 Mayra B. Dorna1*
Mayra B. Dorna1* Pamela F. A. Barbosa1
Pamela F. A. Barbosa1 Andréia Rangel-Santos2
Andréia Rangel-Santos2 Krisztian Csomos3
Krisztian Csomos3 Boglarka Ujhazi3
Boglarka Ujhazi3 Joseph F. Dasso3,4
Joseph F. Dasso3,4 Daniel Thwaites5
Daniel Thwaites5 Joan Boyes5
Joan Boyes5 Sinisa Savic6
Sinisa Savic6 Jolan E. Walter3,7,8*
Jolan E. Walter3,7,8*- 1Department of Pediatrics, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
- 2Laboratory of Medical Investigation (LIM 36), Department of Pediatrics, Faculdade de Medicina da Universidade de São Paulo, Hospital das Clínicas, São Paulo, Brazil
- 3Division of Allergy and Immunology, Department of Pediatrics, Morsani College of Medicine, University of South Florida, Tampa, FL, United States
- 4Department of Biology, University of Tampa, Tampa, FL, United States
- 5School of Molecular and Cellular Biology, University of Leeds, Leeds, United Kingdom
- 6Department of Clinical Immunology and Allergy, Leeds Institute of Rheumatic and Musculoskeletal Medicine, St. James's University Hospital, Leeds, United Kingdom
- 7Johns Hopkins All Children's Hospital, St. Petersburg, FL, United States
- 8Massachusetts General Hospital, Boston, MA, United States
Proteins expressed by recombination activating genes 1 and 2 (RAG1/2) are essential in the process of V(D)J recombination that leads to generation of the T and B cell repertoires. Clinical and immunological phenotypes of patients with RAG deficiencies correlate well to the degree of impaired RAG activity and this has been expanding to variants of combined immunodeficiency (CID) or even milder antibody deficiency syndromes. Pathogenic variants that severely impair recombinase activity of RAG1/2 determine a severe combined immunodeficiency (SCID) phenotype, whereas hypomorphic variants result in leaky (partial) SCID and other immunodeficiencies. We report a patient with novel pathogenic compound heterozygous RAG2 variants that result in a CID phenotype with two distinctive characteristics: late-onset progressive hypogammaglobulinemia and highly elevated B cell count. In addition, the patient had early onset of infections, T cell lymphopenia and expansion of lymphocytes after exposure to herpes family viruses. This case highlights the importance of considering pathogenic RAG variants among patients with preserved B cell count and CID phenotype.
Introduction
RAG1 and RAG2 proteins combine to form a heterotetrameric complex which acts as an endonuclease. The RAG recombinase complex performs the essential first step in recombining variable (V), diversity (D), and joining (J) segments of antigen receptor genes that generate the diversity of T and B cell receptors (TCR, BCR) (1). TCR and BCR are important not only for the function of antigen recognition by T and B cells, but also for their development and survival. Variants in RAG1 and RAG2 are associated with many different immunodeficiencies including T−B−NK+ SCID, Omenn syndrome (OS), leaky SCID (LS) with γδ T cell expansion, and combined immunodeficiency with granulomas and/or autoimmunity (CID-G/AI) (2–5). Increasing access to genetic studies of immunodeficient patients is expanding the spectrum of phenotypes attributed to RAG deficiency. More recently, hypomorphic RAG variants were identified in patients with CD4+ T cell lymphopenia, and phenotypes resembling common variable immunodeficiency (CVID) and specific anti-polysaccharide antibody deficiency affecting all ages (6–10). In this report, we present a patient with CID associated with novel compound heterozygous RAG2 variants. The clinical and immune phenotype includes early onset of infections, T cell lymphopenia, normal B cell count, late onset progressive hypogammaglobulinemia, and evolving expansion of T and B lymphocytes with exposure to herpes family viruses.
Case Presentation
A 14-year-old female with chronic rhinosinusitis and lung disease with bronchiectasis was referred for immunologic investigation in São Paulo, Brazil. She had a history of chronic cough with recurrent wheezing since birth with prolonged use of antimicrobials for lower and upper respiratory tract infections, oral candidiasis and stomatitis. She had one episode of pneumonia and she was never hospitalized. She is an offspring of non-consanguineous parents. One of her sisters died with leukemia at the age of 9 months, and her mother experienced recurrent pneumonias and otitis media in childhood.
