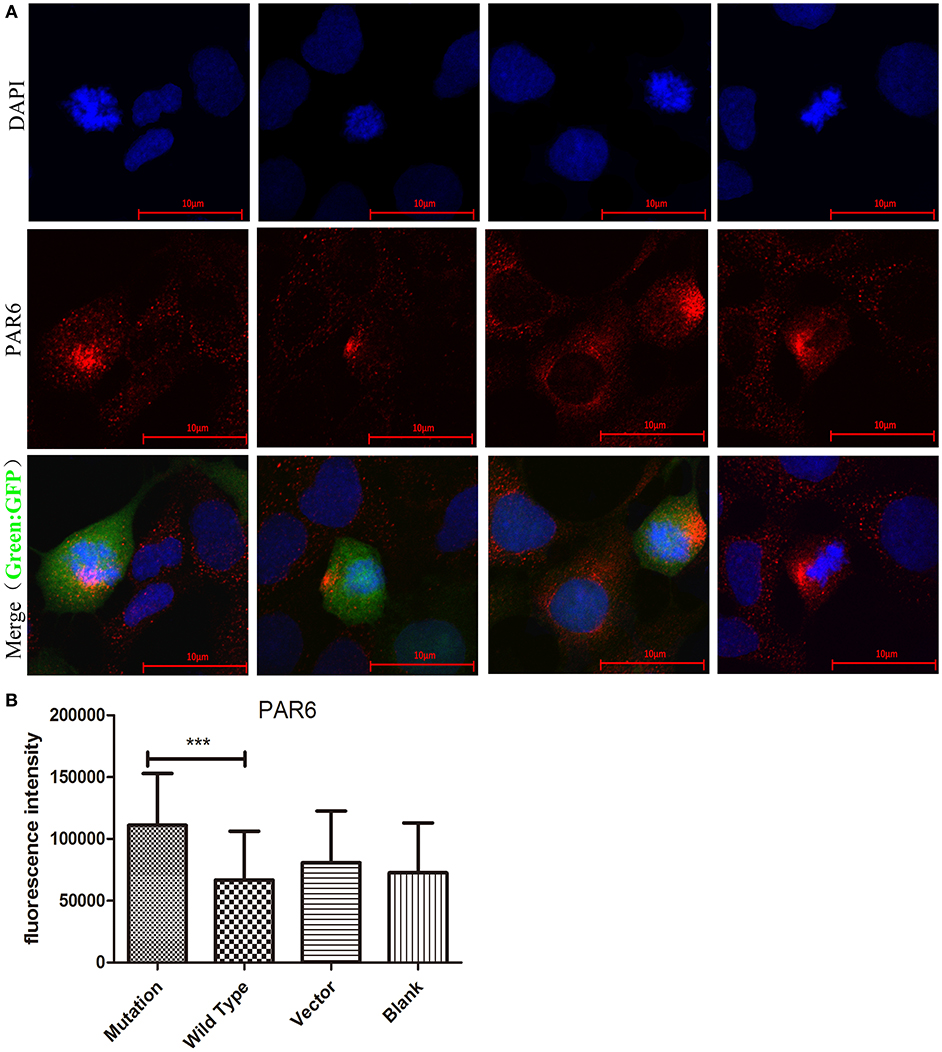
Figure 4. The c. 2617 A->C mutation increased PAR6 protein expression. (A) Images showing PAR6 protein (red) expression and location in four groups in cell division. Nuclei were stained by DAPI (blue). In Merged images, the location of PAR6 protein in c.2617A->C mutation group was abnormal, more partial to the side of daughter cells, compared with wild type group. (B) Quantification of fluorescence intensity of PAR6 in four groups under the conditions described in A. ***P < 0.001, Scale bar = 10 μm. Error bars show mean ± standard deviation.
