 Yves Lecarpentier
Yves Lecarpentier Elizabeth Gourrier2
Elizabeth Gourrier2 Alexandre Vallée
Alexandre Vallée
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 03 May 2019
Sec. Neonatology
Volume 7 - 2019 | https://doi.org/10.3389/fped.2019.00176
Bronchopulmonary dysplasia (BPD) is a serious pulmonary disease which occurs in preterm infants. Mortality remains high due to a lack of effective treatment, despite significant progress in neonatal resuscitation. In BPD, a persistently high level of canonical WNT/β-catenin pathway activity at the canalicular stage disturbs the pulmonary maturation at the saccular and alveolar stages. The excessive thickness of the alveolar wall impairs the normal diffusion of oxygen and carbon dioxide, leading to hypoxia. Transforming growth factor (TGF-β) up-regulates canonical WNT signaling and inhibits the peroxysome proliferator activated receptor gamma (PPARγ). This profile is observed in BPD, especially in animal models. Following a premature birth, hypoxia activates the canonical WNT/TGF-β axis at the expense of PPARγ. This gives rise to the differentiation of fibroblasts into myofibroblasts, which can lead to pulmonary fibrosis that impairs the respiratory function after birth, during childhood and even adulthood. Potential therapeutic treatment could target the inhibition of the canonical WNT/TGF-β pathway and the stimulation of PPARγ activity, in particular by the administration of nebulized PPARγ agonists.
Bronchopulmonary dysplasia (BPD) is a particularly serious pulmonary disease occurring in preterm infants (1). In the USA, 1.6% of all births are premature (<32 weeks of gestation); 1.1% have a birth weight <1.5 kg, of which 25% will develop a og BPD (2). Thus, there are about 10,000–15,000 new cases every year. Current therapies are usually ineffective and, despite all necessary precautions being taken, some are even abandoned due to possible aggravation of the clinical pattern. In BPD, PPARγ has been shown to be downregulated in several animal models, while the canonical WNT/β-catenin pathway has been upregulated. In numerous pathologies, these two pathways are observed to function in an opposite manner (3, 4). PPARγ is upregulated in some diseases, while the WNT/β-catenin system is downregulated (5). However, the inverse is observed in many other diseases (6). The absence of a decrease in the WNT/β-catenin signaling during the canalicular stage of pulmonary development, partly related to inflammatory processes, a hallmark of BPD, will seriously affect normal development in the subsequent saccular and alveolar stages (7). This is responsible for the respiratory distress at birth and the often severe and irreversible sequelae observed in childhood and adulthood. Several animal studies have reported beneficial effects induced by nebulized PPARγ agonists but no studies to date have been performed on human neonates. In the present study, we review the respective roles in BPD of both the canonical WNT pathway and PPARγ, which are determined by TGF-β signaling, and discuss their possible implications for the therapeutic treatment of BPD.
BPD was first described by Northway et al. (1). Most premature infants suffer from respiratory distress and surfactant deficiency with pulmonary lesions resulting from the association of multiple infections and inflammation, and possibly aggravated by mechanical ventilation and oxygen toxicity. In BPD, all lung tissues are impaired and airway inflammation, bronchiolitis, alveolar collapse coexist (8). Surviving premature infants often present an abnormal respiratory function when they become adolescents or adults (9, 10). Over the last 20 years, the survival of preterm infants with BPD has been improved thanks to perinatal care, including the use of surfactant and new strategies for mechanical ventilation and oxygenation, and the cure of ductus arteriosus (11). On the other hand, improved survival rates have resulted in an increased incidence of long-term BPD complications. BPD is characterized by an arrest of the alveolization stage. BDP is secondary to pulmonary aggressions occurring at a strategic moment of lung morphogenesis, particularly at the canalicular stage (7, 12–14). The basic histological description reports a decrease in alveolization and an activation of fibroblasts that differentiate into myofibroblasts (12, 15) which express α-smooth muscle actin (α-SMA) in alveolar septa (16). No therapy consistently enhances lung maturity postnatally (17–19). Myofibroblasts exhibit platelet-derived growth factor (PDGF) receptors and express α-SMA and elastin, which are necessary for alveologenesis (20–26). During tracheal aspiration in children who subsequently develop normal lung function, there are almost no mesenchymal stem cells (MSCs) in the suction fluid (27). In contrast, MSCs are found in the suction fluid of premature infants with respiratory distress. TGF-β induces myofibroblastic differentiation of MSCs. About half of the children in whom MSCs have been isolated will develop a BPD (28). α-SMA is higher in differentiated MSCs in children developing BPD than in those who do not develop it. This reflects a greater amount of differentiated myofibroblasts. The transdifferentiation of pulmonary lipofibroblasts into myofibroblasts also represents an important element in the pathogenesis of BPD (23, 29).
Several cascades are implied in lung organogenesis, such as bone-morphogenic proteins (BMPs), fibroblast growth factors (FGFs), sonic hedgehog (SHH), and the WNT family (30–33). Human lung morphogenesis begins after ~4 weeks of gestation and continues into postnatal life up to early adulthood. At around 4 weeks of gestation, the lung buds originate as an outgrowth from the foregut ventral wall where the lobar division appears. In humans, lung development is traditionally divided into five stages based on changes in the airway tubular structure and epithelial cells as follows (34) (Figure 1):
1° Embryonic stage (4–7 weeks in utero): lung buds are formed and the trachea begins branching in lobar and segmental bronchi.
2° Pseudoglandular stage (7–17 weeks): at about 7 weeks, the airways branch and epithelial cells differentiate into tall pseudostratified epithelium. At ~12 weeks, small tubular structures are surrounded by short columnar cells. About 70% of the total airways are formed at 14 weeks.
3° Canalicular stage (17–27 weeks): cuboidal cells are progressively replaced by a columnar epithelium. All of the conducting airways and terminal bronchioles are formed at 18 weeks. At 21 weeks, the differentiation of pneumocytes appears.
4° Saccular stage (27–36 weeks). At this stage, alveolar ducts and air sacs are present. The number of saccules increases and secondary ridges begin to form with extensive vasculogenesis at the level of the terminal saccules.
5° Alveolar stage (36 weeks of gestation to 2 years of age): the secondary septation occurs with a marked increase in number and size of alveoli and capillaries. There is a rapid increase in lung volume and alveolar area (35). At 36 weeks, all cells are uniformly present, and their number increases exponentially with the gestational age. Lipofibroblasts leading to type II interstitial cells are present at the septa bases and regulate the lipid metabolism for the surfactant phospholipid synthesis.
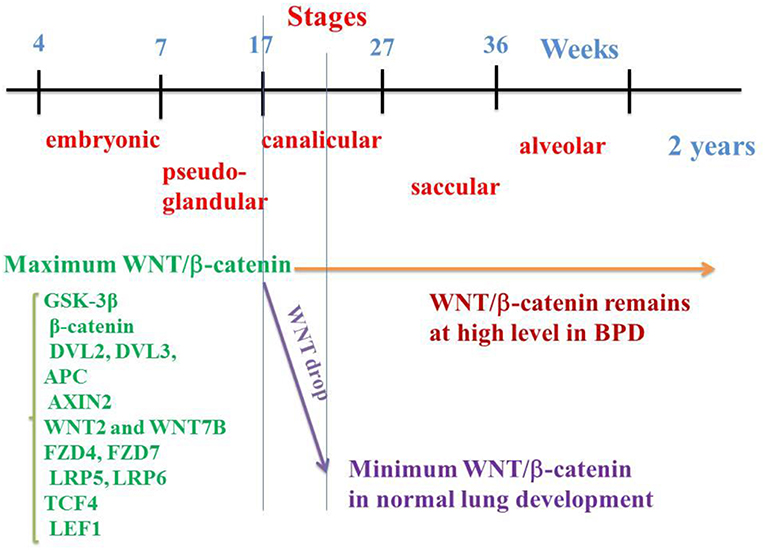
Figure 1. WNT/β-catenin pathway on the embryological pulmonary development in humans. Five stages classically follow one another: embryonic, pseudo-glandular, canalicular, saccular, and alveolar. WNT/β-catenin signaling reaches a maximum activation at the 17th week (end of the pseudo glandular stage or beginning of the canalicular stage). In the middle of the canalicular stage (around the 21st week), canonical WNT pathway activity decreases dramatically, a necessary prerequisite for the correct realization of the alveolar stage. In the event of premature birth, the thick alveolar walls do not allow sufficient gas exchanges, which can lead to pulmonary hypoxia. Hypoxia activates the canonical WNT/β-catenin, impairs the alveolar stage, and promotes the synthesis of myofibroblasts and subsequent fibrosis, with abnormalities in the respiratory function.
Hypoxic episodes occur in BPD preterm infants partly due to immature respiratory control. Relatively short respiratory pauses may precipitate O2 desaturation, bradycardia, and pulmonary hypertension. This can induce retinopathy of prematurity (ROP), sleep disordered breathing, and neurodevelopmental delay (36, 37).
The organogenesis of the respiratory system in premature infants has an adverse effect on pulmonary ventilation and gas exchange. Indeed, the surfactant deficiency maintains the pulmonary compliance at a low level and the alveolar wall thickness limits the diffusion of oxygen and carbon monoxide. For premature infants, an additional supply of oxygen and mechanical ventilation is proposed at this stage, especially as the compliance of the chest wall is low, helping to lower the functional residual capacity and to increase the likelihood of respiratory failure. With such prematurity, the alveolar stage cannot be carried out correctly. Once born, the premature infant must breathe independently. Thickened alveolar walls and vascular pulmonary abnormalities impair effective gas diffusion leading to lung tissue hypoxia, which impairs lung development (38, 39). Importantly, hypoxia is probably the primum movens which activates the canonical WNT pathway and inactivates PPARγ (40). During hypoxia, the respiratory chain increases the ROS production leading to increase hypoxia-induced factor (HIF)-dependent gene expression which represents an important regulator of glycolysis energy metabolism involved in the pathogenesis of lung fibrosis (41).
Conversely, the oxygen therapy that is sometimes necessary can induce a hyperoxia which also activates the canonical WNT signaling and inactivates PPARγ.
The role of hyperoxia in BPD is now well-established (42, 43). In animals, hyperoxia alone can stop the pulmonary septation at the saccular stage of lung development (44, 45). Neonatal resuscitation of premature infants between 24 and 26 weeks (canalicular stage) with 30% O2 instead of 90% O2 decreases the incidence of BDP at 36 weeks by a factor of about 2 (46). In a newborn rat model of BPD induced by intra-amniotic LPS administration and postnatal hyperoxia, there is an arrest in alveolar and pulmonary vascular development, which is a hallmark of BPD (47–50). Hyperoxia-induced neonatal lung injury is associated with activation of Wnt/β-catenin signaling and inhibition of WNT/β-catenin signaling attenuates hyperoxia-induced pulmonary hypertension in neonatal rats (51, 52). Hyperoxia-induced neonatal lung injury is mediated via TGF-β activation (53, 54). Thus, p-Smad3 and 7, and TGF-β R2 receptor protein levels increase after hyperoxia exposure. Hyperoxia leads to pro-inflammatory factors such as TNF-α and IL-8 and reactive oxygen species (ROSs). Likewise, high tidal-volume mechanical ventilation increases the appearance of several pro-inflammatory agents (IL-1b, IL-6, IL-8) (55). This has led to the use of anti-oxidant agents in BPD as a preventive therapy (56, 57). Lungs exposed to hyperoxia show an increase in interstitial myofibroblasts that produce α-SMA and TGF-β (58–61). In the neonatal mouse, hyperoxia induces the expression of periostin in the alveolar wall and particularly in thickened interstitial areas (62). This is also observed on histological sections of infants who died from BPD. Moreover, periostin knockout mice are protected from alveolar complications generated by hyperoxia and do not show interstitial myofibroblasts. Hyperoxia induces an increase in Connective Tissue Growth Factor (CTGF or CCN2) mRNA and CTGF protein in lung epithelial cells and thickening of the alveolar wall (63). In animals, mechanical ventilation induces a pulmonary phenotype close to BPD (64–66). Paracrine effects between lung fibroblasts and epithelial cells are impaired after exposure to hyperoxia. This increases the likelihood of lipofibroblast transdifferentiation into myofibroblasts (54). Thus, both hyperoxia and hypoxia lead to impaired alveolization, myofibroblast differentiation, irreversible pulmonary fibrosis, and major alterations in lung function.