At 8.5 years of age, pulmonary symptoms worsened, bronchiectasis was detected on computed tomography and pulmonary function assessment showed mild obstructive lung disease. Cystic fibrosis and ciliary dyskinesia were excluded. She was treated with inhaled corticosteroids, azithromycin and chest physical therapy for 2 years with poor clinical response.
Immune evaluation was performed at several time points in the period of 8.5–14.3 years of age (Tables 1, 2). Total lymphocyte count was grossly preserved. Immunoglobulin (Ig) levels were variable with low IgA, low to normal IgG and low to high IgM initially. By 10 years of age, laboratory evaluation showed low levels of all Ig isotypes and low CD4+ and CD8+ T cells with low fraction of CD45RA+ naïve cells and skewing to activated memory T cell phenotype. Lymphocyte proliferation was normal with mitogens but impaired with antigen stimulation (Table 1). As Ig levels decreased, treatment with intravenous Ig (IVIG) was initiated at 11 years of age (Table 2). There was no evidence of protein loss. The B cell developmental subsets were significantly skewed with a marked decline in the switched memory B cell compartment (Table 3). B cell dysfunction is also reflected by decreased total IgG, IgM, and IgA levels with increased age (Table 2). Anti-thyroglobulin and anti-thyroperoxidase antibodies were persistently positive with normal thyroid function.
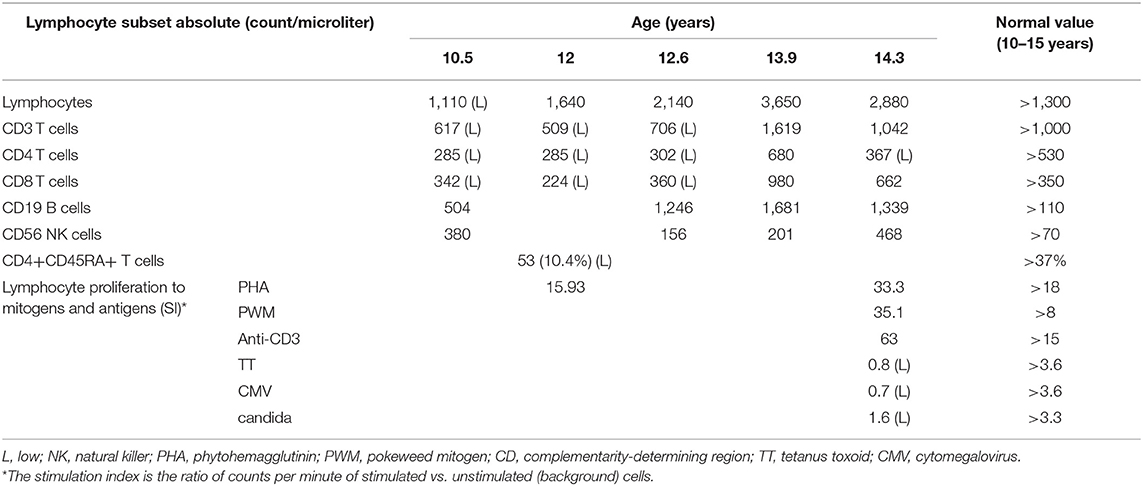
Table 1. Lymphocytes subsets profile (count/microliter) and lymphocyte proliferation to mitogens and antigens over 4 years [stimulation index (SI*)].
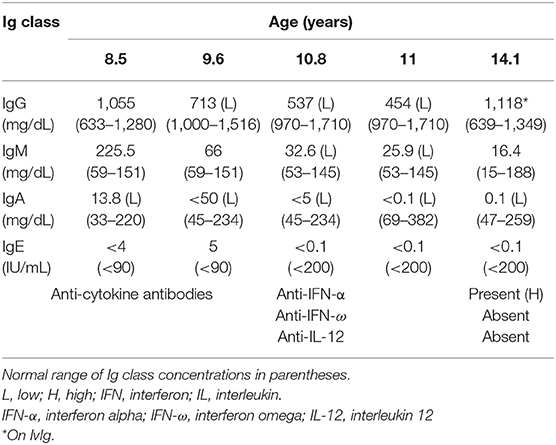
Table 2. Immunoglobulin (Ig) levels and anti-cytokine antibodies before and after IgG replacement therapy, which was begun at 11 years of age.