The c. 2617A->C Mutation Regulated PAR6 Protein Expression in Association With Rac1/Cdc42 Signaling
Rac1/Cdc42, the subfamily members of the Rho GTPase family, affect cell polarity, migration, and differentiation (24). Some studies have reported that Rac1/Cdc42 signaling plays a crucial role in adjusting the formation of PAR6-PAR3-aPKC complexes (25). Phosphorylated Rac1/Cdc42 inhibits GTP binding to Rac1/Cdc42, thereby weakening the downstream signal transduction pathway (26). Our study revealed that PAR6 protein levels were subject to Rac1/Cdc42 phosphorylation levels. Western blot analysis confirmed significantly lower phosphorylated Rac1/Cdc42 protein expression in the mutation group compared to the wild type and blank groups (P = 0.003, P < 0.01, Figures 5A,B). These findings indicate that PAR6 protein expression in the mutation group was regulated by the phosphorylation level of Rac1/Cdc42.
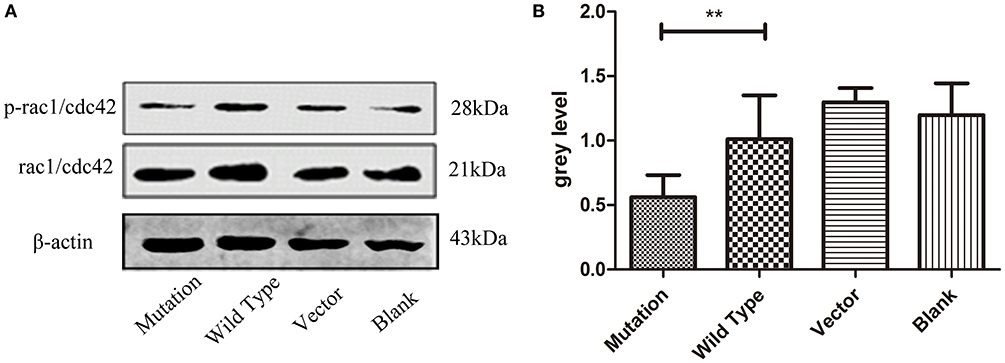
Figure 5. The c. 2617 A->C mutation regulated PAR6 protein expression in association with Rac1/Cdc42 signaling. (A) Western blotting analysis of phosphorylated Rac1/Cdc42 protein in four groups. Total Rac1/cdc42 is shown as a loading control. In cell division, the expression level of phosphorylated Rac1/Cdc42 was decreased in c. 2617A->C mutation than that in wild type group. (B) Quantification of western blot measurements of p-Rac1/Cdc42 level (total Rac1/Cdc42 as control) in four groups under the conditions described in A. **P < 0.01; Error bars show mean ± standard deviation in (B).
High Expression of PAR6 Protein in the c. 2617A->C Mutation-Carrying Cells Altered the Morphology of Daughter Cells
Cellular polarity cannot be established in the absence of the polarity protein complex. Disruption of the polarity protein complex results in aberrant mitotic spindle alignment and the loss of polarization of cells during cell division (27). Furthermore, changing the polarity of cells can lead to abnormal cell morphology and spindle localization (28). To further investigate the effect of increased expression of PAR6 protein in the mutation group on daughter cell morphology, we stained HEK-293 cells in each group for α-tubulin. We then examined the difference in the stained area in daughter cells and the distance between the spindle and cytoplasmic membrane in the daughter cells during cell division by immunofluorescence with confocal microscopy. The area in the daughter cells staining positive for α-tubulin in the mutation group was significantly larger than in the other groups (P < 0.001, Figures 6A,C). The distance between the spindle and cytoplasmic membrane of the daughter cells in the mutation group was significantly larger than in the wild type group (P < 0.01, Figures 4A,B). These results indicate that the c. 2617A->C mutation changed the morphology of the daughter cells after cell division by affecting the polarity of the cells.
