 Anja Schnabel
Anja Schnabel Christian M. Hedrich
Christian M. Hedrich
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr. , 10 January 2019
Sec. Pediatric Immunology
Volume 6 - 2018 | https://doi.org/10.3389/fped.2018.00421
This article is part of the Research Topic Childhood Vasculitis View all 19 articles
The term vasculitis covers heterogeneous disorders that share the presence of inflammation of blood vessel walls. Immune cell infiltrates can vary significantly and involve granulocytes or mononuclear cells. Vasculitis can be a symptom of other underlying disorders or the underlying cause of organ specific or systemic disease. Classification of childhood vasculitis is based on clinic, the size of predominantly affected vessels, and the histopathology of inflammatory infiltrates. Timely and accurate diagnosis and (where necessary) treatment initiation determine disease progression and outcomes. In light of new developments and the identification of autoinflammatory conditions with vasculitis, new classification tools may be discussed.
Vasculitis is an umbrella term for various and heterogeneous disorders sharing the presence of inflammation of blood vessel walls. Immune cell infiltrates can vary significantly and involve granulocytes (neutrophils, eosinophils) or mononuclear cells (monocytes/macrophages, lymphocytes). Vasculitis can be a symptom of other underlying disorders (secondary vasculitis: infections, medication intake, malignancies, autoimmune/inflammatory conditions, etc.) or the cause of organ specific or systemic disease. In the latter case, the term “primary vasculitis” is used (1).
It has been an ongoing challenge to classify childhood vasculitis, which is usually based on clinical phenotypes (e.g., single organ vs. systemic vasculitis), the size of predominantly affected vessels (small/medium/large), and the histopathology of inflammatory infiltrates (e.g., granulomatous vs. non-granulomatous). The most commonly applied classifications are the updated Chapel Hill consensus conference criteria for systemic vasculitis (2012) (2), which have not been developed specifically for children, and the EULAR (European League against Rheumatism)/PRES (Pediatric Rheumatology European Society)/PRINTO (Pediatric Rheumatology International Trials Organization) classification criteria for childhood vasculitis (3–5). Both sets of criteria are largely identical for primary systemic vasculitis (Figure 1) (with the exception for giant cell arteritis), but show some differences for the classification of secondary forms (Table 1) and vasculitis limited to single organ systems.
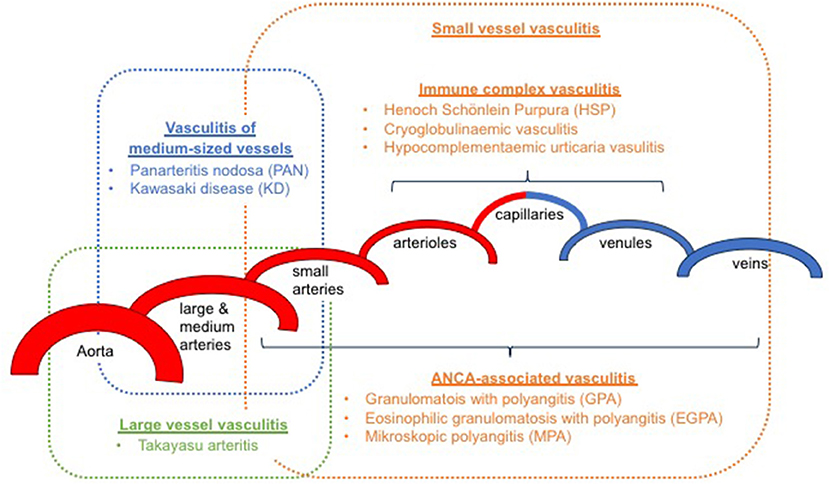
Figure 1. Classification of primary childhood vasculitis based on revised Chapel Hill criteria (2012) and the EULAR/PRES classification (2–5).
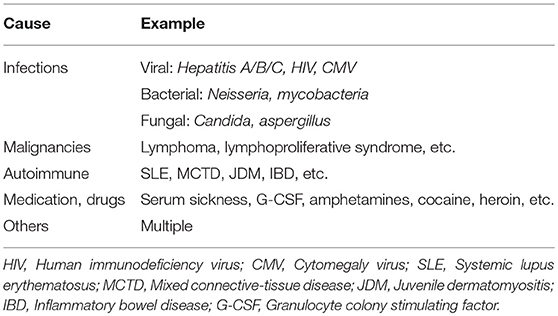
Table 1. Selection of underlying causes in secondary vasculitis.