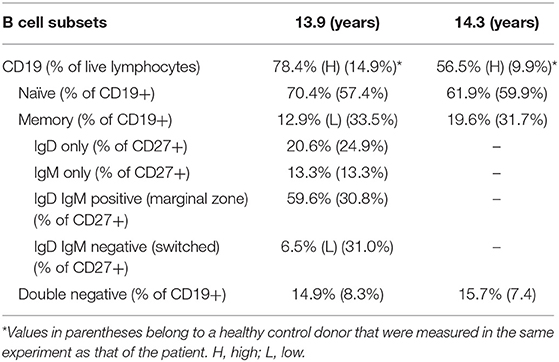
Table 3. B cells subset percentages at 13.9 and 14.3 years of age.
Regarding infectious complications, symptoms of respiratory infections improved on intravenous immunoglobulin G (IVIG) replacement therapy. However, recurrent candidiasis continued to occur as well as episodes of oral ulcers. At 13 years of age, she was hospitalized with bilateral pneumonia and stomatitis with positive polymerase chain reaction (PCR) for herpes simplex virus (HSV) on oral lesion biopsy. She responded well to intravenous antibiotics and acyclovir. Persistently, Epstein-Barr virus (EBV) and intermittently, cytomegalovirus (CMV) viral loads were detected without obvious clinical manifestations of lymphoproliferation or acute viral distress since 11.8 years for EBV and 13.4 years of age for CMV (Figure 1). A progressive increase in CD8+ T cells, B cells (CD19+) and natural killer (NK) cells (CD16+CD56+) was noticed in the same period (Figure 1). Testing at 14 years of age revealed the presence of antibodies targeting interferon-alpha (anti-IFN-α) (Figure 1).
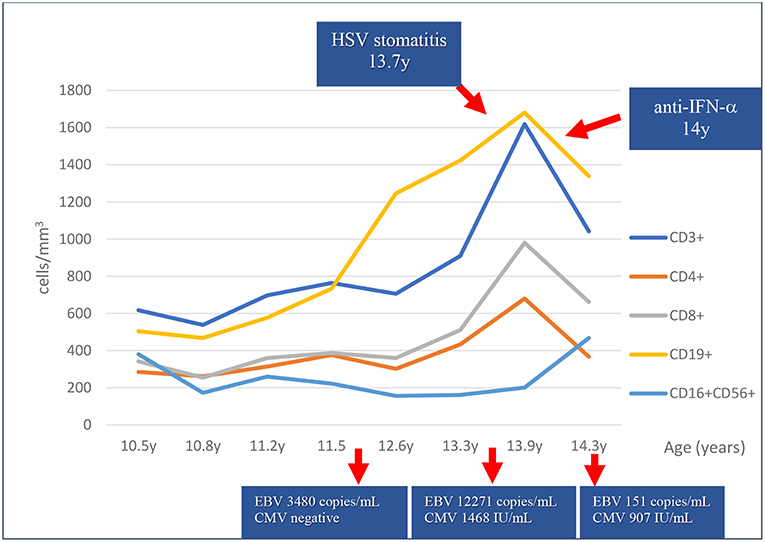
Figure 1. Lymphocyte subset counts and viral copy numbers over 4 years. An increase of T and B cell counts has been observed in the last 2.5 years coincidently with cytomegalovirus (CMV) and Epstein-Barr virus (EBV) viremia, and herpes simplex virus (HSV) stomatitis as shown by polymerase chain reaction. Anti-interferon-alpha antibodies (anti-IFN-α) were present at 14 years of age.
Genetic studies using targeted sequencing of 16 SCID genes identified a heterozygous compound variant in RAG2 (c.509A > G: p.E170G and c.829insT, p.Y277fs) in the coding regions. Both components of the compound variant were novel. The heterozygous variant was confirmed by Sanger sequencing. The c.509A > G, p.E170G variant was present in the mother. The father was unavailable for study.