Oxygen toxicity is in great part due to reactive oxygen species (ROSs) generated by mitochondrial respiratory chain, inflammation, hypoxia, ischemia, and hyperoxia (67). Antioxidant capacity is decreased in preterm newborns with deficiency of antioxidant factors (68, 69). ROSs following hyperoxia are responsible for injuries in lungs, central nervous system, retina, and red blood. Severe ROP is due to susceptibility of the phospholipid-rich retina to ROSs (70). To research the safest level of O2 saturation in preterm newborns requiring supplemental oxygenation is important to reduce the incidence of ROP. For some authors, the setting up oxygen alarms are below 85% of oxygen saturation and above 93% in newborns <32 weeks of gestation (71). Other authors have reported a decrease of incidence of ROP in newborns treated with lower O2 saturation (70–90%. rather 88–98%) and cognitive outcomes after 10 years in newborns treated with lower O2 saturation level (72). Several randomized controlled trials have studied the question of what are the optimal O2 saturation levels to reduce outcomes and especially incidence of ROP, chronic lung diseases and hospitalization duration (73, 74). In other randomized controlled trials, the same ranges of O2 saturation has been used in two different groups, i.e., 85–89% in the lower group vs. 91–95% in the higher group (75–77). Based on these randomized controlled trials, mechanical ventilation may be lifesaving but can cause lung injury. Supplemental oxygenation protocols must avoid mechanical ventilation when it is possible (78). In preterm newborns requiring supplemental oxygenation, the range of optimal O2 saturation remains elusive, due numerous different clinical conditions and gestational ages. It is important to avoid both hypoxia and hyperoxia and is recommended to maintain an intended optimal O2 saturation of 90–95% (70).
The canonical WNT pathway plays a key role in embryogenesis, cell fate, metabolism and epithelial-mesenchymal transition (EMT) (79–81). In the presence of canonical WNT ligands (Figures 2, 3), the canonical WNT receptor is linked with Frizzled (FZD) and LDL receptor-related protein 5/6 (LRP5/6). FZD linked to Disheveled (DSH) disrupts the destruction complex [tumor suppressor adenomatous polyposis coli (APC), AXIN and glycogen synthase kinase-3β (GSK-3β)]. Then, β-catenin translocates to the nucleus and interacts with the T-cell /lymphoid enhancer (TCF/LEF) transcription factors to activate β-catenin target genes (82, 83). With no WNT ligands, β-catenin is phosphorylated by the destruction complex and degraded in the proteasome. β-catenin regulates the expression of numerous target genes such as cyclin D1, MMP7, c-Myc, fibronectin, etc., through interactions with TCF and LEF (84).
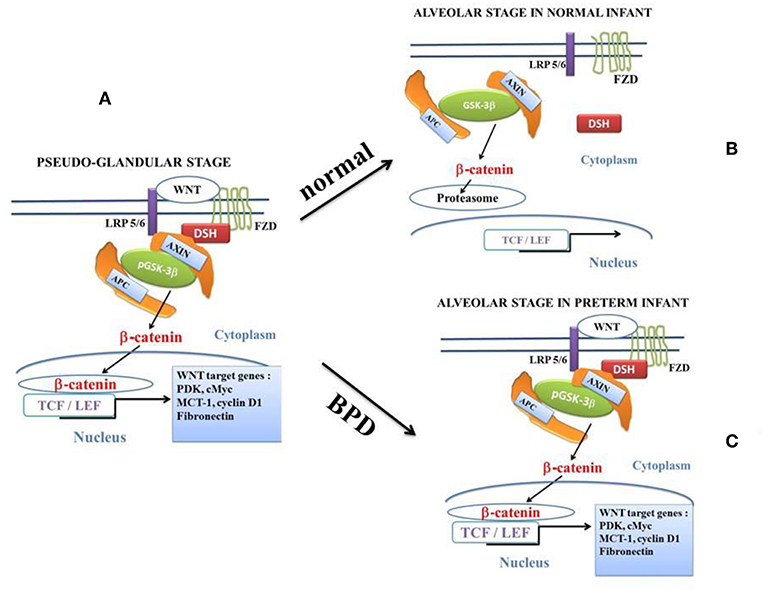
Figure 2. The canonical β-catenin/WNT pathway: “on” and “off” states. The hallmark of the canonical β-catenin/WNT pathway activation is the elevation of the cytoplasmic β-catenin protein level, the subsequent nuclear translocation of β-catenin and further activation of β-catenin specific gene transcription. The canonical β-catenin/WNT pathway can be either in “on-state” or in “off-state.” The pathway is in “on-state” in the presence of a WNT ligand that binds both Frizzled (FZD) and LRP5/6receptors. This leads to activation of the phosphoprotein Disheveled (DSH). DSH recruits the destruction complex (pGSK-3β + AXIN + APC) to the plasma membrane, where AXIN directly binds the cytoplasmic tail of LRP5/6. APC is the adenomatous polyposis coli and GSK-3β is the glycogen synthase kinase-3β. In “on-state,” pGSK-3β is inactivated which corresponds to the phosphorylated state (pGSK-3β). Activation of DSH leads to the inhibition of both phosphorylation and degradation of beta-catenin. Beta-catenin accumulates into the cytosol and then translocates to the nucleus to bind lymphoid-enhancing/T cell (LEF-TCF) co-transcription factors. This induces the WNT-response gene transcription. In the “off state,” in the absence of WNT ligand or in the presence of the active form of GSK-3β (i.e., the unphosphorylated form of GSK-3β), cytosolic β-catenin is phosphorylated by the active form of GSK-3β. Beta-catenin undergoes the destruction process into the proteasome. (A): at the pseudo-glandular stage of the pulmonary development, the canonical WNT/β-catenin pathway is in “on-state.” (B): at the saccular and alveolar states, and in normal infants, the canonical WNT/β-catenin pathway is in “off-state.” The pulmonary development is normal. (C): at the saccular and alveolar states, in preterm infants with BPD, the canonical WNT/β-catenin pathway is in “on-state” and the pulmonary development is dramatically impaired.
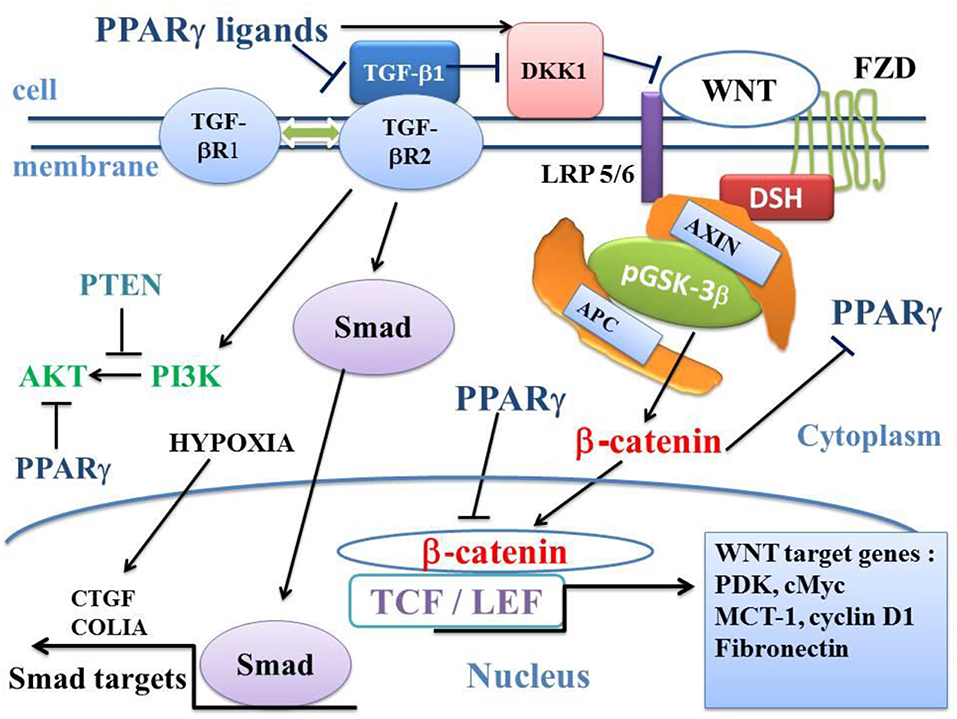
Figure 3. Influence of TGF-β1 on the balance between the canonical WNT/β-catenin signaling and PPARγ. In the presence of the WNT ligands, the WNT receptor binds both LRP5/6 and FZD receptors to initiate LRP phosphorylation and DSH-mediated Frizzled internalization. This leads to the dissociation of the GSK-3 β/AXIN/APC destruction complex. Phosphorylation of β-catenin is inhibited and β-catenin accumulates in the cytosol and then translocates to the nucleus to bind TCF-LEF transcription factors. This leads to the WNT-response gene transcription (PDK, MCT-1, cMyc, and Cyclin D1). PPARγ inhibits the β-catenin/TCF-LEF-induced activation of WNT target genes. TGF-β also enhances WNT signaling through the inhibition of DKK1. DKK1 is activated by PPARγ. TGF-β1 binds type 2 TGF-βR2 receptor (TGF-βR2), which recruits type 1 TGF-βR1 receptor (TGF-βR1). This results in the formation of a heterotetramer that phosphorylates Smad. The Smad complex then translocates to the nucleus and regulates the transcription of target genes (CTGF, COL1A). A non-Smad pathway also occurs through PI3K-AKT. PTEN inhibits PI3K-AKT and PPARγ inhibits AKT. APC, adenomatous polyposis coli; CTGF, Connective tissue growth factor; DKK1, Dickkopf-1; DSH, Disheveled; FZD, Frizzled; GSK-3β, glycogen synthase kinase-3β; LRP5/6, low-density lipoprotein receptor-related protein 5/6; MCT-1, monocarboxylate lactate transporter-1; PPARγ, peroxisome proliferator-activated receptor gamma; PI3K, phosphatidylinositol 3-kinase and AKT, AKT/Protein Kinase B; PTEN, Phosphatase and tensin homolog; PDK, pyruvate; dehydrogenase kinase; TCF/LEF, T-cell factor/lymphoid enhancer factor; TGF, Transforming Growth Factor.
TGF-βs are three proteins, TGF-β1, TGF-β2, and TGF-β3, and receptors are the transmembrane proteins Type I (TGFβRI) and Type II (TGFβRII). TGF-β1 activates the Smad pathway and non-Smad pathways such as MAPK, Rho, PI3K-AKT and downregulates PPARγ expression via the SMAD pathway (85) (Figure 3). In alveolar epithelial cells (AEC), TGF-β1 mediates epithelial-mesenchymal transition (EMT) and this leads to lung fibrosis. In idiopathic pulmonary fibrosis, myofibroblasts induce lung fibrosis (86–88). TGF-β1-induced EMT of AEC type II results in myofibroblast differentiation in fibrotic lungs (89–93).
PPARγ is a ligand-dependent transcriptional factor belonging to the nuclear hormone receptor superfamily. PPARγ regulates glucose and lipid homeostasis, insulin sensitivity, inflammation, innate immune responses and cell fate (94, 95). PPARγ is activated by natural agents such as 15d- prostaglandin J2 (15d-PGJ2) and synthetic ligands including thiazolidinediones (TZDs). TZDs improve glucose tolerance and insulin sensitivity in type 2 diabetes. PPARγ dedifferentiates myofibroblasts, increases collagen uptake by lung alveolar macrophages, and may reverse fibrosis in an animal model (96). PPARγ plays an important role in normal lung development via epithelial-mesenchymal signaling (97).