This manuscript provides an overview on current classification of childhood vasculitis, focusing on primary systemic vasculitis. We deliver a brief summary on clinical signs, basic pathomechanisms involved and treatment options. More comprehensive discussion of individual forms of vasculitis, their molecular pathophysiology and treatment available is discussed in focused manuscripts of this special topic.
Large vessel vasculitis (LVV) mainly affects large arteries and includes giant cell arteritis (2). In childhood, primary LVV is limited to Takayasu arteritis (TA). Temporal giant cell arteritis, that predominantly involves the temporal artery, carotid and/or vertebral arteries, does not occur in children (1, 3, 5). Takayasu arteritis (TA) is characterized by granulomatous vasculitis of the aorta and/or its main branches, but also coronary and/or pulmonary arteries (6, 7). While TA usually manifests in the third or fourth decade, children (including infants) and adolescents can develop disease. The exact incidence and prevalence of TA in childhood is not known and variation has been reported between geographical regions for adults (UK: 0.8/million vs. Asia 2/million; likely 10–20% of who are children) (8, 9). Girls and young women are more frequently affected when compared to boys (3:1) (10). The molecular pathophysiology of TA remains unclear, and genetic factors, humoral, and cellular autoimmunological factors, as well as infections have been discussed as contributors. Chronic inflammation results in scarring and stenosis of large vessels (6, 7).
Clinical presentation of TA is highly variable, ranging from mild symptoms (e.g., arterial hypertension and/or weak peripheral pulses) to cardiac and/or respiratory failure, and further organ involvement (CNS, gastrointestinal system, etc.). Disease stages prior to the development of key clinical symptoms, such as “pulselessness,” are characterized by non-specific symptoms associated with systemic inflammation and include fever, night sweats, malaise, arthralgia, and myalgia. As inflammation progresses, vascular stenosis develop and may result in intermittent claudication, vascular bruits and/or hypertension (11).
Diagnosis is usually based on EULAR/PRINTO/PRES classification criteria (Table 2) and informed by imaging (usually MRI angiography; Figure 2). Biopsies may be helpful in unclear cases (10, 12, 13).
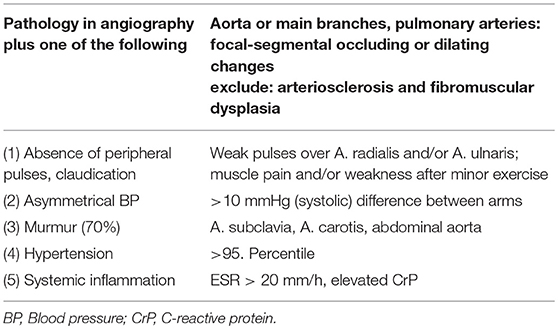
Table 2. EULAR/PRINTO/PRES criteria for TA.
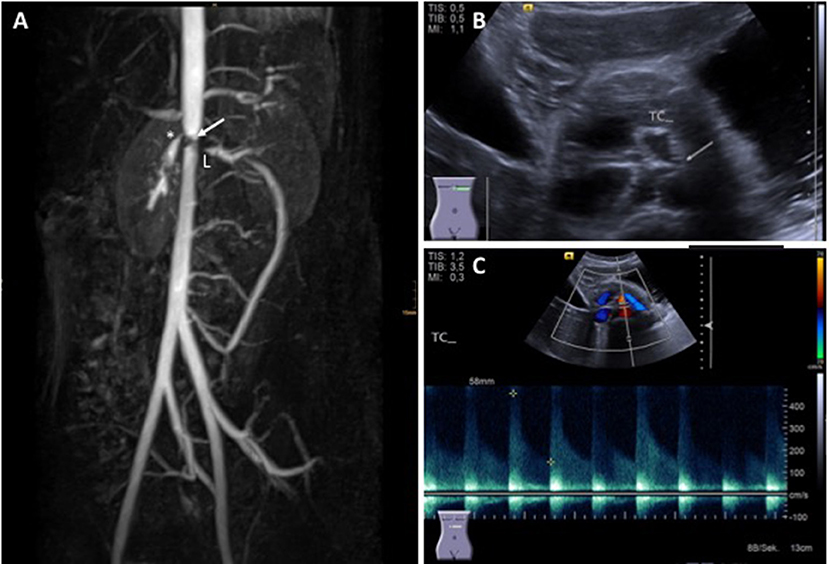
Figure 2. Takayasu arteritis in a 12-year-old girl. (A) MRI angiography showing significant stenosis of the coeliac truncus (*) and lower aorta (→); artifacts are caused by stent implants in renal arteries (L) (provided by Dr. Gabriele Hahn, Pädiatrische Radiologie, Universitätsklinikum Carl Gustav Carus, TU Dresden), (B) transversal abdominal ultrasonography indicating significant stenosis of the coeliac truncus, (C) Stenosis of coeliac truncus resulting in pathologically increased arterial blood flow velocity (provided by Dr. Heike Taut, Klinik und Poliklinik für Kinder- und Jugendmedizin, Universitätsklinikum Carl Gustav Carus, TU Dresden). Consent of the patient was obtained to publish these images.