Based on prediction analysis and structural modeling, it is expected that the mutated RAG2 allele with Y277fs will not contribute any recombinase activity as truncation of RAG2 further than amino acid 350 leads to a non-functioning protein that is unlikely to form any complex with RAG1 (11). Mutation in the RAG2 E170 is likely important in RAG1/2 dimerization as it contacts an arginine residue in RAG1 (R561). Mutation of RAG1 R561 to histidine has been previously reported in Omenn patients and shown to have reduced DNA binding and cleavage activity, but retains some activity in vitro (12). Therefore, we predicted that RAG2 E170G will perform all of the recombinase activity in the patient.
The relative recombinase activity of the RAG2 variants in vitro individually and in a bi-cistronic system was tested using methods previously reported (13). The relative recombinase activity level of the protein expressed by RAG2 p.E170G variant was 14.2% ± 1.6 standard error of mean (SEM) whereas the p.Y227fs variant had zero activity and their combined activity was 16.2% ± 2.5 SEM in an in vitro system (Figure 2). Thus, the RAG2 E170G variant solely contributed to the partial recombinase activity of the patient.
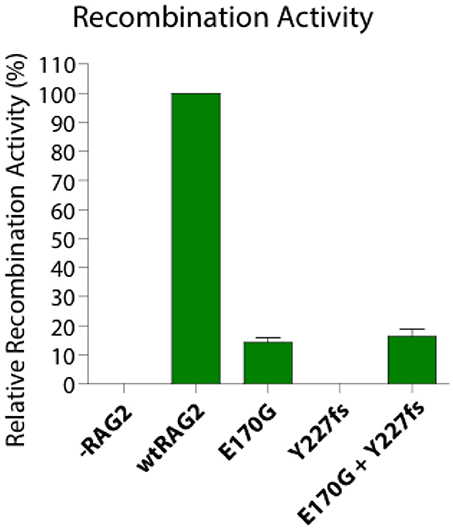
Figure 2. Recombination activity of two heterozygous RAG2 pathogenic variants (E170G, Y227fs) alone and in combination relative to the wild type allele (wtRAG2). -RAG2 is a negative control.
Although considered, the family elected to wait with hematopoietic stem cell transplantation. Regarding donor selection, there is no matched sibling available and we await results of human leukocyte antigen testing to search for a matched unrelated donor.
Methods
Targeted genes sequencing was performed using genomic DNA extracted from peripheral blood with Qiagen columns (QIAamp DNA Mini Kit; Qiagen, Hilden, Germany) according to the manufacturer's instructions. PCR was carried out by a set of primers designed using www.bioinformatics.nl/primer3plus for all genes (IL2RG:NM_000206, ADA:NM_000022, JAK3:NM_000215, ORAI1:NM_032790, RAG1:NM_000448, RAG2:NM_001243785, GATA2:NM_001145662, CD247:NM_000734, CD3D:NM_000732, CD3E:NM_000733, CD3G:NM_000073, LCK:NM_005356, NHEJ1:NM_024782, IL7R:NM_002185, LIG4:NM_206937, and PNP:NM_000270). The genes were sequenced using targeted sequencing performed with Nextera®XT DNA Library Preparation Kit (Illumina, San Diego, CA, USA) according to the manufacturer's instructions. Variants were identified using Platypus software (Welcome Centre for Human Genetics, Oxford, UK) and annotated using Annovar, an open source software tool. In order to predict the impact of these variants on the protein function, we employed in silico prediction tools: PhyloP, SIFT, PolyPhen-2, LRT and VariantTaster. The variants considered important were Sanger sequenced. PCR products were directly sequenced with BigDye Terminators (version 3.1) and analyzed on a Genetic Analyzer (Applied Biosystems. Foster City, CA, USA). B and T cells subset populations were determined by flow cytometry and analyzed using FlowJo software (Treestar, Ashland, OR, USA). Anti-cytokine antibodies were measured by enzyme-linked immunosorbent assay as previously described (4). Recombinase activity level was determined using bi-cistronic in vitro method performed in triplicate as described by Thwaites et al. (13).
Discussion
Impaired RAG function is associated with different clinical and immunological phenotypes and usually presents with variable degrees of B and T cell lymphopenia and antibody deficiency. The reported RAG deficient patient has a CID phenotype with two distinctive characteristics: late-onset progressive hypogammaglobulinemia and normal B cell count. The patient does not meet the criteria for LS or OS as mitogen proliferation are preserved, and has higher relative RAG activity [16% in combined variants (Figure 2)] compared to LS/OS patients. This is consistent with most CID patients having RAG activity ranging from 10 to 60% of wild type RAG activity (12, 14).