There is a strong link between TGF-β1, canonical WNT/β-catenin and PPARγ (98, 99). TGF-β1 upregulates the WNT/β-catenin pathway, and inhibits PPARγ. TGF-β1 induces differentiation of human lung fibroblasts into myofibroblasts. Conversely, PPARγ inhibits the TGF-β1/WNT/β-catenin pathway. PPARγ ligands repress TGF-β1-induced myofibroblast differentiation via the PI3K/AKT signaling (100) (Figure 3). TGF-β1 induces fibrosis and represents a key therapeutic target in fibrotic processes (101, 102).
In humans, the WNT/β-catenin pathway plays a key role in the patterning of early lung organogenesis, especially during the canalicular stage (7, 103–107). Regional specialization of the epithelium and mesenchyme and branching morphogenesis have been investigated by means of various techniques, such as reporter gene activity, misexpression, lineage tracing, β-catenin deletion/stabilization, WNT/ β-catenin in patterning of the mesenchyme and morphogenesis of proximal structures (108). During the pseudoglandular stage, most of the canonical WNT/β-catenin components appear to be already present in the lung buds. They are involved in the branching and division of conduction airways. The WNT pathway is not required for the lung primary branching pattern but is necessary for a correct branching morphogenesis (103). The canonical WNT/β-catenin components are mainly expressed in the alveolar and bronchial epithelium. An appropriate patterning of the developing lung is dependent on epithelial-mesenchymal cell interactions (32).
In humans, M. Zhang et al. (7) have determined the spatio-temporal patterns of the main components of the WNT/β-catenin signaling by means of qRT-PCR and in situ hybridization at 7, 12, 17, and 21 weeks of gestation. Among them are the GSK-3β, β-catenin, DVL2, DVL3, APC and AXIN2 components, WNT2 and WNT7B ligands, FZD4, FZD7, LRP5, and LRP6 receptors, and TCF4 and LEF1 transcription factors. Importantly, most of them are detected at 7 weeks, reach their maximum value at 17 weeks (onset of the canalicular stage) and dramatically decrease at 21 weeks (middle of the canalicular stage) (Figure 1). This is corroborated by other studies that show firstly, in the normal lung, the presence of β-catenin in the majority of epithelial and mesenchymal cell nuclei at 18 weeks of gestation with a decrease at the 21st week of gestation (14) and, secondly, a second-trimester β-catenin spike with attenuation of nuclear β-catenin at the alveolar stage of lung development (109). Other studies have shown a peak of the WNT signal in epithelial and mesenchymal cells toward the onset of the canalicular stage with a sharp decline toward the middle of the canalicular stage, and a low level during the saccular and alveolar stages (110). In human lung at 15 weeks (pseudo-glandular stage) and after exposure to CHIR 99021, a selective GSK-3β inhibitor (111), an increased expression of β-catenin transcription factors (TCF4, LEF1) and target genes (Cyclin D1, MMP7) have been observed. In human lung tissues in vitro, WNT3A induces an increase in mRNA levels of the WNT target gene Cyclin D1 (112). There is therefore a regulation of the WNT canonical system both temporally and spatially, which helps explain the pathophysiology of BPD.
PPARγ has been shown to play a pivotal role in normal lung development and homeostasis by stimulating alveolar interstitial lipofibroblast maturation (113–115). Lipofibroblasts induce alveolar epithelial-mesenchymal paracrine interactions through the stimulation of surfactant phospholipid and by favoring endogenous antioxidants (113, 116, 117). PPARγ is critical for normal lung organogenesis and homeostasis of lung epithelium and mesenchyme. PPARγ is involved in normal lung development via EMT (97). PPARγ induces lipofibroblast differentiation which exerts a cytoprotective effect against oxidant injury (113, 116, 118). PPARγ stimulates trans-differentiation of myofibroblasts into lipofibroblasts, which helps normal alveolarization (119). PPARγ also favors the lipofibroblastic phenotype, which leads to the alveolar type II cell.
BPD results from pulmonary airway aggregates during the saccular stage (27–36 W), after the peak of WNT/β-catenin activity which occurs in the normal lung at 17W, i.e., at the onset of the canalicular stage (7). In BPD, nuclear β-catenin in lung tissues is quantitatively close to that observed in week 17–18 of the normal lung (which corresponds to the peak of β-catenin in the normal fetus (7, 14). The WNT/β-catenin pathway remains in the “on-state” in the lung of premature infants in the saccular and alveolar stages, and in the postnatal period. Instead of dropping drastically at 21 weeks (i.e., in the middle of the canalicular stage), WNT/β-catenin signaling remains high, which leads to major alterations in pulmonary development, with significant abnormalities being observed at the alveolar stage. This makes the WNT/β-catenin pathway a potential therapeutic target for BPD (7, 14, 110, 120).
In BPD infants, the resting metabolic expenditure is elevated with growth failure and expression of some genes of oxidative phosphorylation is decreased (121). Overactivation of the Wnt/β-catenin pathway induces aerobic glycolysis where a part of the glucose supply is fermented into lactate with activation of pyruvate dehydrogenase kinase1 (PDK-1), lactic dehydrogenase (LDH-A) and monocarboxylate lactate transporter (MCT-1). This results in pyruvate being diverted toward lactate (6, 122). In the cytosol, pyruvate is converted into lactate through activation of lactic dehydrogenase (LDH-A). In cancers, this phenomenon is referred to as aerobic glycolysis or Warburg effect (123). PDK1 phosphorylates the pyruvate dehydrogenase complex which is inhibited and prevents the conversion of pyruvate into acetyl-CoA in mitochondria. This leads to decreases acetyl-CoA entering the tricarboxylic acid cycle. In addition, the WNT/β-catenin pathway induces the transcription of the genes Cyclin D1 and cMyc (124) (Figure 3). cMyc activates LDH-A (125) and increases the hypoxia-inducible factor-1α which controls PDK-1 (126). Conversely, PPARγ activation selectively decreases PDK mRNA (127). To date, complex cellular metabolism through canonical WNT/β-catenin and PPARγ pathways is not fully detailed in BPD.
GSK-3β (inactive state corresponding to the WNT on-state) is involved in the differentiation of mesenchymal stem cells (MSCs) into myofibroblasts (128). GSK-3β is down-regulated in mesenchymal myofibroblastic cells obtained by means of tracheal aspiration in infants with BPD (120). GSK-3β kinase activity can be inhibited by several factors such as WNTs, TGF-β, BMPs, serotonin, endothelin, cardiotrophin, and connective tissue growth factor (CTGF) (129). In addition, CTGF activates the TGF-β receptor. During lung development, hyper-expression of both TGF-β (130) and CTGF leads to a pulmonary phenotype similar to that of BPD (63). During the neonatal period, hyper-expression of CTGF induces thickening of alveolar septa and differentiation of MSCs into myofibroblasts, decreases the formation of secondary septa (63), and increases the risk of pulmonary hypertension (131). Both TGF-β and CTGF are likely to activate each other (132–134) (Figure 3). In mice lung, hyper-expression of CTGF induces phosphorylation of Ser9 GSK-3β and the translocation of β-catenin to the nucleus of type II alveolar cells (131).
Stretch on alveolar type II cells induces the expression of parathyroid-hormone-related protein (PTHrP) which binds to the PTHrP receptor. PTHrP is expressed in lipofibroblasts and upregulates PPARγ via protein kinase activation. PPARγ induces lung transdifferentiation of myofibroblasts into lipofibroblasts. Moreover, lipofibroblasts stimulate the proliferation and differentiation of alveolar type II cells, thus contributing to alveolarization (113). Lipofibroblasts synthesize leptin, which leads to stimulation of the synthesis of de novo surfactant phospholipid. The decrease in PTHrP in alveolar type II cells, after exposure to hyperoxia, volutrauma, inflammation and infections, downregulates the alveolar lipofibroblast PPARγ expression (118, 119, 135) and induces lipofibroblast-myofibroblast transdifferentiation (97, 113, 136). PPARγ prevents these deleterious effects (97), reducing inflammation processes by inhibiting NF-κB and rosiglitazone (RGZ) by decreasing lung infiltration by neutrophils (137). PPARγ induces angiogenesis after direct stimulation of endothelial cells through angiogenic growth factors and cytokines that stimulate endothelial cells that participate in the maintenance of alveolar structures (138, 139). Moreover, the pulmonary vasculature is dramatically impaired in human premature infants dying from BPD. Thus, VEGF and VEGF receptors are diminished in their lungs (59). Disruption of the VEGF signaling stops the alveolar and pulmonary vascular development (140). This strongly suggests that PPARγ plays an important role in the microvascular development in the lungs (115, 141).
Numerous studies have shown that PPARγ agonists can play a positive role in animal models of BPD. Angiogenesis is enhanced by VEGF which improves both alveolar and pulmonary vascular development in neonatal rats (142, 143). VEGF plays a major role in the microvascular development of the lungs (138). In rat exposed to intra-amniotic LPS and postnatal hyperoxia, the angiogenic growth factor is decreased, a situation that is alleviated by RGZ (144). Hyperoxia induces a decrease in VEGF and the platelet endothelial cell adhesion molecule (PECAM-1or CD31) and an increase in fibronectin. This is blocked after pioglitazone (PGZ) nebulization. PGZ nebulization also blocks modifications in lung interleukins (IL-6 and IL-1β), chemokine ligand 2 (CCL-2), pro-inflammatory cytokine MIF, the BcL2/Bax protein ratio and lung morphometry (radial alveolar count). RGZ restores alveolar and pulmonary vascular development and lessens pulmonary hypertension (145). In a similar rat model, RGZ has also been found to restore VEGF and its receptor VEGF R-2 in the lungs (144). This protective effect of RGZ may be induced via VEGF enhancing the angiogenic activity. In an animal model, the systemic administration of RGZ, either antenatally or postnatally, enhances lung maturation and can prevent hyperoxia-induced acute neonatal lung injury. This strongly suggests that PPARγ agonists could potentially play an important role in preventing and/or treating BPD (53, 115, 146, 147).
In rat pup lung, RGZ increases the expression of the PTHrP receptor and surfactant protein-B (115). The inhibitory effects of intra-amniotic LPS and postnatal hyperoxia on alveolarization are alleviated by RGZ, which induces a protective effect on it via an increased PPARγ expression in the alveolar lipofibroblasts. RGZ significantly enhances lung vascular maturation during normal lung morphogenesis in rat pups (115). In rat, after exposure to hyperoxia, the level of PPARγ protein decreases but increases after PGZ nebulization (145). PPARγ ligands inhibit fibroblast activation through TGF-β1. RGZ and ciglitazone (CGZ) inhibit profibrotic changes in TGF-β1-stimulated serum-deprived A549 cells (from the human AEC cell line), independently of the inhibition of the Smad pathway. Their inhibitory effects on changes in collagen I and E-cadherin (epithelial cell marker) appear to be PPARγ-dependent. PPARγ agonists inhibit pulmonary myofibroblast differentiation by TGF-β and collagen synthesis (148). In newborn rat, nebulized PGZ significantly improves hyperoxia-induced abnormalities via TGF-β pathway activation. PGZ-nebulization blocks Smad3 and 7, and TGF-β II receptor protein levels increased after hyperoxia exposure.
In newborn rat, the nebulized PPARγ agonists RGZ and PGZ increase the expression of alveolar epithelial (apolipoproteins SPB-SPC, surfactant phospholipid) and mesenchymal (adipose differentiation-related protein) markers of lung maturation and decrease markers of lung injury (BcL2, Bax), and markers of TGF-β activation (145). Human fibroblasts treated with PPARγ agonists blocked the TGF-β signaling. SMAD3 or SMAD4 knockdown suppresses the effects of TGF-β on PPARγ mRNA and protein expression (149). In cultured human lung fibroblasts, the increases in α-SMA expression and the fibrillary collagen content induced by TGF-β are prevented by RGZ, TGZ and CGZ (148, 150). TGF-β1 induces EMT, and epithelial cells acquire a phenotype of mesenchymal cells (89, 151, 152). RGZ and CGZ inhibit several TGF-β1-induced changes in EMT markers as well as lung fibrosis (153).