Treatment is guided by organ involvement and pre-existing damage and is currently not standardized (14). Pharmaceutical treatment involves corticosteroids as first-line induction treatment (15), disease-modifying antirheumatic drugs methotrexate (MTX), azathioprine (AZA), cyclosporine A (CsA), and mycophenolate mofetil (MMF), TNF inhibitors (infliximab/IFX, adalimumab/ADA, etanercept/ETA), IL-6 inhibitors (Tocilizumab), and B cell depleting agents (Rituximab/RTX) (14, 16–19). Furthermore, Cyclophosphamide (CPM) can be discussed but should be reserved for otherwise treatment refractory or acutely life-threatening cases. If hemodynamically relevant vessel stenosis occur, interventional or surgical interventions may be necessary, but should be avoided if possible, since they are discussed to promote inflammation (14).
This group includes two forms of primary childhood vasculitis: Kawasaki disease and polyarteritis nodosa.
Kawasaki disease (KD) is a systemic necrotizing vasculitis of small and medium-sized vessels (20). A relatively common and severe complication is the involvement of coronary arteries that can result in aneurysms and cardiac infarction. KD is a rare condition in adults. More than 90% of KD cases affect children and infants (21). Globally, KD is the most common primary childhood vasculitis (Japan 239/100,000 children under 5 years), in predominantly Caucasian populations (9/100,000 children under 5 years), Henoch Schönlein purpura (HSP) is more common (see below). The molecular pathophysiology of KD is unclear (22–24). Genetic predisposition (BLK, CASP3, CD40, FCGR2A, HLA class II, IPTKC), infectious triggers, super antigens, humoral factors and immune complexes have been suggested to contribute. In the acute phase of KD, monocytes/macrophages and T cells produce pro-inflammatory mediators that result in endothelial inflammation and the clinical picture of KD (22, 23).
Most (75%) of patients develop KD before their 5th birthday. By definition, fever without focus is present in all KD patients. In children under 5 years-of-age, KD should be considered in the case of unexplained fevers over 4–5 days (22, 23, 25). Infants are less frequently but more severely (increased risk for aneurysm development) affected and tend to exhibit “incomplete” clinical pictures that can be challenging to diagnose (26). The term “atypical KD” is reserved for children who fulfill diagnostic criteria for KD, but experience a not typical disease course, which may, amongst other symptoms include exudative pharyngitis and/or conjunctivitis, aseptic meningitis, arthritis, and/or anterior uveitis. Without timely establishment of treatment, 15–25% of KD patients will develop coronary aneurysms, which define prognosis and long-term outcomes (27). Diagnosis is based on criteria summarized in Table 3 and Figure 3 (28, 29).

Table 3. Clinical criteria for the diagnosis of “classical” KD (22).
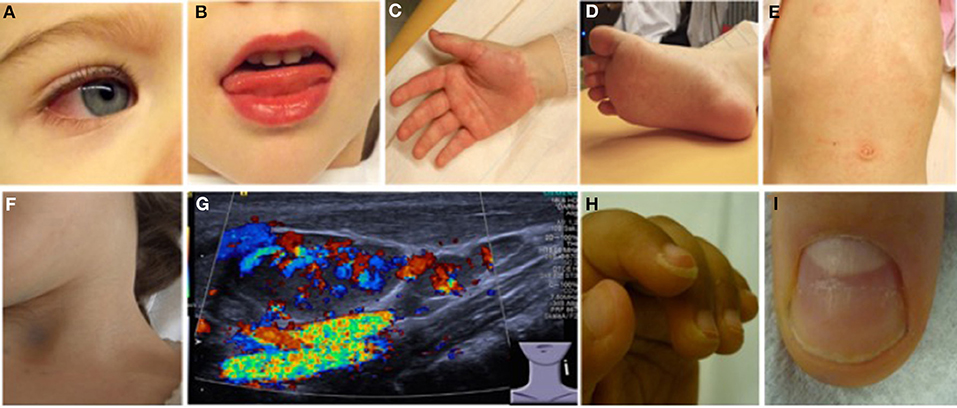
Figure 3. Clinical criteria in KD. (A) Bilateral non-purulent conjunctivitis (80–90%), (B) changes to oropharyngeal mucous membranes, including injected and/or fissured lips, strawberry tongue (80–90%), (C) Palmar, and/or (D) plantar erythema (E) polymorphous exanthema, primarily truncal, not vesicular (>90%), and (F,G) cervical lymphadenopathy (>1.5 cm) (50%). (G) Ultrasound of enlarged cervical lymph nodes with increased perfusion (provided by Dr. Heike Taut, Klinik und Poliklinik für Kinder- und Jugendmedizin, Universitätsklinikum Carl Gustav Carus, TU Dresden). (H) Periungual desquamation (in covalescent phase) (80%), (I) Beau lines; images from (22).