Late onset progressive hypogammaglobulinemia has been previously identified in patients with pathogenic RAG variants. Buchbinder et al. (8) reported two patients who not only had decreasing Ig levels but also a later onset of clinical symptoms, both of which contributed to the delay in correct diagnosis. Patients with late onset CID (LOCID) may be misdiagnosed with CVID due to similarities in clinical and immunologic characteristics (8). Nevertheless, patients with LOCID usually present with lower T cell counts and a marked decrease in CD4+ T cells in particular naïve T cells, compared to CVID patients (15). The findings that led to the hypothesis of CID in our patient and motivated the search for the underlying genetic defect were early onset of recurrent bacterial, viral and fungal infections associated with marked reduction of CD4+ and CD45RA naïve T cells with skewing to activated memory phenotype.
Another interesting immunologic finding in this patient is the presence of a normal B cell count. Due to the importance of the recombinase activity of RAG1/2 for B cell development and repertoire diversity, RAG variants are usually associated with marked B cell lymphopenia. However, normal B cell count has been found in other young patients with RAG variants, including patients with CID, ages 2.5 months and 2 years, respectively (16, 17). In both cases, the patients had active herpes virus infections (CMV and varicella specifically). The oldest RAG deficient patient reported with normal B cell count has been recently published by our group [Patient #12 in Lawless et al. (10)]. This 64 year-old female patient with RAG1 compound heterozygous variants (M435V and M1006V) and relative recombinase activity of 28%, had a normal B cell count of 216 (cells/μl) (10% of total lymphocytes). This patient had no history of major infections and had a sole autoimmune manifestation of pernicious anemia. She is currently on Ig replacement therapy with no inflammatory lung complications.
The B cell count may be preserved because of the lymphoproliferation (17). Kuijpers et al. (6) found a disturbed B cell population distribution in a RAG deficient patient with CD4+ T cell lymphopenia; an extensive antigen-independent proliferation of the transitional, naïve and non-switched B cells apparently accounted for the normal B cell count in peripheral blood. Our patient shows similar phenomena in B cell developmental abnormalities.
Expansion of gamma-delta T lymphocytes driven by severe herpes family viral infection have been described in LS patients with RAG variants (18, 19). Interestingly, we noticed that our patient presented a marked increase in both B cell and CD8+ T cells counts and a lesser increase in CD4+ T cells during herpes family virus asymptomatic viremia (CMV and EBV) and stomatitis (HSV) (Table 1, Figure 1). The reduction of EBV and CMV copies in peripheral blood and the resolution of the stomatitis were followed by a reduction in B and T cell counts.
Late onset progressive hypogammaglobulinemia is less common in combined immunodeficiencies that present early in life with profound antibody deficiency. However, key feature of late onset progressive hypogammaglobulinemia occurs among patients with IKAROS deficiency (20). Large studies have not been published on naïve IgG levels of RAG deficient patients in different age groups as most published patients receive Ig replacement therapy early in life. Therefore, it is yet to be determined how large is the subgroup with late onset progressive hypogammaglobulinemia.
Indication of hematopoietic stem cell transplantation (HSCT) are unclear for late onset CID and RAG deficiency. The patient is clinically well on Ig replacement therapy. There is still concern for progressive inflammatory lung disease, or occurrence of autoimmune complication, as reported by our group (10). Only a selected number of adolescent and adults with partial RAG deficiency received HSCT with variable success (21, 22). Allogeneic HSCT is a viable option for our patient; we are discussing with the family whether it is worthwhile given the patient's good response to current treatment vs. the risk of potential inflammatory and/or autoimmune sequela. With recent advances in gene therapy, our patient may be a candidate for this approach as well. The compound heterozygous variant, however, may create another layer of complexity in this process.