Targeting the canonical WNT/β-catenin-TGF-β-PPARγ axis by upregulating PPARγ and downregulating the canonical WNT/TGF-β pathways might be of interest with respect to improving the development of the alveolar stage of BPD at birth and minimizing pulmonary fibrosis later during childhood and adulthood. Numerous studies have demonstrated the potential role of PPARγ agonists and the inhibition of canonical WNT/TGF-β pathways in many pathologies leading to pulmonary fibrosis outside of BPD.
PPARγ agonists are considered part of a potential future strategy that would target PPARγ activity or expression as a therapy for controlling fibrosis (154). In human lung, PPARγ agonists inhibit profibrotic phenotype fibroblasts and pulmonary fibrosis induced by bleomycin (150). In rat, PGZ improves bleomycin-induced acute lung injury and fibrosis (155). In a bleomycin-induced model of lung fibrosis, PPARγ agonists induce an anti-fibrotic activity (96, 148, 150) and RGZ attenuates fibrotic effects in rats (156). In mouse, bleomycin-induced lung injury, RGZ and 15-deoxy-Δ12,14-prostaglandin J2 significantly reduce lung abnormalities (157). In scleroderma, the skin fibrotic phenotype of fibroblasts is alleviated by RGZ (158). Bleomycin-induced scleroderma is abrogated by RGZ and this blocks profibrotic effects (159). RGZ induces an antifibrotic effect in scleroderma lung fibroblasts (160). In systemic sclerosis, PPARγ activation induces protection against an excessive fibrosis and may represent a therapeutic target (161). The PPARγ agonist PGZ inhibits fibrosis in rat liver after a choline-deficient L-amino acid defined diet (162, 163). PPARγ ligands exert notable antifibrotic effects in human lung fibroblasts (164). RGZ inhibits differentiation, migration, and proliferation in cultured human lung fibroblasts (165). In a murine model of neutrophilic asthma, RGZ diminishes airway inflammation by inhibiting T cell proliferation (166). PPARγ is expressed in airways and inhibits features of airway remodeling in a mouse asthma model (167). TZDs increase the WNT inhibitor Dickkopf-1 in adipocytes (168). In fetal rat lung exposed to LPS, RGZ exerts protective effects against inflammation through PTHrP-driven epithelial-mesenchymal interactions (169). RGZ increases angiogenesis and may represent a new potential therapy for improving lung development in extremely premature infants. In rat pups, nebulized PPARγ agonists RGZ and PGZ augment neonatal lung maturation and injury repair by increasing the expression of markers of alveolar epithelial and mesenchymal maturation via stimulation of alveolar interstitial lipofibroblast maturation and by providing protection against hyperoxia-induced lung injury (145). In lung epithelial cells in chronic obstructive pulmonary disease, down-regulated PPARγ increases the likelihood of a proinflammatory phenotype (170).
In mesangial cells, TGF-β upregulates the type I collagen and this is blocked by PPARγ activation (171). In hypertrophic scar fibroblasts, induction of CTGF and extracellular matrix by TGF-β1 is inhibited by PPARγ (172). Fibrosis may be attenuated by blocking TGF-β1 by means of PPARγ agonists (173). PPARγ abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator (174). In murine lung fibroblasts, TGF-β1 controls PPARγ expression, transcription and activity through Smad3 signaling (175). Several drugs targeting the TGF-β pathway have been used in phase III clinical trials in oncology and against fibrosis and radiation lesions (176). In human hepatic stellate cells, the TGF-β1/Smad3-pathway is prevented by PPARγ (177). In systemic sclerosis, abnormal TGF-β expression is involved in fibrosis. Small-molecule inhibitors of TGF-β-receptor activity are effective in animal models of fibrosis. Imatinib mesylate and related tyrosine kinase inhibitors block TGF-β signaling and abrogate fibrotic processes (178). In systemic sclerosis, the aberrant PPARγ function is involved in fibrosis in skin and lungs. The antifibrotic effects seem to be related to the inhibition of TGF-β/Smad signal transduction (99). In fibroblasts, TGF-β downregulates PPARγ in systemic sclerosis (102). The TGF-β1 gene is repressed by PPARγ through the PTEN-mediated p70 ribosomal S6 kinase-1 inhibition (179). TGF-β suppresses PPARγ expression via SMAD binding (149) (Figure 3). The canonical WNT/β-catenin pathway represents a potential therapeutic target for fibrosis (180). In systemic sclerosis, the canonical WNT/β-catenin pathway is upregulated and induces a Smad-dependent fibrotic effect in mesenchymal cells (98). Inhibition of the β-catenin pathway improves renal interstitial fibrosis (181). Activation of the canonical Wnt/β-catenin signaling inhibits the GSK3-β and induces dermal fibrosis (182).
In very premature infants, the alveolar stage is very abnormal and the thickening of the alveolar walls makes gaseous exchanges difficult, leading to hypoxia and hypercapnia. However, chronic hypoxia itself impairs lung development (38, 39). In the newborn lung animal model, hypoxia enhances the TGF-β pathway and inhibits alveolar development, resulting in pulmonary arterial abnormalities. PPARγ can be reduced by increasing TGF-β (85, 183). Hypoxia inhibits PPARγ (40, 184), but increases TGF-β signaling, which is attenuated by RGZ. Importantly, RGZ attenuates hypoxia-induced inhibition of lung development (40). In newborn mouse lung, chronic hypoxia diminishes the levels of PPARγ mRNA expression and PPARγ protein. After hypoxia exposure the oral administration of RGZ restores PPAR-γ mRNA and protein levels (40). RGZ diminishes hypoxia-induced inhibition of alveolar development and prevents hypoxia-induced impairment of the respiratory function. PPARγ is decreased by hypoxia, a decrease that is prevented by inhibition of the TGF-β pathway.
TZDs can have adverse effects. The clinical administration of PGZ and RGZ has revealed several adverse effects, including weight gain, bone fractures, fluid retention and congestive heart failure (185–187). In the USA, TGZ was removed from the market in 2000 due to liver failure in diabetic patients (188). RGZ has been withdrawn due to hepatotoxicity which has not been observed with RGZ and PGZ (189). RGZ is associated with an increased risk of strokes, myocardial infarction, heart failure and even death (190, 191). However, it has been shown that there is no increase in cardiovascular incidents with RGZ therapy (192, 193). As for PGZ, it has been associated with bladder cancer, bone fracture and heart failure (194). Few selective PPARγ modulators have been synthesized and these have been tested for safety and therapeutic efficacy. Thus, the PPARγ ligand SR1664 has been found to improve insulin sensitivity and does not induce adverse effects with regard to fluid retention or bone formation (195). SR1664 binds PPARγ with high affinity and blocks the PPARγ-Cdk5-mediated phosphorylation. Great efforts are done to improve the safety of the first-generation of PPARγ agonists. It appears important to understand the complex PPARγ regulation by integrating the role of coactivators and corepressors and the role of posttranslational mechanisms. It would seem important to develop new tissue-specific PPARγ agonists. In the case of BPD, lung-specific nebulized PPARγ agonists could enhance the therapeutic benefits and reduce the deleterious effects.
Significant prematurity predisposes to general hypoxia, particularly in the pulmonary tissue. Both hypoxia and hyperoxia promote upregulation of the canonical WNT/β-catenin system and TGF-β and downregulation of PPARγ. This stops lung maturation and gives rise to the transdifferentiation of lipofibroblasts into myofibroblasts, which may lead to pulmonary fibrosis and severe pulmonary sequelae. Since hypoxia and hyperoxia have similar consequences for the TGF-β/WNT/β-catenin/PPARγ pathways, this requires a fine tuning of oxygen use in very premature infants. Numerous studies have shown PPARγ agonists have a beneficial effect on lung maturation and may provide a potentially safe lung therapy (53, 54, 115, 146, 147, 157, 196, 197). Nebulized PPARγ agonists may represent a potential strategy for improving postnatal lung maturation, both by inhibition of the TGF-β/WNT/β-catenin pathway and by preventing neonatal hyperoxia-induced lung injury in premature BPD infants, thereby reducing morbidity and mortality.
All the authors contributed equally to the final version of the manuscript and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Dr. Christophe Locher, Director of the Clinical Research Center, Grand Hôpital de l'Est Francilien, Meaux, France, for his valuable support in making the necessary research facilities available for this study. Prof. Brian Keogh substantially improved the manuscript.
1. Northway WH Jr, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N Engl J Med. (1967) 276:357–68.
2. Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. (2001) 163:1723–29. doi: 10.1164/ajrccm.163.7.2011060
3. Lecarpentier Y, Claes V, Duthoit G, Hebert JL. Circadian rhythms, wnt/beta-catenin pathway and PPAR alpha/gamma profiles in diseases with primary or secondary cardiac dysfunction. Front Physiol. (2014) 5:429. doi: 10.3389/fphys.2014.00429
4. Lu D, Carson DA. Repression of beta-catenin signaling by PPAR gamma ligands. Eur J Pharmacol. (2010) 636:198–202. doi: 10.1016/j.ejphar.2010.03.010
5. Vallee A, Lecarpentier Y. Alzheimer disease: crosstalk between the canonical wnt/beta-catenin pathway and PPARs alpha and gamma. Front Neurosci. (2016) 10:459. doi: 10.3389/fnins.2016.00459
6. Lecarpentier Y, Claes V, Vallee A, Hebert JL. Thermodynamics in cancers: Opposing interactions between PPAR gamma and the canonical wnt/beta-catenin pathway. Clin Transl Med. (2017) 6:14. doi: 10.1186/s40169-017-0144-7
7. Zhang M, Shi J, Huang Y, Lai L. Expression of canonical wnt/beta-catenin signaling components in the developing human lung. BMC Dev Biol. (2012) 12:21. doi: 10.1186/1471-213X-12-21
8. Bonikos DS, Bensch KG, Northway WH Jr, Edwards DK. Bronchopulmonary dysplasia: the pulmonary pathologic sequel of necrotizing bronchiolitis and pulmonary fibrosis. Hum Pathol. (1976) 7:643–66.
9. Kennedy JD. Lung function outcome in children of premature birth. J Paediatr Child Health. (1999) 35:516–21.
10. Eber E, Zach MS. Long term sequelae of bronchopulmonary dysplasia (chronic lung disease of infancy). Thorax. (2001) 56:317–23. doi: 10.1136/thorax.56.4.317
11. Fanaroff AA, Stoll BJ, Wright LL, Carlo WA, Ehrenkranz RA, Stark AR, et al. Trends in neonatal morbidity and mortality for very low birthweight infants. Am J Obstet Gynecol. (2007) 196:147 e141–8. doi: 10.1016/j.ajog.2006.09.014
12. Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med. (2007) 357:1946–55. doi: 10.1056/NEJMra067279
13. Hadchouel A, Franco-Montoya ML, Delacourt C. Altered lung development in bronchopulmonary dysplasia. Birth Defects Res Part A Clin Mol Teratol. (2014) 100:158–67. doi: 10.1002/bdra.23237
14. Sucre JM, Vijayaraj P, Aros CJ, Wilkinson D, Paul M, Dunn B, et al. Posttranslational modification of beta-catenin is associated with pathogenic fibroblastic changes in bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L186–95. doi: 10.1152/ajplung.00477.2016
15. McEvoy CT, Jain L, Schmidt B, Abman S, Bancalari E, Aschner JL. Bronchopulmonary dysplasia: Nhlbi workshop on the primary prevention of chronic lung diseases. Ann Am Thorac Soc. (2014) 11(Suppl 3):S146–53. doi: 10.1513/AnnalsATS.201312-424LD
16. Benjamin JT, Smith RJ, Halloran BA, Day TJ, Kelly DR, Prince LS. Fgf-10 is decreased in bronchopulmonary dysplasia and suppressed by toll-like receptor activation. Am J Physiol Lung Cell Mol Physiol. (2007) 292:L550–8. doi: 10.1152/ajplung.00329.2006
17. Cerny L, Torday JS, Rehan VK. Prevention and treatment of bronchopulmonary dysplasia: contemporary status and future outlook. Lung. (2008) 186:75–89. doi: 10.1007/s00408-007-9069-z
18. Massaro D, Massaro GD. Dexamethasone accelerates postnatal alveolar wall thinning and alters wall composition. Am J Physiol. (1986) 251(2 Pt 2):R218–24.