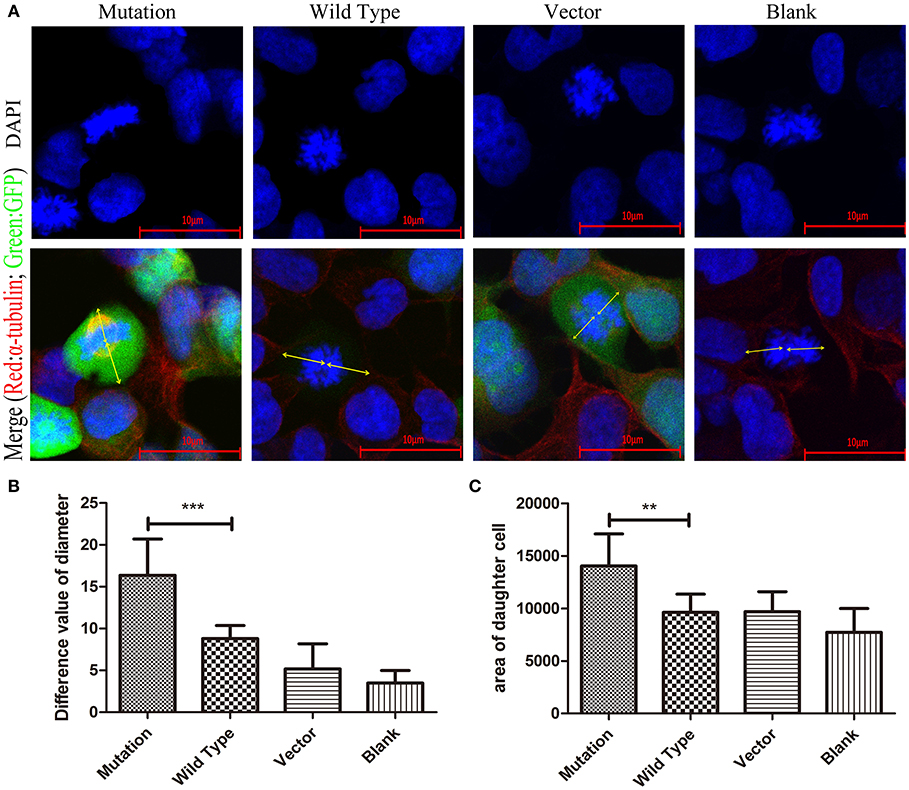
Figure 6. The morphology of daughter cells in HEK293 cells transfected with c. 2617A->C mutation. (A) In the merge images, the arrows (yellow) represent the distance from the spindle to the daughter cell membrane respectively at the stage of cell division. (B) Quantification of the distance from the spindle to the daughter cell membrane respectively at the stage of cell division in four groups under the conditions described in (A). (C) The area of daughter cells in mutation group was obviously significantly larger than other groups. Blue, DAPI; Red, α-tubulin; Green, GFP. **P < 0.01; ***P < 0.001. Scale bar = 10 μm. Error bars show mean ± standard deviation in (B,C).
Discussion
NVM is a rare congenital cardiomyopathy resulting from an arrest in normal endomyocardial embryogenesis. The characteristic echocardiographic findings of NVM consist of multiple, prominent myocardial trabeculations, and deep intertrabecular recesses communicating with the left ventricular cavity (29, 30). The genetic causes and pathogenic mechanisms underlying this disease are largely unknown (31), though it has been described as an inherited cardiomyopathy with both familial and sporadic forms. Since the first genetic cause of NVM was identified as mutations in the X-linked TAZ gene, an increasing number of related genes have been found, including the sarcomere-encoding genes MYH7, ACTC, TNNT2, MYBPC3, TMP1, and TNNI3, Z-line protein-encoding ZASP/LDB3 gene, sodium channel gene SCN5A, and calcium-handling genes TAZ and LMNA (4, 32, 33). However, there have been few publications reporting an association between NVM and the gene encoding microtubules.
In the present study, we demonstrated a de novo mutation in MTUS1, a gene encoding microtubule-associated protein, caused decreased expression of α-tubulin, increased expression of α/β-tubulin heterodimer and PAR6 protein, enlarged the area of the daughter cells and the distance between the spindle and cytoplasmic membrane in daughter cells, and decreased levels of phosphorylated Rac1/Cdc42. These data are consistent with our hypothesis that the de novo c. 2617A->C mutation in MTUS1 decreased the stability of microtubules and increased cell polarity, which might be through the Rac1/Cdc42 pathway, thus partly elucidating the protective mechanism in NVM.