In conclusion, we found a novel compound heterozygous RAG2 variants that results in 16% relative recombinase activity and CID phenotype with normal B cell count but skewed B cells subsets and late-onset hypogammaglobulinemia. RAG deficiency may present with many different phenotypes and should be considered in patients with early-onset infections. This is especially true with T cell lymphopenia and low naïve T cell counts, even with normal B cell counts and late-onset hypogammaglobulinemia as in our case study. The immune phenotype can be modified by herpes viral infection including skewed B cell expansion and generation of anti-cytokine antibodies in temporal association of chronic herpes viral infections as seen in our case and previous reports (4, 17). Ig levels can decrease with time and should be monitored for early intervention with Ig therapy. Identifying the underlying genetic defect can greatly facilitate diagnosis of new phenotypes and enable effective therapy, including HSCT.
Ethics Statement
This study was performed in accord with the recommendations of the ethics committee of Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo. The mother gave written informed consent for the laboratory investigations in this case report and for publication of the study.
Author Contributions
MD, PB, and JW wrote the manuscript. MD, PB performed clinical and laboratory data collection and analysis. MD followed the patient. AR-S performed genetic studies. KC and BU performed B cell and anti-cytokine antibody studies. JW contributed to the interpretation of genetic data. DT performed and JB and SS gave advice on recombinase activity. JD and JW provided a critical revision of the article, and JW gave final approval of the version to be published.
Funding
This work is partly funded by the National Institute of Allergy and Infectious Diseases, National Institutes of Health [grant no. 5K08AI103035 and R01 AI100887-05 (sub) to JW], the Jeffrey Modell Foundation (to JW) and Robert A. Good Endowment at the University of South Florida (JW).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Laboratory of Medical Investigation (LIM 36), Department of Pediatrics, Hospital das Clínicas, Faculdade de Medicina da Universidade de São Paulo, SP, Brazil.
References
1. Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. (2016) 16:234–46. doi: 10.1038/nri.2016.28
2. Niehues T, Perez-Becker R, Schuetz C. More than just SCID—the phenotypic range of combined immunodeficiencies associated with variants in the recombinase activating genes (RAG) 1 and 2. Clin Immunol. (2010) 135:183–92. doi: 10.1016/j.clim.2010.01.013
3. Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG variants and granulomas. N Engl J Med. (2008) 358:2030–8. doi: 10.1056/NEJMoa073966
4. Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest. (2015) 125:4135–48. doi: 10.1172/JCI80477
5. De Ravin SS, Cowen EW, Zarember KA, Whiting-Theobald NL, Kuhns DB, Sandler NG, et al. Hypomorphic Rag variants can cause destructive midline granulomatous disease. Blood. (2008) 116:1263–71. doi: 10.1182/blood-2010-02-267583
6. Kuijpers TW, Ijspeert H, van Leeuwen EM, Jansen MH, Hazenberg MD, Weijer KC, et al. Idiopathic CD4+ T lymphopenia without autoimmunity or granulomatous disease in the slipstream of RAG variants. Blood. (2011) 117:5892–6. doi: 10.1182/blood-2011-01-329052
7. Abolhassani H, Wang N, Aghamohammadi A, Rezaei N, Lee YN, Frugoni F, et al. A hypomorphic recombination-activating gene 1 (RAG1) variant resulting in a phenotype resembling common variable immunodeficiency. J Allergy Clin Immunol. (2014) 134:1375–80. doi: 10.1016/j.jaci.2014.04.042
8. Buchbinder D, Baker R, Lee YN, Ravell J, Zhang Y, McElwee J, et al. Identification of patients with RAG variants previously diagnosed with common variable immunodeficiency disorders. Clin Immunol. (2015) 35:119–24. doi: 10.1007/s10875-014-0121-5