19. Massaro D, Teich N, Massaro GD. Postnatal development of pulmonary alveoli: modulation in rats by thyroid hormones. Am J Physiol. (1986) 250(1 Pt 2):R51–5.
20. Noguchi A, Reddy R, Kursar JD, Parks WC, Mecham RP. Smooth muscle isoactin and elastin in fetal bovine lung. Exp Lung Res. (1989) 15:537–52.
21. Bostrom H, Willetts K, Pekny M, Leveen P, Lindahl P, Hedstrand H, et al. Pdgf-a signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell. (1996) 85:863–73.
22. Lindahl P, Karlsson L, Hellstrom M, Gebre-Medhin S, Willetts K, Heath JK, et al. Alveogenesis failure in pdgf-a-deficient mice is coupled to lack of distal spreading of alveolar smooth muscle cell progenitors during lung development. Development. (1997) 124:3943–53.
23. McGowan SE, Torday JS. The pulmonary lipofibroblast (lipid interstitial cell) and its contributions to alveolar development. Annu Rev Physiol. (1997) 59:43–62.
24. McGowan SE, Grossmann RE, Kimani PW, Holmes AJ. Platelet-derived growth factor receptor-alpha-expressing cells localize to the alveolar entry ring and have characteristics of myofibroblasts during pulmonary alveolar septal formation. Anatom Rec. (2008) 291:1649–61. doi: 10.1002/ar.20764
25. Wright C, Strauss S, Toole K, Burt AD, Robson SC. Composition of the pulmonary interstitium during normal development of the human fetus. Pediatr Dev Pathol. (1999) 2:424–31.
26. Yamada M, Kurihara H, Kinoshita K, Sakai T. Temporal expression of alpha-smooth muscle actin and drebrin in septal interstitial cells during alveolar maturation. J Histochem Cytochem. (2005) 53:735–44. doi: 10.1369/jhc.4A6483.2005
27. Hennrick KT, Keeton AG, Nanua S, Kijek TG, Goldsmith AM, Sajjan US, et al. Lung cells from neonates show a mesenchymal stem cell phenotype. Am J Respir Crit Care Med. (2007) 175:1158–64. doi: 10.1164/rccm.200607-941OC
28. Popova AP, Bozyk PD, Bentley JK, Linn MJ, Goldsmith AM, Schumacher RE, et al. Isolation of tracheal aspirate mesenchymal stromal cells predicts bronchopulmonary dysplasia. Pediatrics. (2010) 126:e1127–33. doi: 10.1542/peds.2009-3445
29. Torday JS, Rehan VK. The evolutionary continuum from lung development to homeostasis and repair. Am J Physiol Lung Cell Mol Physiol. (2007) 292:L608–11. doi: 10.1152/ajplung.00379.2006
30. Whitsett JA, Clark JC, Picard L, Tichelaar JW, Wert SE, Itoh N, et al. Fibroblast growth factor 18 influences proximal programming during lung morphogenesis. J Biol Chem. (2002) 277:22743–9. doi: 10.1074/jbc.M202253200
31. Cardoso WV, Lu J. Regulation of early lung morphogenesis: questions, facts and controversies. Development. (2006) 133:1611–24. doi: 10.1242/dev.02310
32. Shannon JM, Hyatt BA. Epithelial-mesenchymal interactions in the developing lung. Annu Rev Physiol. (2004) 66:625–45. doi: 10.1146/annurev.physiol.66.032102.135749
33. Okubo T, Hogan BL. Hyperactive wnt signaling changes the developmental potential of embryonic lung endoderm. J Biol. (2004) 3:11. doi: 10.1186/jbiol3
34. Joshi S, Kotecha S. Lung growth and development. Early Human Dev. (2007) 83:789–94. doi: 10.1016/j.earlhumdev.2007.09.007
35. Langston C, Kida K, Reed M, Thurlbeck WM. Human lung growth in late gestation and in the neonate. Am Rev Respir Dis. (1984) 129:607–13.
36. Martin RJ, Di Fiore JM, Walsh MC. Hypoxic episodes in bronchopulmonary dysplasia. Clin Perinatol. (2015) 42:825–38. doi: 10.1016/j.clp.2015.08.009
37. Raffay TM, Dylag AM, Sattar A, Abu Jawdeh EG, Cao S, Pax BM, et al. Neonatal intermittent hypoxemia events are associated with diagnosis of bronchopulmonary dysplasia at 36 weeks postmenstrual age. Pediatr Res. (2019) 85:318–23. doi: 10.1038/s41390-018-0253-z
38. Blanco LN, Massaro D, Massaro GD. Alveolar size, number, and surface area: Developmentally dependent response to 13% o2. Am J Physiol. (1991) 261(6 Pt 1):L370–7.
39. Massaro D, Massaro GD. Invited review: pulmonary alveoli: formation, the “call for oxygen,” and other regulators. Am J Physiol Lung Cell Mol Physiol. (2002) 282:L345–58. doi: 10.1152/ajplung.00374.2001
40. Nicola T, Ambalavanan N, Zhang W, James ML, Rehan V, Halloran B, et al. Hypoxia-induced inhibition of lung development is attenuated by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Am J Physiol Lung Cell Mol Physiol. (2011) 301:L125–34. doi: 10.1152/ajplung.00074.2011
41. Vallee A, Lecarpentier Y, Vallee JN. Thermodynamic aspects and reprogramming cellular energy metabolism during the fibrosis process. Int J Mol Sci. (2017) 18:E2537. doi: 10.3390/ijms18122537
42. Bhandari V, Bizzarro MJ, Shetty A, Zhong X, Page GP, Zhang H, et al. Familial and genetic susceptibility to major neonatal morbidities in preterm twins. Pediatrics. (2006) 117:1901–6. doi: 10.1542/peds.2005-1414
43. Saugstad OD. Oxygen and oxidative stress in bronchopulmonary dysplasia. J Perinat Med. (2010) 38:571–7. doi: 10.1515/jpm.2010.108
44. Coalson JJ, Winter V, deLemos RA. Decreased alveolarization in baboon survivors with bronchopulmonary dysplasia. Am J Respir Crit Care Med. (1995) 152:640–6.
45. Warner BB, Stuart LA, Papes RA, Wispe JR. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol. (1998) 275(1 Pt 1):L110–7.
46. Vento M, Moro M, Escrig R, Arruza L, Villar G, Izquierdo I, et al. Preterm resuscitation with low oxygen causes less oxidative stress, inflammation, and chronic lung disease. Pediatrics. (2009) 124:e439–49. doi: 10.1542/peds.2009-0434
47. Lee HJ, Kim BI, Choi ES, Choi CW, Kim EK, Kim HS, et al. Effects of postnatal dexamethasone or hydrocortisone in a rat model of antenatal lipopolysaccharide and neonatal hyperoxia exposure. J Korean Med Sci. (2012) 27:395–401. doi: 10.3346/jkms.2012.27.4.395
48. Lee HJ, Choi CW, Kim BI, Kim EK, Kim HS, Choi JH, et al. Serial changes of lung morphology and biochemical profiles in a rat model of bronchopulmonary dysplasia induced by intra-amniotic lipopolysaccharide and postnatal hyperoxia. J Perinat Med. (2010) 38:675–81. doi: 10.1515/jpm.2010.091
49. Choi CW, Kim BI, Hong JS, Kim EK, Kim HS, Choi JH. Bronchopulmonary dysplasia in a rat model induced by intra-amniotic inflammation and postnatal hyperoxia: morphometric aspects. Pediatr Res. (2009) 65:323–7. doi: 10.1203/PDR.0b013e318193f165
50. Kim DH, Choi CW, Kim EK, Kim HS, Kim BI, Choi JH, et al. Association of increased pulmonary interleukin-6 with the priming effect of intra-amniotic lipopolysaccharide on hyperoxic lung injury in a rat model of bronchopulmonary dysplasia. Neonatology. (2010) 98:23–32. doi: 10.1159/000263056
51. Alapati D, Rong M, Chen S, Lin C, Li Y, Wu S. Inhibition of lrp5/6-mediated wnt/beta-catenin signaling by mesd attenuates hyperoxia-induced pulmonary hypertension in neonatal rats. Pediatr Res. (2013) 73:719–25. doi: 10.1038/pr.2013.42
52. Alapati D, Rong M, Chen S, Hehre D, Hummler SC, Wu S. Inhibition of beta-catenin signaling improves alveolarization and reduces pulmonary hypertension in experimental bronchopulmonary dysplasia. Am J Respir Cell Mol Biol. (2014) 51:104–13. doi: 10.1165/rcmb.2013-0346OC
53. Rehan VK, Sakurai R, Corral J, Krebs M, Ibe B, Ihida-Stansbury K, et al. Antenatally administered PPAR-gamma agonist rosiglitazone prevents hyperoxia-induced neonatal rat lung injury. Am J Physiol Lung Cell Mol Physiol. (2010) 299:L672–80. doi: 10.1152/ajplung.00240.2010
54. Dasgupta C, Sakurai R, Wang Y, Guo P, Ambalavanan N, Torday JS, et al. Hyperoxia-induced neonatal rat lung injury involves activation of TGF-β and wnt signaling and is protected by rosiglitazone. Am J Physiol Lung Cell Mol Physiol. (2009) 296:L1031–41. doi: 10.1152/ajplung.90392.2008
55. Hillman NH, Moss TJ, Kallapur SG, Bachurski C, Pillow JJ, Polglase GR, et al. Brief, large tidal volume ventilation initiates lung injury and a systemic response in fetal sheep. Am J Respir Crit Care Med. (2007) 176:575–81. doi: 10.1164/rccm.200701-051OC
56. Asikainen TM, White CW. Pulmonary antioxidant defenses in the preterm newborn with respiratory distress and bronchopulmonary dysplasia in evolution: Implications for antioxidant therapy. Antioxid Redox Signal. (2004) 6:155–67. doi: 10.1089/152308604771978462
57. Welty SE. Antioxidants and oxidations in bronchopulmonary dysplasia: there are no easy answers. J Pediatr. (2003) 143:697–8. doi: 10.1067/S0022-3476(03)00626-7
58. Toti P, Buonocore G, Tanganelli P, Catella AM, Palmeri ML, Vatti R, et al. Bronchopulmonary dysplasia of the premature baby: an immunohistochemical study. Pediatr Pulmonol. (1997) 24:22–8.
59. Bhatt AJ, Pryhuber GS, Huyck H, Watkins RH, Metlay LA, Maniscalco WM. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, flt-1, and tie-2 in human infants dying with bronchopulmonary dysplasia. Am J Respir Crit Care Med. (2001) 164(10 Pt 1):1971–80. doi: 10.1164/ajrccm.164.10.2101140
60. Kaarteenaho-Wiik R, Kinnula VL, Herva R, Soini Y, Pollanen R, Paakko P. Tenascin-c is highly expressed in respiratory distress syndrome and bronchopulmonary dysplasia. J Histochem Cytochem. (2002) 50:423–31. doi: 10.1177/002215540205000313
61. Kaarteenaho-Wiik R, Paakko P, Herva R, Risteli J, Soini Y. Type i and iii collagen protein precursors and mrna in the developing human lung. J Pathol. (2004) 203:567–74. doi: 10.1002/path.1547
62. Bozyk PD, Bentley JK, Popova AP, Anyanwu AC, Linn MD, Goldsmith AM, et al. Neonatal periostin knockout mice are protected from hyperoxia-induced alveolar simplication. PLoS ONE. (2012) 7:e31336. doi: 10.1371/journal.pone.0031336
63. Wu S, Platteau A, Chen S, McNamara G, Whitsett J, Bancalari E. Conditional overexpression of connective tissue growth factor disrupts postnatal lung development. Am J Respir Cell Mol Biol. (2010) 42:552–63. doi: 10.1165/rcmb.2009-0068OC
64. Coalson JJ, Winter VT, Siler-Khodr T, Yoder BA. Neonatal chronic lung disease in extremely immature baboons. Am J Respir Crit Care Med. (1999) 160:1333–46.