The cytoskeleton is well known to be a dynamic three-dimensional structure that fills the cytoplasm of cells and is responsible for cell movement, cytokinesis, and the organization of organelles or organelle-like structures within cells. The major components of the cytoskeleton include microfilaments, microtubules, and intermediate filament systems. As one of the components of the cytoskeleton, microtubules may cause abnormalities in cardiac compacting during embryogenesis (34). In our study, we found that the fluorescence intensities in the mutated MTUS1 group differed from the wild type group, indicating the microtubule stability in the mutated MTUS1 group may have been destroyed. These results suggest the mutated MTUS1 may alter myocardial densification by affecting microtubules during cardiac development.
Cell polarity refers to the unequal distribution of some cytoplasmic components in a cell in a certain spatial order, thus forming a concentration gradient of various intracellular components. Cellular biologic functions related to cell polarity include asymmetric cell division, migration, and proliferation. Some studies have found cardiomyocyte polarity to be disordered and cardiomyocyte morphology abnormal in heart slices from NVM model mice compared with normal heart sections, suggesting destruction of myocardial cell polarity may be involved in the occurrence of myocardial insufficiency during cardiac development (35, 36). The presence of cell polarity is inseparable from the formation of polar protein complexes and regulation of the Rho GTPase family, which are key regulatory factors associated with the cytoskeleton or microtubule stability (37, 38). The polar protein is transported from the cell cytoplasm to one side of the cell membrane by microtubule heterodimers to form the polar protein complexes that mediate polar cellular division. In the present study, we demonstrated that the de novo c. 2617A->C mutation in MTUS1 increased cell polarity and concurrently decreased levels of phosphorylated Rac1/Cdc42. In addition, we found expression of polar protein PAR6, one of the members of the front-end polar protein complex, enhanced cell division, and located on the side of cell in the mutant group during cell division. These outcomes suggest that mutation of MTUS1 increased polarity of HEK-293 cells by first lowering the phosphorylation level of Rac1/Cdc42 and then increasing expression of the polar protein PAR6, which is a downstream target gene of Rac1/Cdc42.
Biological functions related to cell polarization include asymmetric cell division and cell migration. However, cellular contents are unequally distributed when cell polarity is present (39). In this study, we found the area in daughter cells staining for α-tubulin and the distance between the spindle and cytoplasmic membrane of the daughter cells during cell division in the mutant group were obviously significantly larger than in other groups. It is suggested that the HEK-293 cells with the c. 2617A->C mutation underwent more asymmetric division than the wild type group due to changes in PAR6 protein levels. Furthermore, these results further suggest the de novo c. 2617A->C mutation in MTUS1 promotes cell polarity in patients with NVM during cardiac development, increasing resistance to the disorder of adult cardiomyocytes that will occur in the development of NVM.
Although the present study was conducted in a cell culture system, the data had clinical relevance because the acquired mutation studied originated from a clinical patient. However, future studies are warranted to verify the effect of the de novo c. 2617A->C mutation in MTUS1 on development of NVM and the underlying mechanism(s) using animal models and living human tissue. To date, there are no well-established animal models of NVM. Furthermore, it is prohibited to use living human tissue for ethical reasons. To overcome this challenge, we have already obtained the patient urine cells and generated induced pluripotent stem cell-derived cardiomyocytes, which carry the same genetic background as the patient (data not published). Next, to alleviate the phenotype of NVM mice model, the adeno associated virus (AAV) vectors containing mutation MTUS1 will be injected into the pregnant mice of NVM model. Through these technologies, we will elucidate the roles of the MTUS1 mutation in the NVM pathogenesis, as well as the underlying mechanism(s).