Laboratory findings reflect systemic inflammation and include elevated CRP (≥30 mg/l) and ESR (>40/h), elevated liver enzymes (ALT ≥50 U/l), hypoproteinemia, elevated cholestasis parameters, thrombocytosis, leukocytosis, and/or anemia (22).
Early and aggressive treatment of KD aims at symptom alleviation and the prevention of vasculitis-associated damage (such as aneurysms). Standard treatment includes salicylic acid (initially during febrile period 30–50(−80) mg/kg/day, followed by 3–5 mg/kg/day for 6–8 weeks), and intravenous immunoglobulins (IVIG; usually 2 g/kg over 8–12 h). IVIG should be applied within the first 7–10 days of fever to reduce the risk for coronary aneurysms. Concomitant treatment with prednisolone should be considered in KD patients with a high-risk for the development of coronary aneurysms or individuals who do not respond to a first course of IVIG. In otherwise treatment refractory cases, cytokine-blocking strategies and other have been discussed (22, 23, 27, 28, 30). Evidence for the usefulness of risk scores exists in Asian populations, but their value in predominantly Caucasian populations remains unclear (22, 31–33).
Panarteritis nodosa (PAN) is a necrotizing systemic vasculitis of medium-sized vessels (3). Globally, after KD and HSP, PAN is the third most common childhood primary vasculitis (34). Recently, a rare disorder caused by loss-of-function mutations in the ADA2 (or formerly CECR1) gene encoding for adenosine deaminase 2 (ADA2) has been described and accounts for a subset of particularly early-onset cases of PAN (Deficiency of ADA2: DADA2) (35–37). The molecular pathophysiology of remaining PAN cases is currently unclear. Interestingly, increased incidence and prevalence was recorded in individuals who experience Familial Mediterranean Fever (FMF), one of the most common autoinflammatory disorders in childhood (38). The result of transmural vessel inflammation is scarring and (name giving) nodular changes to vessel walls and aneurysm formation (24). For the diagnosis of PAN, classical angiography and (at least in some patients) angio-MRI can be applied to demonstrate vasal microaneurysms in affected organs (3, 39).
Though generally any organ system can be involved, most patients develop vasculitis of the skin, musculoskeletal system, kidneys and/or gastrointestinal system. Less frequently, the heart and peripheral and/or central nervous system can be affected. Initially, non-specific general symptoms, including fever and malaise are common (5, 34). Diagnosis can be made bases on EULAR/PRINTO/PRES criteria (Table 4) (3).

Table 4. EULAR/PRINTO/PRES criteria for PAN (3).
Because of the rarity of childhood PAN, widely accepted and evidence-based treatment recommendations do not exist. Treatment is usually informed by adult rheumatology and includes corticosteroids, IVIG, DMARDS for maintenance treatment, and/or CPM for severe cases (40, 41). Recently, anti-TNF agents have been demonstrated effective at least in a subset of DADA2 patients (35).
Small vessel vasculitis (SVV) predominantly affects parenchymal arteries, arterioles, capillaries, and venules. Medium-sized arteries and veins may also be affected. This group can be subdivided into immune complex mediated and ANCA-associated vasculitis.
While Henoch Schönlein purpura (HSP) is the most common form of primary childhood vasculitis in Europe and North America, it is significantly less common in adults (3–14 cases per million) (42). Exact numbers on prevalence and incidence do not exist secondary to very variable presentations and resulting variability in medical needs and providers involved.