65. Albertine KH, Jones GP, Starcher BC, Bohnsack JF, Davis PL, Cho SC, et al. Chronic lung injury in preterm lambs. Disordered respiratory tract development. Am J Respir Crit Care Med. (1999) 159:945–58.
66. Mokres LM, Parai K, Hilgendorff A, Ertsey R, Alvira CM, Rabinovitch M, et al. Prolonged mechanical ventilation with air induces apoptosis and causes failure of alveolar septation and angiogenesis in lungs of newborn mice. Am J Physiol Lung Cell Mol Physiol. (2010) 298:L23–35. doi: 10.1152/ajplung.00251.2009
67. Mach WJ, Thimmesch AR, Pierce JT, Pierce JD. Consequences of hyperoxia and the toxicity of oxygen in the lung. Nurs Res Pract. (2011) 2011:260482. doi: 10.1155/2011/260482
68. Patel A, Lakshminrusimha S, Ryan RM, Swartz DD, Wang H, Wynn KA, et al. Exposure to supplemental oxygen downregulates antioxidant enzymes and increases pulmonary arterial contractility in premature lambs. Neonatology. (2009) 96:182–92. doi: 10.1159/000211667
69. Kumar VH, Patel A, Swartz DD, Wang H, Wynn KA, Nielsen LC, et al. Exposure to supplemental oxygen and its effects on oxidative stress and antioxidant enzyme activity in term newborn lambs. Pediatr Res. (2010) 67:66–71. doi: 10.1203/PDR.0b013e3181bf587f
70. Perrone S, Bracciali C, Di Virgilio N, Buonocore G. Oxygen use in neonatal care: a two-edged sword. Front Pediatr. (2016) 4:143. doi: 10.3389/fped.2016.00143
71. Chow LC, Wright KW, Sola A, Group COAS. Can changes in clinical practice decrease the incidence of severe retinopathy of prematurity in very low birth weight infants? Pediatrics. (2003) 111:339–45. doi: 10.1542/peds.111.2.339
72. Tin W, Gupta S. Optimum oxygen therapy in preterm babies. Arch Dis Child Fetal Neonatal Ed. (2007) 92:F143–7. doi: 10.1136/adc.2005.092726
73. Supplemental therapeutic oxygen for prethreshold retinopathy of prematurity (stop-rop) a randomized controlled trial. I: Primary outcomes. Pediatrics. (2000) 105:295–310. doi: 10.1542/peds.105.2.295
74. Askie LM, Henderson-Smart DJ, Irwig L, Simpson JM. Oxygen-saturation targets and outcomes in extremely preterm infants. N Engl J Med. (2003) 349:959–67. doi: 10.1056/NEJMoa023080
75. Tarnow-Mordi WO, Darlow B, Doyle L. Target ranges of oxygen saturation in extremely preterm infants. N Engl J Med. (2010) 363:1285; author reply 1285–6. doi: 10.1056/NEJMc1007912
76. Schmidt B, Whyte RK, Asztalos EV, Moddemann D, Poets C, Rabi Y, et al. Effects of targeting higher vs lower arterial oxygen saturations on death or disability in extremely preterm infants: a randomized clinical trial. JAMA. (2013) 309:2111–20. doi: 10.1001/jama.2013.5555
77. Khadawardi E, Al Hazzani F. Oxygen saturation and outcomes in preterm infants the boost II United Kingdom, Australia, and New Zealand collaborative groups. J Clin Neonatol. (2013) 2:73–5. doi: 10.4103/2249-4847.116404
78. Sweet DG, Carnielli V, Greisen G, Hallman M, Ozek E, Plavka R, et al. European consensus guidelines on the management of neonatal respiratory distress syndrome in preterm infants-2010 update. Zhonghua er ke za zhi Chin J Pediatr. (2011) 49:27–33. doi: 10.1159/000297773
79. Moon RT, Kohn AD, De Ferrari GV, Kaykas A. Wnt and beta-catenin signalling: diseases and therapies. Nat Rev Genet. (2004) 5:691–701. doi: 10.1038/nrg1427
80. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. (2006) 127:469–80. doi: 10.1016/j.cell.2006.10.018
81. Heuberger J, Birchmeier W. Interplay of cadherin-mediated cell adhesion and canonical wnt signaling. Cold Spring Harbor Perspect Biol. (2010) 2:a002915. doi: 10.1101/cshperspect.a002915
82. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, et al. The cyclin d1 gene is a target of the beta-catenin/lef-1 pathway. Proc Natl Acad Sci USA. (1999) 96:5522–7.
83. Angers S, Moon RT. Proximal events in wnt signal transduction. Nat Rev Mol Cell Biol. (2009) 10:468–77. doi: 10.1038/nrm2717
84. Konigshoff M, Eickelberg O. Wnt signaling in lung disease: a failure or a regeneration signal? Am J Respir Cell Mol Biol. (2010) 42:21–31. doi: 10.1165/rcmb.2008-0485TR
85. Fu M, Zhang J, Lin Y, Zhu X, Zhao L, Ahmad M, et al. Early stimulation and late inhibition of peroxisome proliferator-activated receptor gamma (PPAR gamma) gene expression by transforming growth factor beta in human aortic smooth muscle cells: role of early growth-response factor-1 (Egr-1), activator protein 1 (Ap1) and Smads. Biochem J. (2003) 370(Pt 3):1019–25. doi: 10.1042/bj20021503
86. Phan SH. The myofibroblast in pulmonary fibrosis. Chest. (2002) 122(6 Suppl):286S−9S. doi: 10.1378/chest.122.6_suppl.286S
87. Zhang K, Rekhter MD, Gordon D, Phan SH. Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis. A combined immunohistochemical and in situ hybridization study. Am J Pathol. (1994) 145:114–25.
88. Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res. (2000) 257:180–9. doi: 10.1006/excr.2000.4869
89. Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. (2005) 166:1321–32. doi: 10.1016/S0002-9440(10)62351-6
90. Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. (2009) 119:213–24. doi: 10.1172/JCI36940
91. Willis BC, Borok Z. TGF-beta-induced emt: Mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. (2007) 293:L525–34. doi: 10.1152/ajplung.00163.2007
92. Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. (2006) 3:377–82. doi: 10.1513/pats.200601-004TK
93. Goldmann T, Zissel G, Watz H, Dromann D, Reck M, Kugler C, et al. Human alveolar epithelial cells type ii are capable of TGFbeta-dependent epithelial-mesenchymal-transition and collagen-synthesis. Respir Res. (2018) 19:138. doi: 10.1186/s12931-018-0841-9
94. Elbrecht A, Chen Y, Cullinan CA, Hayes N, Leibowitz M, Moller DE, et al. Molecular cloning, expression and characterization of human peroxisome proliferator activated receptors gamma 1 and gamma 2. Biochem Biophys Res Commun. (1996) 224:431–7.
95. Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. (1999) 20:649–88.
96. Reddy AT, Lakshmi SP, Zhang Y, Reddy RC. Nitrated fatty acids reverse pulmonary fibrosis by dedifferentiating myofibroblasts and promoting collagen uptake by alveolar macrophages. FASEB J. (2014) 28:5299–310. doi: 10.1096/fj.14-256263
97. Rehan VK, Torday JS. PPARgamma signaling mediates the evolution, development, homeostasis, and repair of the lung. PPAR Res. (2012) 2012:289867. doi: 10.1155/2012/289867
98. Wei J, Fang F, Lam AP, Sargent JL, Hamburg E, Hinchcliff ME, et al. Wnt/beta-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum. (2012) 64:2734–45. doi: 10.1002/art.34424
99. Dantas AT, Pereira MC, de Melo Rego MJ, da Rocha LF Jr, Pitta Ida R, Marques CD, et al. The role of PPAR gamma in systemic sclerosis. PPAR Res. (2015) 2015:124624. doi: 10.1155/2015/124624
100. Kulkarni AA, Thatcher TH, Olsen KC, Maggirwar SB, Phipps RP, Sime PJ. PPAR-gamma ligands repress TGFbeta-induced myofibroblast differentiation by targeting the pi3k/akt pathway: implications for therapy of fibrosis. PLoS ONE. (2011) 6:e15909. doi: 10.1371/journal.pone.0015909
101. Rosenbloom J, Castro SV, Jimenez SA. Narrative review: fibrotic diseases: cellular and molecular mechanisms and novel therapies. Ann Int Med. (2010) 152:159–66. doi: 10.7326/0003-4819-152-3-201002020-00007
102. Wei J, Ghosh AK, Sargent JL, Komura K, Wu M, Huang QQ, et al. PPARgamma downregulation by TGFss in fibroblast and impaired expression and function in systemic sclerosis: a novel mechanism for progressive fibrogenesis. PLoS ONE. (2010) 5:e13778. doi: 10.1371/journal.pone.0013778
103. De Langhe SP, Reynolds SD. Wnt signaling in lung organogenesis. Organogenesis. (2008) 4:100–8. doi: 10.4161/org.4.2.5856
104. Pongracz JE, Stockley RA. Wnt signalling in lung development and diseases. Respir Res. (2006) 7:15. doi: 10.1186/1465-9921-7-15
105. Goss AM, Morrisey EE. Wnt signaling and specification of the respiratory endoderm. Cell Cycle. (2010) 9:10–1. doi: 10.4161/cc.9.1.10272
106. Shu W, Guttentag S, Wang Z, Andl T, Ballard P, Lu MM, et al. Wnt/beta-catenin signaling acts upstream of N-myc, BMP4, and FGF signaling to regulate proximal-distal patterning in the lung. Dev Biol. (2005) 283:226–39. doi: 10.1016/j.ydbio.2005.04.014
107. Snowball J, Ambalavanan M, Cornett B, Lang R, Whitsett J, Sinner D. Mesenchymal wnt signaling promotes formation of sternum and thoracic body wall. Dev Biol. (2015) 401:264–75. doi: 10.1016/j.ydbio.2015.02.014
108. De Langhe SP, Carraro G, Tefft D, Li C, Xu X, Chai Y, et al. Formation and differentiation of multiple mesenchymal lineages during lung development is regulated by beta-catenin signaling. PLoS ONE. (2008) 3:e1516. doi: 10.1371/journal.pone.0001516
109. Eberhart CG, Argani P. Wnt signaling in human development: beta-catenin nuclear translocation in fetal lung, kidney, placenta, capillaries, adrenal, and cartilage. Pediatr Dev Pathol. (2001) 4:351–7. doi: 10.1007/s10024001-0037-y
110. Ota C, Baarsma HA, Wagner DE, Hilgendorff A, Konigshoff M. Linking bronchopulmonary dysplasia to adult chronic lung diseases: role of wnt signaling. Mol Cell Pediatr. (2016) 3:34. doi: 10.1186/s40348-016-0062-6