Conclusion
Taken together, the data from the present study demonstrate that the Rac1/Cdc42 pathway might be involved in the control of cell polarity by the de novo c. 2617A->C mutation in MTUS1, thus may promote densification of the myocardium and reduce the occurrence of myocardial densification arrest. Therefore, the de novo c. 2617A->C mutation in MTUS1 may be protective during cardiac development and may decrease the risk of NVM. This is the first study assessing the effects of MTUS1 gene polymorphisms on cell polarity in NVM. These findings may help understand the genetic basis of cell polarity and provide insight for developing new approaches in the pathogenesis of NVM and reducing the prevalence of NVM through early gene intervention.
Ethics Statement
This study was approved by the Institutional Review Board of Children's Hospital of Chongqing Medical University, China (Approval Notice 24/2015) and abided by the ethical principles outlined in the Declaration of Helsinki.
Author Contributions
All authors contributed substantially to the conception and design of the study, and to the critical review of the manuscript. NO collected the samples and evaluated the WES result. YZ did the WES data analysis. XB completed the whole basic experiment and wrote the manuscript. LL, JT, XH, and TL reviewed the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Number: 81570218).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank professor Weinian Shou from Indiana University School of Medicine for his guidance to our research. We also thank Beijing deyi Oriental translational medicine research center for their help with DNA extraction and whole exome sequencing for samples.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2019.00247/full#supplementary-material
Abbreviations
NVM, non-compaction of ventricular myocardium; MTUS1, Microtubule-associated tumor suppressor 1; AGTR2, type-2 angiotensin II receptor; ERK2, extracellular signal-regulated kinase 2; WES, whole exome sequencing; ANOVA, one-way analysis of variance; RHD, rheumatic heart disease.
References
1. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. (2008) 29:270–6. doi: 10.1093/eurheartj/ehm342
2. Anderson RH, Ho SY, Sanchez-Quintana D, Redmann K, Lunkenheimer PP. Heuristic problems in defining the three-dimensional arrangement of the ventricular myocytes. Anat Rec A Discov Mol Cell Evol Biol. (2006) 288:579–86. doi: 10.1002/ar.a.20330
3. Pankuweit S, Richter A, Ruppert V, Maisch B. Classification of cardiomyopathies and indication for endomyocardial biopsy revisited. Herz. (2009) 34:55–62. doi: 10.1007/s00059-009-3195-8
4. Luxan G, Casanova JC, Martinez-Poveda B, Prados B, D'Amato G, MacGrogan D, et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med. (2013) 19:193–201. doi: 10.1038/nm.3046
5. Aras D, Tufekcioglu O, Ergun K, Ozeke O, Yildiz A, Topaloglu S, et al. Clinical features of isolated ventricular noncompaction in adults long-term clinical course, echocardiographic properties, and predictors of left ventricular failure. J Card Fail. (2006) 12:726–33. doi: 10.1016/j.cardfail.2006.08.002
6. Hussein A, Karimianpour A, Collier P, Krasuski RA. Isolated noncompaction of the left ventricle in adults. J Am Coll Cardiol. (2015) 66:578–85. doi: 10.1016/j.jacc.2015.06.017
7. Towbin JA, Lorts A, Jefferies JL. Left ventricular non-compaction cardiomyopathy. Lancet. (2015) 386:813–25. doi: 10.1016/S0140-6736(14)61282-4
8. Zhang W, Chen H, Qu X, Chang CP, Shou W. Molecular mechanism of ventricular trabeculation/compaction and the pathogenesis of the left ventricular noncompaction cardiomyopathy (LVNC). Am J Med Genet C Semin Med Genet. (2013) 163C:144–56. doi: 10.1002/ajmg.c.31369