9. Geier CB, Piller A, Linder A, Sauerwein KM, Eibl MM, Wolf HM, et al. Leaky RAG Deficiency in adult patients with impaired antibody production against bacterial polysaccharide antigens. PLoS ONE. (2015) 10:e0133220. doi: 10.1371/journal.pone.0133220
10. Lawless D, Geier CB, Farmer JR, Lango Allen H, Thwaites D, Atschekzei F, et al. Prevalence and clinical challenges among adults with primary immunodeficiency and recombination-activating gene deficiency. J Allergy Clin Immunol. (2018) 141:2303–6. doi: 10.1016/j.jaci.2018.02.007
11. Coussens MA, Wendland RL, Deriano L, Lindsay CR, Arnal SM, Roth DB. RAG2's acidic hinge restricts repair-pathway choice and promotes genomic stability. Cell Rep. (2013) 4:870–8. doi: 10.1016/j.celrep.2013.07.041
12. Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol. (2014) 133:1099–108. doi: 10.1016/j.jaci.2013.10.007
13. Thwaites DT, Carter C, Lawless D, Savic S, Boyes JM. A novel RAG1 mutation reveals a critical in vivo role for HMGB1/2 during V(D)J recombination. Blood. (2018) 133:820–9. doi: 10.1182/blood-2018-07-866939
14. Lee YN, Frugoni F, Dobbs K, Tirosh I, Du L, Ververs FA, et al. Characterization of T and B cell repertoire diversity in patients with RAG deficiency. Sci Immunol. (2016) 1:eaah6109. doi: 10.1126/sciimmunol.aah6109
15. Malphettes M, Gérard L, Carmagnat M, Mouillot G, Vince N, Boutboul D, et al. DEFI study group. Late-onset combined immune deficiency: a subset of common variable immunodeficiency with severe T cell defect. Clin Infect Dis. (2009) 49:1329–38. doi: 10.1086/606059
16. Ohm-Laursen L, Nielsen C, Fisker N, Lillevang ST, Barington T. Lack of nonfunctional B-cell receptor rearrangements in a patient with normal B cell numbers despite partial RAG1 deficiency and atypical SCID/Omenn syndrome. J Clin Immunol. (2008) 28:588–92. doi: 10.1007/s10875-008-9210-7
17. Goda V, Malik A, Kalmar T, Maroti Z, Patel B, Ujhazi B, et al. Partial RAG deficiency in a patient with varicella infection, autoimmune cytopenia, and anticytokine antibodies. J Allergy Clin Immunol Pract. (2018) 6:1769–71.e2. doi: 10.1016/j.jaip.2018.01.015
18. de Villartay JP, Lim A, Al-Mousa H, Dupont S, Déchanet-Merville J, Coumau-Gatbois E, et al. A novel immunodeficiency associated with hypomorphic RAG1 variants and CMV infection. J Clin Invest. (2005) 115:3291–9. doi: 10.1172/JCI25178
19. Ehl S, Schwarz K, Enders A, Duffner U, Pannicke U, Kühr J, et al. A variant of SCID with specific immune responses and predominance of gamma delta T cells. J Clin Invest. (2005) 115:3140–8. doi: 10.1172/JCI25221
20. Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, et al. Loss of B cells in patients with heterozygous mutations in IKAROS. N Engl J Med. (2016) 374:1032–43. doi: 10.1056/NEJMoa1512234
21. John T, Walter JE, Schuetz C, Chen K, Abraham RS, Bonfim C, et al. Unrelated hematopoietic cell transplantation in patient with combined immunodeficiency with granulomatous disease and autoimmunity secondary to RAG deficiency. J Clin Immunol. (2016). 36:725–32. doi: 10.1007/s10875-016-0326-x
Keywords: RAG deficiency, RAG2, combined immunodeficiency, primary immunodeficiency, hypomorphic variant, compound heterozygous variant
Citation: Dorna MB, Barbosa PFA, Rangel-Santos A, Csomos K, Ujhazi B, Dasso JF, Thwaites D, Boyes J, Savic S and Walter JE (2019) Combined Immunodeficiency With Late-Onset Progressive Hypogammaglobulinemia and Normal B Cell Count in a Patient With RAG2 Deficiency. Front. Pediatr. 7:122. doi: 10.3389/fped.2019.00122
Received: 23 January 2019; Accepted: 12 March 2019;
Published: 16 April 2019.
Edited by:
Ivan K. Chinn, Baylor College of Medicine, United StatesReviewed by:
Andrew R. Gennery, Newcastle University, United KingdomOskar A. Haas, St. Anna Children's Cancer Research Institute (CCRI), Austria
Copyright © 2019 Dorna, Barbosa, Rangel-Santos, Csomos, Ujhazi, Dasso, Thwaites, Boyes, Savic and Walter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mayra B. Dorna, bWF5cmFkb3JuYUB1b2wuY29tLmJy
Jolan E. Walter, am9sYW53YWx0ZXJAaGVhbHRoLnVzZi5lZHU=