111. Ring DB, Johnson KW, Henriksen EJ, Nuss JM, Goff D, Kinnick TR, et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes. (2003) 52:588–95. doi: 10.2337/diabetes.52.3.588
112. Konigshoff M, Balsara N, Pfaff EM, Kramer M, Chrobak I, Seeger W, Eickelberg O. Functional wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS ONE. (2008) 3:e2142. doi: 10.1371/journal.pone.0002142
113. Torday JS, Torres E, Rehan VK. The role of fibroblast transdifferentiation in lung epithelial cell proliferation, differentiation, and repair in vitro. Pediatr Pathol Mol Med. (2003) 22:189–207. doi: 10.1080/pdp.22.3.189.207
114. Simon DM, Arikan MC, Srisuma S, Bhattacharya S, Tsai LW, Ingenito EP, et al. Epithelial cell PPAR[gamma] contributes to normal lung maturation. FASEB J. (2006) 20:1507–9. doi: 10.1096/fj.05-5410fje
115. Wang Y, Santos J, Sakurai R, Shin E, Cerny L, Torday JS, et al. Peroxisome proliferator-activated receptor gamma agonists enhance lung maturation in a neonatal rat model. Pediatr Res. (2009) 65:150–5. doi: 10.1203/PDR.0b013e3181938c40
116. Torday JS, Torday DP, Gutnick J, Qin J, Rehan V. Biologic role of fetal lung fibroblast triglycerides as antioxidants. Pediatr Res. (2001) 49:843–9. doi: 10.1203/00006450-200106000-00021
117. Simon DM, Arikan MC, Srisuma S, Bhattacharya S, Andalcio T, Shapiro SD, et al. Epithelial cell PPARgamma is an endogenous regulator of normal lung maturation and maintenance. Proc Am Thorac Soc. (2006) 3:510–1. doi: 10.1513/pats.200603-034MS
118. Torday JS, Rehan VK. Stretch-stimulated surfactant synthesis is coordinated by the paracrine actions of pthrp and leptin. Am J Physiol Lung Cell Mol Physiol. (2002) 283:L130–5. doi: 10.1152/ajplung.00380.2001
119. Rehan VK, Wang Y, Patel S, Santos J, Torday JS. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma agonist, prevents hyperoxia-induced neonatal rat lung injury in vivo. Pediatric pulmonology. (2006) 41:558–69. doi: 10.1002/ppul.20407
120. Popova AP, Bentley JK, Anyanwu AC, Richardson MN, Linn MJ, Lei J, et al. Glycogen synthase kinase-3beta/beta-catenin signaling regulates neonatal lung mesenchymal stromal cell myofibroblastic differentiation. Am J Physiol Lung Cell Mol Physiol. (2012) 303:L439–48. doi: 10.1152/ajplung.00408.2011
121. Zhao H, Dennery PA, Yao H. Metabolic reprogramming in the pathogenesis of chronic lung diseases, including bpd, copd, and pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L544–54. doi: 10.1152/ajplung.00521.2017
122. Pate KT, Stringari C, Sprowl-Tanio S, Wang K, TeSlaa T, Hoverter NP, et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. (2014) 33:1454–73. doi: 10.15252/embj.201488598
124. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. (2012) 149:1192–205. doi: 10.1016/j.cell.2012.05.012
125. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. (2008) 105:18782–7. doi: 10.1073/pnas.0810199105
126. Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. (2007) 27:7381–93. doi: 10.1128/MCB.00440-07
127. Abbot EL, McCormack JG, Reynet C, Hassall DG, Buchan KW, Yeaman SJ. Diverging regulation of pyruvate dehydrogenase kinase isoform gene expression in cultured human muscle cells. FEBS J. (2005) 272:3004–14. doi: 10.1111/j.1742-4658.2005.04713.x
128. Lecarpentier Y, Schussler O, Claes V, Vallée A. The myofibroblast: TGFβ-1, a conductor which plays a key role in fibrosis by regulating the balance between PPARγ and the canonical wnt pathway. Nucl Recept Res. (2017) 4:101299. doi: 10.11131/2017/101299
129. Deng H, Dokshin GA, Lei J, Goldsmith AM, Bitar KN, Fingar DC, et al. Inhibition of glycogen synthase kinase-3beta is sufficient for airway smooth muscle hypertrophy. J Biol Chem. (2008) 283:10198–207. doi: 10.1074/jbc.M800624200
130. Vicencio AG, Lee CG, Cho SJ, Eickelberg O, Chuu Y, Haddad GG, et al. Conditional overexpression of bioactive transforming growth factor-beta1 in neonatal mouse lung: a new model for bronchopulmonary dysplasia? Am J Respir Cell Mol Biol. (2004) 31:650–56. doi: 10.1165/rcmb.2004-0092OC
131. Whitehead GS, Burch LH, Berman KG, Piantadosi CA, Schwartz DA. Genetic basis of murine responses to hyperoxia-induced lung injury. Immunogenetics. (2006) 58:793–804. doi: 10.1007/s00251-006-0147-9
132. Abreu JG, Ketpura NI, Reversade B, De Robertis EM. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat Cell Biol. (2002) 4:599–604. doi: 10.1038/ncb826
133. Shi-wen X, Stanton LA, Kennedy L, Pala D, Chen Y, Howat SL, et al. Ccn2 is necessary for adhesive responses to transforming growth factor-beta1 in embryonic fibroblasts. J Biol Chem. (2006) 281:10715–26. doi: 10.1074/jbc.M511343200
134. Sohn M, Tan Y, Wang B, Klein RL, Trojanowska M, Jaffa AA. Mechanisms of low-density lipoprotein-induced expression of connective tissue growth factor in human aortic endothelial cells. Am J Physiol Heart Circul Physiol. (2006) 290:H1624–34. doi: 10.1152/ajpheart.01233.2004
135. Rehan VK, Fong J, Lee R, Sakurai R, Wang ZM, Dahl MJ, et al. Mechanism of reduced lung injury by high-frequency nasal ventilation in a preterm lamb model of neonatal chronic lung disease. Pediatr Res. (2011) 70:462–6. doi: 10.1203/PDR.0b013e31822f58a1
136. Torday JS, Rehan VK. Up-regulation of fetal rat lung parathyroid hormone-related protein gene regulatory network down-regulates the sonic hedgehog/wnt/betacatenin gene regulatory network. Pediatr Res. (2006) 60:382–8. doi: 10.1203/01.pdr.0000238326.42590.03
137. Cuzzocrea S, Pisano B, Dugo L, Ianaro A, Patel NS, Di Paola R, et al. Rosiglitazone, a ligand of the peroxisome proliferator-activated receptor-gamma, reduces the development of nonseptic shock induced by zymosan in mice. Crit Care Med. (2004) 32:457–66. doi: 10.1097/01.CCM.0000109446.38675.61
138. Stenmark KR, Abman SH. Lung vascular development: implications for the pathogenesis of bronchopulmonary dysplasia. Ann Rev Physiol. (2005) 67:623–61. doi: 10.1146/annurev.physiol.67.040403.102229
139. Thebaud B, Ladha F, Michelakis ED, Sawicka M, Thurston G, Eaton F, et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation. (2005) 112:2477–86. doi: 10.1161/CIRCULATIONAHA.105.541524
140. Thebaud B, Abman SH. Bronchopulmonary dysplasia: where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med. (2007) 175:978–85. doi: 10.1164/rccm.200611-1660PP
141. Yamakawa K, Hosoi M, Koyama H, Tanaka S, Fukumoto S, Morii H, et al. Peroxisome proliferator-activated receptor-gamma agonists increase vascular endothelial growth factor expression in human vascular smooth muscle cells. Biochem Biophys Res Commun. (2000) 271:571–4. doi: 10.1006/bbrc.2000.2665
142. Hosford GE, Olson DM. Effects of hyperoxia on VEGF, its receptors, and HIF-2alpha in the newborn rat lung. Am J Physiol Lung Cell Mol Physiol. (2003) 285:L161–8. doi: 10.1152/ajplung.00285.2002
143. Kunig AM, Balasubramaniam V, Markham NE, Seedorf G, Gien J, Abman SH. Recombinant human vegf treatment transiently increases lung edema but enhances lung structure after neonatal hyperoxia. Am J Physiol Lung Cell Mol Physiol. (2006) 291:L1068–78. doi: 10.1152/ajplung.00093.2006
144. Lee HJ, Lee YJ, Choi CW, Lee JA, Kim EK, Kim HS, Kim BI, Choi JH. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma agonist, restores alveolar and pulmonary vascular development in a rat model of bronchopulmonary dysplasia. Yonsei Med J. (2014) 55:99–106. doi: 10.3349/ymj.2014.55.1.99
145. Morales E, Sakurai R, Husain S, Paek D, Gong M, Ibe B, et al. Nebulized PPARgamma agonists: a novel approach to augment neonatal lung maturation and injury repair in rats. Pediatr Res. (2014) 75:631–40. doi: 10.1038/pr.2014.8
146. Sierra H, Sakurai R, Lee WN, Truong NC, Torday JS, Rehan VK. Prenatal rosiglitazone administration to neonatal rat pups does not alter the adult metabolic phenotype. PPAR Res. (2012) 2012:604216. doi: 10.1155/2012/604216
147. Truong NC, Abbasi A, Sakurai R, Lee WN, Torday JS, Rehan VK. Postnatal rosiglitazone administration to neonatal rat pups does not alter the young adult metabolic phenotype. Neonatology. (2012) 101:217–24. doi: 10.1159/000331772
148. Burgess HA, Daugherty LE, Thatcher TH, Lakatos HF, Ray DM, Redonnet M, et al. PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am J Physiol Lung Cell Mol Physiol. (2005) 288:L1146–53. doi: 10.1152/ajplung.00383.2004
149. Lakshmi SP, Reddy AT, Reddy RC. Transforming growth factor beta suppresses peroxisome proliferator-activated receptor gamma expression via both Smad binding and novel TGF-beta inhibitory elements. Biochem J. (2017) 474:1531–46. doi: 10.1042/BCJ20160943
150. Milam JE, Keshamouni VG, Phan SH, Hu B, Gangireddy SR, Hogaboam CM, et al. PPAR-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2008) 294:L891–901. doi: 10.1152/ajplung.00333.2007
151. Kasper M, Haroske G. Alterations in the alveolar epithelium after injury leading to pulmonary fibrosis. Histol Histopathol. (1996) 11:463–83.
152. Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir Res. (2005) 6:56. doi: 10.1186/1465-9921-6-56
153. Tan X, Dagher H, Hutton CA, Bourke JE. Effects of PPAR gamma ligands on TGF-beta1-induced epithelial-mesenchymal transition in alveolar epithelial cells. Respir Res. (2010) 11:21. doi: 10.1186/1465-9921-11-21
154. Wei J, Bhattacharyya S, Jain M, Varga J. Regulation of matrix remodeling by peroxisome proliferator-activated receptor-gamma: a novel link between metabolism and fibrogenesis. Open Rheumatol J. (2012) 6:103–15. doi: 10.2174/1874312901206010103
155. Aoki Y, Maeno T, Aoyagi K, Ueno M, Aoki F, Aoki N, et al. Pioglitazone, a peroxisome proliferator-activated receptor gamma ligand, suppresses bleomycin-induced acute lung injury and fibrosis. Respiration. (2009) 77:311–9. doi: 10.1159/000168676
156. Samah M, El-Aidy Ael R, Tawfik MK, Ewais MM. Evaluation of the antifibrotic effect of fenofibrate and rosiglitazone on bleomycin-induced pulmonary fibrosis in rats. Eur J Pharmacol. (2012) 689:186–93. doi: 10.1016/j.ejphar.2012.05.026
157. Genovese T, Cuzzocrea S, Di Paola R, Mazzon E, Mastruzzo C, Catalano P, et al. Effect of rosiglitazone and 15-deoxy-delta12,14-prostaglandin j2 on bleomycin-induced lung injury. Eur Respir J. (2005) 25:225–34. doi: 10.1183/09031936.05.00049704
158. Shi-wen X, Eastwood M, Stratton RJ, Denton CP, Leask A, Abraham DJ. Rosiglitazone alleviates the persistent fibrotic phenotype of lesional skin scleroderma fibroblasts. Rheumatology. (2010) 49:259–63. doi: 10.1093/rheumatology/kep371
159. Wu M, Melichian DS, Chang E, Warner-Blankenship M, Ghosh AK, Varga J. Rosiglitazone abrogates bleomycin-induced scleroderma and blocks profibrotic responses through peroxisome proliferator-activated receptor-gamma. Am J Pathol. (2009) 174:519–33. doi: 10.2353/ajpath.2009.080574
160. Bogatkevich GS, Highland KB, Akter T, Silver RM. The PPARgamma agonist rosiglitazone is antifibrotic for scleroderma lung fibroblasts: mechanisms of action and differential racial effects. Pulm Med. (2012) 2012:545172. doi: 10.1155/2012/545172
161. Wei J, Bhattacharyya S, Varga J. Peroxisome proliferator-activated receptor gamma: innate protection from excessive fibrogenesis and potential therapeutic target in systemic sclerosis. Curr Opin Rheumatol. (2010) 22:671–6. doi: 10.1097/BOR.0b013e32833de1a7
162. Kawaguchi K, Sakaida I, Tsuchiya M, Omori K, Takami T, Okita K. Pioglitazone prevents hepatic steatosis, fibrosis, and enzyme-altered lesions in rat liver cirrhosis induced by a choline-deficient l-amino acid-defined diet. Biochem Biophys Res Commun. (2004) 315:187–95. doi: 10.1016/j.bbrc.2004.01.038
163. Uto H, Nakanishi C, Ido A, Hasuike S, Kusumoto K, Abe H, et al. The peroxisome proliferator-activated receptor-gamma agonist, pioglitazone, inhibits fat accumulation and fibrosis in the livers of rats fed a choline-deficient, l-amino acid-defined diet. Hepatol Res. (2005) 32:235–42. doi: 10.1016/j.hepres.2005.05.008
164. Ferguson HE, Kulkarni A, Lehmann GM, Garcia-Bates TM, Thatcher TH, Huxlin KR, et al. Electrophilic peroxisome proliferator-activated receptor-gamma ligands have potent antifibrotic effects in human lung fibroblasts. Am J Respir Cell Mol Biol. (2009) 41:722–30. doi: 10.1165/rcmb.2009-0006OC
165. Lin Q, Fang LP, Zhou WW, Liu XM. Rosiglitazone inhibits migration, proliferation, and phenotypic differentiation in cultured human lung fibroblasts. Exp Lung Res. (2010) 36:120–8. doi: 10.3109/01902140903214659
166. Zhao Y, Huang Y, He J, Li C, Deng W, Ran X, et al. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma agonist, attenuates airway inflammation by inhibiting the proliferation of effector T cells in a murine model of neutrophilic asthma. Immunol Lett. (2014) 157:9–15. doi: 10.1016/j.imlet.2013.11.004
167. Honda K, Marquillies P, Capron M, Dombrowicz D. Peroxisome proliferator-activated receptor gamma is expressed in airways and inhibits features of airway remodeling in a mouse asthma model. J Allergy Clin Immunol. (2004) 113:882–8. doi: 10.1016/j.jaci.2004.02.036
168. Gustafson B, Eliasson B, Smith U. Thiazolidinediones increase the wingless-type MMTV integration site family (WNT) inhibitor dickkopf-1 in adipocytes: a link with osteogenesis. Diabetologia. (2010) 53:536–40. doi: 10.1007/s00125-009-1615-1
169. Rehan VK, Dargan-Batra SK, Wang Y, Cerny L, Sakurai R, Santos J, et al. A paradoxical temporal response of the PTHrP/PPARgamma signaling pathway to lipopolysaccharide in an in vitro model of the developing rat lung. Am J Physiol Lung Cell Mol Physiol. (2007) 293:L182–90. doi: 10.1152/ajplung.00319.2006
170. Lakshmi SP, Reddy AT, Zhang Y, Sciurba FC, Mallampalli RK, Duncan SR, et al. Down-regulated peroxisome proliferator-activated receptor gamma (PPARgamma) in lung epithelial cells promotes a PPARgamma agonist-reversible proinflammatory phenotype in chronic obstructive pulmonary disease (COPD). J Biol Chem. (2014) 289:6383–93. doi: 10.1074/jbc.M113.536805
171. Zheng F, Fornoni A, Elliot SJ, Guan Y, Breyer MD, Striker LJ, et al. Upregulation of type I collagen by TGF-beta in mesangial cells is blocked by PPARgamma activation. Am J Physiol Renal Physiol. (2002) 282:F639–48. doi: 10.1152/ajprenal.00189.2001
172. Zhang GY, Cheng T, Zheng MH, Yi CG, Pan H, Li ZJ, et al. Activation of peroxisome proliferator-activated receptor-gamma inhibits transforming growth factor-beta1 induction of connective tissue growth factor and extracellular matrix in hypertrophic scar fibroblasts in vitro. Arch Dermatol Res. (2009) 301:515–22. doi: 10.1007/s00403-009-0959-1
173. Deng YL, Xiong XZ, Cheng NS. Organ fibrosis inhibited by blocking transforming growth factor-beta signaling via peroxisome proliferator-activated receptor gamma agonists. Hepatobil Pancreat Dis Int. (2012) 11:467–78. doi: 10.1016/S1499-3872(12)60210-0
174. Ghosh AK, Bhattacharyya S, Wei J, Kim S, Barak Y, Mori Y, et al. Peroxisome proliferator-activated receptor-gamma abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB J. (2009) 23:2968–77. doi: 10.1096/fj.08-128736
175. Ramirez A, Ballard EN, Roman J. TGFbeta1 controls PPARgamma expression, transcriptional potential, and activity, in part, through Smad3 signaling in murine lung fibroblasts. PPAR Res. (2012) 2012:375876. doi: 10.1155/2012/375876
176. Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. (2012) 11:790–811. doi: 10.1038/nrd3810
177. Zhao C, Chen W, Yang L, Chen L, Stimpson SA, Diehl AM. PPARgamma agonists prevent TGFbeta1/Smad3-signaling in human hepatic stellate cells. Biochem Biophys Res Commun. (2006) 350:385–91. doi: 10.1016/j.bbrc.2006.09.069
178. Varga J, Pasche B. Transforming growth factor beta as a therapeutic target in systemic sclerosis. Nat Rev Rheumatol. (2009) 5:200–6. doi: 10.1038/nrrheum.2009.26
179. Lee SJ, Yang EK, Kim SG. Peroxisome proliferator-activated receptor-gamma and retinoic acid X receptor alpha represses the TGFbeta1 gene via PTEN-mediated p70 ribosomal S6 kinase-1 inhibition: role for Zf9 dephosphorylation. Mol Pharmacol. (2006) 70:415–25. doi: 10.1124/mol.106.022954
180. Lam AP, Gottardi CJ. Beta-catenin signaling: a novel mediator of fibrosis and potential therapeutic target. Curr Opin Rheumatol. (2011) 23:562–7. doi: 10.1097/BOR.0b013e32834b3309
181. Hao S, He W, Li Y, Ding H, Hou Y, Nie J, et al. Targeted inhibition of beta-catenin/cbp signaling ameliorates renal interstitial fibrosis. J Am Soc Nephrol. (2011) 22:1642–53. doi: 10.1681/ASN.2010101079
182. Bergmann C, Akhmetshina A, Dees C, Palumbo K, Zerr P, Beyer C, et al. Inhibition of glycogen synthase kinase 3beta induces dermal fibrosis by activation of the canonical wnt pathway. Ann Rheum Dis. (2011) 70:2191–8. doi: 10.1136/ard.2010.147140
183. Han J, Hajjar DP, Tauras JM, Feng J, Gotto AM Jr, Nicholson AC. Transforming growth factor-beta1 (TGF-beta1) and TGF-beta2 decrease expression of CD36, the type b scavenger receptor, through mitogen-activated protein kinase phosphorylation of peroxisome proliferator-activated receptor-gamma. J Biol Chem. (2000) 275:1241–6. doi: 10.1074/jbc.275.2.1241
184. Gong K, Xing D, Li P, Aksut B, Ambalavanan N, Yang Q, et al. Hypoxia induces downregulation of PPAR-gamma in isolated pulmonary arterial smooth muscle cells and in rat lung via transforming growth factor-beta signaling. Am J Physiol Lung Cell Mol Physiol. (2011) 301:L899–907. doi: 10.1152/ajplung.00062.2011
185. Nesto RW, Bell D, Bonow RO, Fonseca V, Grundy SM, Horton ES, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the american heart association and american diabetes association. October 7, 2003. Circulation. (2003) 108:2941–8. doi: 10.1161/01.CIR.0000103683.99399.7E
186. Betteridge DJ. Thiazolidinediones and fracture risk in patients with type 2 diabetes. Diabet Med. (2011) 28:759–71. doi: 10.1111/j.1464-5491.2010.03187.x
187. Kota BP, Huang TH, Roufogalis BD. An overview on biological mechanisms of PPARs. Pharmacol Res. (2005) 51:85–94. doi: 10.1016/j.phrs.2004.07.012
188. Kohlroser J, Mathai J, Reichheld J, Banner BF, Bonkovsky HL. Hepatotoxicity due to troglitazone: report of two cases and review of adverse events reported to the united states food and drug administration. Am J Gastroenterol. (2000) 95:272–6. doi: 10.1111/j.1572-0241.2000.01707.x
190. Nissen SE, Nicholls SJ, Wolski K, Nesto R, Kupfer S, Perez A, et al. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the periscope randomized controlled trial. JAMA. (2008) 299:1561–73. doi: 10.1001/jama.299.13.1561
191. Graham DJ, Ouellet-Hellstrom R, MaCurdy TE, Ali F, Sholley C, Worrall C, et al. Risk of acute myocardial infarction, stroke, heart failure, and death in elderly medicare patients treated with rosiglitazone or pioglitazone. JAMA. (2010) 304:411–8. doi: 10.1001/jama.2010.920
192. Mahaffey KW, Hafley G, Dickerson S, Burns S, Tourt-Uhlig S, White J, et al. Results of a reevaluation of cardiovascular outcomes in the record trial. Am Heart J. (2013) 166:240–9 e241. doi: 10.1016/j.ahj.2013.05.004
193. Nissen SE, Tardif JC, Nicholls SJ, Revkin JH, Shear CL, Duggan WT, et al. Effect of torcetrapib on the progression of coronary atherosclerosis. N Engl J Med. (2007) 356:1304–16. doi: 10.1056/NEJMoa070635
194. Vallarino C, Perez A, Fusco G, Liang H, Bron M, Manne S, et al. Comparing pioglitazone to insulin with respect to cancer, cardiovascular and bone fracture endpoints, using propensity score weights. Clin Drug Invest. (2013) 33:621–31. doi: 10.1007/s40261-013-0106-9
195. Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, et al. Antidiabetic actions of a non-agonist PPARgamma ligand blocking cdk5-mediated phosphorylation. Nature. (2011) 477:477–81. doi: 10.1038/nature10383
196. Liu D, Zeng BX, Zhang SH, Yao SL. Rosiglitazone, an agonist of peroxisome proliferator-activated receptor gamma, reduces pulmonary inflammatory response in a rat model of endotoxemia. Inflamm Res. (2005) 54:464–70. doi: 10.1007/s00011-005-1379-0
Keywords: bronchopulmonary dysplasia, canonical WNT/β-catenin, TGF-β, PPARγ, fibrosis, myofibroblast, nebulized thiazolidinediones
Citation: Lecarpentier Y, Gourrier E, Gobert V and Vallée A (2019) Bronchopulmonary Dysplasia: Crosstalk Between PPARγ, WNT/β-Catenin and TGF-β Pathways; The Potential Therapeutic Role of PPARγ Agonists. Front. Pediatr. 7:176. doi: 10.3389/fped.2019.00176
Received: 24 January 2019; Accepted: 16 April 2019;
Published: 03 May 2019.
Edited by:
David Warburton, Children's Hospital Los Angeles, United StatesReviewed by:
Martin Post, Hospital for Sick Children, CanadaCopyright © 2019 Lecarpentier, Gourrier, Gobert and Vallée. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yves Lecarpentier, eXZlcy5jLmxlY2FycGVudGllckBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.