9. Passer D, van de Vrugt A, Atmanli A, Domian IJ. Atypical protein kinase C-dependent polarized cell division is required for myocardial trabeculation. Cell Rep. (2016) 14:1662–72. doi: 10.1016/j.celrep.2016.01.030
10. Raman R, Pinto CS, Sonawane M. Polarized organization of the cytoskeleton: regulation by cell polarity proteins. J Mol Biol. (2018) 430:3565–84. doi: 10.1016/j.jmb.2018.06.028
11. Bryant DM, Mostov KE. From cells to organs: building polarized tissue. Nat Rev Mol Cell Biol. (2008) 9:887–901. doi: 10.1038/nrm2523
12. Tepass U. The apical polarity protein network in Drosophila epithelial cells: regulation of polarity, junctions, morphogenesis, cell growth, and survival. Annu Rev Cell Dev Biol. (2012) 28: 655–85. doi: 10.1146/annurev-cellbio-092910-154033
13. Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays. (2007) 29:356–70. doi: 10.1002/bies.20558
14. Munro E, Nance J, Priess JR. Cortical flows powered by asymmetrical contraction transport PAR proteins to establish and maintain anterior-posterior polarity in the early C. elegans embryo. Dev Cell. (2004) 7:413–24. doi: 10.1016/j.devcel.2004.08.001
15. Badano JL, Katsanis N. Life without centrioles: cilia in the spotlight. Cell. (2006) 125:1228–30. doi: 10.1016/j.cell.2006.06.013
16. Seibold S, Rudroff C, Weber M, Galle J, Wanner C, Marx M. Identification of a new tumor suppressor gene located at chromosome 8p21.3-22. FASEB J. (2003) 17:1180–2. doi: 10.1096/fj.02-0934fje
17. Rodrigues-Ferreira S, Di TA, Dimitrov A, Cazaubon S, Gruel N, Colasson H, et al. 8p22 MTUS1 gene product ATIP3 is a novel anti-mitotic protein underexpressed in invasive breast carcinoma of poor prognosis. PLoS ONE. (2009) 4:e7239. doi: 10.1371/journal.pone.0007239
18. Zuern C, Krenacs L, Starke S, Heimrich J, Palmetshofer A, Holtmann B, et al. Microtubule associated tumor suppressor 1 deficient mice develop spontaneous heart hypertrophy and SLE-like lymphoproliferative disease. Int J Oncol. (2012) 40:1079–88. doi: 10.3892/ijo.2011.1311
19. Zhang W, Chen H, Wang Y, Yong W, Zhu W, Liu Y, et al. Tbx20 transcription factor is a downstream mediator for bone morphogenetic protein-10 in regulating cardiac ventricular wall development and function. J Biol Chem. (2011) 286:36820–9. doi: 10.1074/jbc.M111.279679
20. Wu X, Pan B, Wang Y, Liu L, Huang X, Tian J. The protective role of low-concentration alcohol in high-fructose induced adverse cardiovascular events in mice. Biochem Biophys Res Commun. (2018) 495:1403–10. doi: 10.1016/j.bbrc.2017.11.141
21. Boczonadi V, Gillespie R, Keenan I, Ramsbottom SA, Donald-Wilson C, Al Nazer M, et al. Scrib:Rac1 interactions are required for the morphogenesis of the ventricular myocardium. Cardiovasc Res. (2014) 104:103–15. doi: 10.1093/cvr/cvu193
22. Molina A, Velot L, Ghouinem L, Abdelkarim M, Bouchet BP, Luissint AC, et al. ATIP3, a novel prognostic marker of breast cancer patient survival, limits cancer cell migration and slows metastatic progression by regulating microtubule dynamics[J]. Cancer Res. (2013) 73:2905–15. doi: 10.1158/0008-5472.CAN-12-3565
23. Berika M, Elgayyar ME, El-Hashash AH. Asymmetric cell division of stem cells in the lung and other systems. Front Cell Dev Biol. (2014) 2:33. doi: 10.3389/fcell.2014.00033
24. Liu Y, Wang J, Li J, Wang R, Tharakan B, Zhang SL, et al. Deletion of Cdc42 in embryonic cardiomyocytes results in right ventricle hypoplasia. Clin Transl Med. (2017) 6:40. doi: 10.1186/s40169-017-0171-4
25. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. (2002) 420:629–35. doi: 10.1038/nature01148
26. Huang Q, Shen HM, Ong CN. Emodin inhibits tumor cell migration through suppression of the phosphatidylinositol 3-kinase-Cdc42/Rac1 pathway. Cell Mol Life Sci. (2005) 62:1167–75. doi: 10.1007/s00018-005-5050-2
27. Gomez-Lopez S, Lerner RG, Petritsch C. Asymmetric cell division of stem and progenitor cells during homeostasis and cancer. Cell Mol Life Sci. (2014) 71:575–97. doi: 10.1007/s00018-013-1386-1
28. Derivery E, Seum C, Daeden A, Loubéry S, Holtzer L, Jülicher F, et al. Polarized endosome dynamics by spindle asymmetry during asymmetric cell division. Nature. (2015) 528:280–5. doi: 10.1038/nature16443
29. Weiford BC, Subbarao VD, Mulhern KM. Noncompaction of the ventricular myocardium. Circulation. (2004) 109:2965–71. doi: 10.1161/01.CIR.0000132478.60674.D0
30. Engberding R, Yelbuz TM, Breithardt G. Isolated noncompaction of the left ventricular myocardium – a review of the literature two decades after the initial case description. Clin Res Cardiol. (2007) 96:481–8. doi: 10.1007/s00392-007-0528-6
31. Arbustini E, Weidemann F, Hall JL. Left ventricular noncompaction: a distinct cardiomyopathy or a trait shared by different cardiac diseases. J Am Coll Cardiol. (2014) 64:1840–50. doi: 10.1016/j.jacc.2014.08.030
32. Pentassuglia L, Sawyer DB. ErbB/integrin signaling interactions in regulation of myocardial cell-cell and cell-matrix interactions. Biochim Biophys Acta. (2013) 1833:909–16. doi: 10.1016/j.bbamcr.2012.12.007
33. Field LJ, Shou W, Markham L. 2017 riley heart center symposium on cardiac development: development and repair of the ventricular wall. Pediatr Cardiol. (2018) 39:1067–8. doi: 10.1007/s00246-018-1942-4
34. Chen H, Zhang W, Sun X, Yoshimoto M, Chen Z, Zhu W, et al. Fkbp1a controls ventricular myocardium trabeculation and compaction by regulating endocardial Notch1 activity. Development. (2013) 140:1946–57. doi: 10.1242/dev.089920
35. Ajima R, Bisson JA, Helt JC, Nakaya MA, Habas R, Tessarollo L, et al. DAAM1 and DAAM2 are co-required for myocardial maturation and sarcomere assembly. Dev Biol. (2015) 408:126–39. doi: 10.1016/j.ydbio.2015.10.003
36. Leung C, Liu Y, Lu X, Kim M, Drysdale TA, Feng Q. Rac1 signaling is required for anterior second heart field cellular organization and cardiac outflow tract development. J Am Heart Assoc. (2015) 5:1. doi: 10.1161/JAHA.115.002508
37. Wu G, Huang X, Hua Y, Mu D. Roles of planar cell polarity pathways in the development of neural [correction of neutral] tube defects. J Biomed Sci. (2011) 18:66. doi: 10.1186/1423-0127-18-66
38. Gönczy P. Mechanisms of asymmetric cell division: flies and worms pave the way. Nat Rev Mol Cell Biol. (2008) 9:355–66. doi: 10.1038/nrm2388
Keywords: non-compaction of ventricular myocardium, MTUS1, microtubule, cell polarity, Rac1/Cdc42
Citation: Bai X, Zhou Y, Ouyang N, Liu L, Huang X, Tian J and Lv T (2019) A de novo Mutation in the MTUS1 Gene Decreases the Risk of Non-compaction of Ventricular Myocardium via the Rac1/Cdc42 Pathway. Front. Pediatr. 7:247. doi: 10.3389/fped.2019.00247
Received: 12 January 2019; Accepted: 30 May 2019;
Published: 02 July 2019.
Edited by:
Antonio Francesco Corno, University of Leicester, United KingdomReviewed by:
Junbao Du, Peking University First Hospital, ChinaShiwei Yang, Children's Hospital of Nanjing Medical University, China
Copyright © 2019 Bai, Zhou, Ouyang, Liu, Huang, Tian and Lv. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiewei Lv, bHR3MjAwMTQ1JiN4MDAwNDA7MTYzLmNvbQ==
†These authors have contributed equally to this work