Henoch Schönlein purpura (HSP) is an immune complex vasculitis with (mainly) IgA containing immune depositions in small vessels. It can generally affect all age groups (20/100,000/year), but is most common in 4–6 year-old children (70/100,000/year). Boys are slightly more frequently affected as compared to girls (1.2–2:1) (43). While the exact pathophysiology of HSP is unknown, an association with previous infections within the past 2–4 weeks has been recorded (44). As the result of an unknown trigger mechanism, IgA containing immune complexes deposit in small vessels and cause complement activation, immune cell invasion, endothelial activation, and lastly vasculitis (Figure 4).
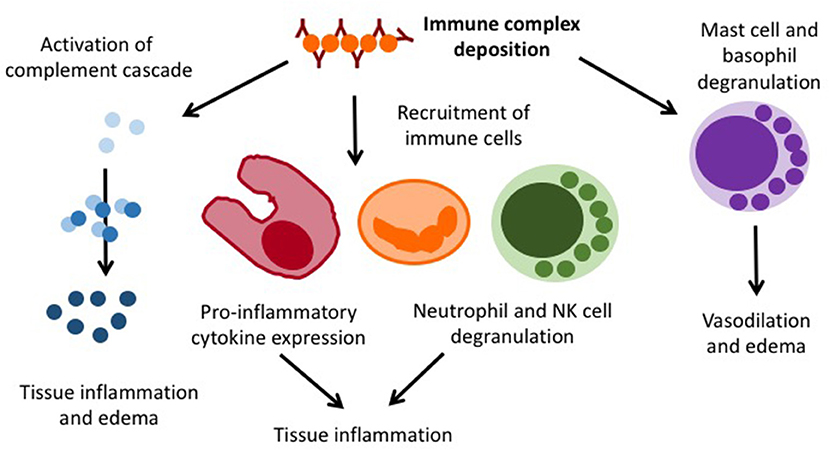
Figure 4. Pro-inflammatory mechanisms in immune complex vasculitis.
Henoch Schönlein purpura (HSP) is a diagnosis of exclusion and no specific diagnostic tests are available. Secondary immune complex vasculitis and thrombocytopenic purpura require to be excluded. To diagnose HSP, EULAR/PRINO/PRES classification criteria are commonly used (Table 5) (3). Clinical presentation is variable and, in addition to palpable purpura, includes relatively common symptoms of abdominal pain, arthritis or nephritis, as well as additional organ involvement and associated complications (Table 6). Laboratory findings are unspecific and include (usually) mildly elevated inflammatory parameters (ESR and CrP), normal or slightly reduced serum C3 and/or C4, the absence of high-titer autoantibodies (particularly ANCA and ANA) and, in 50% of all cases, elevated serum IgA and/or IgM (46). Fecal occult blood tests and urinary specimens are recommended at diagnosis. Since glomerulonephritis can manifest after the initial phase, regular urinary analysis at least for the first 6 months after disease-onset are recommended.
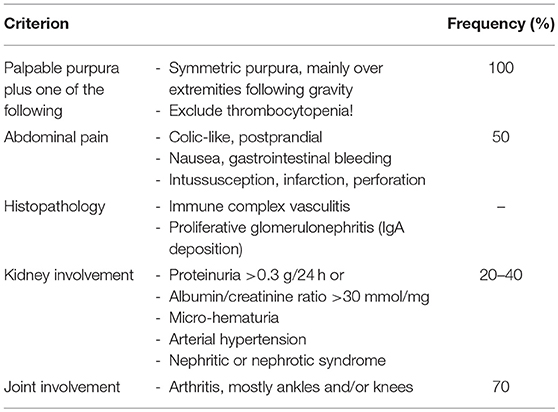
Table 5. EULAR/PRINTO/PRES criteria for HSP (3).

Table 6. Non-cutaneous complications in HSP (45).
Treatment depends on clinical presentation and organ involvement. Non-steroidal anti-inflammatory drugs (NSAIDs) or acetaminophen/paracetamol can be considered for analgesia in cases with arthritis or arthralgia. For gastrointestinal involvement treatment with corticosteroids (usually 1–2 mg/kg/day for 1 week, followed by taper over 2–3 weeks) can be considered. However, intestinal perforations have been seen under treatment (47, 48). Substitution of factor XIII can be discussed for gastrointestinal bleedings. Of note, studies indicate that early corticosteroids are not effective in preventing HSP-associated nephritis (45, 49). Evidence for treatment is weak, and expert recommendations have been provided e.g., by the German Society for Pediatric Nephrology (GPN) (Table 7) (50). Bed rest can be considered in severe cases.
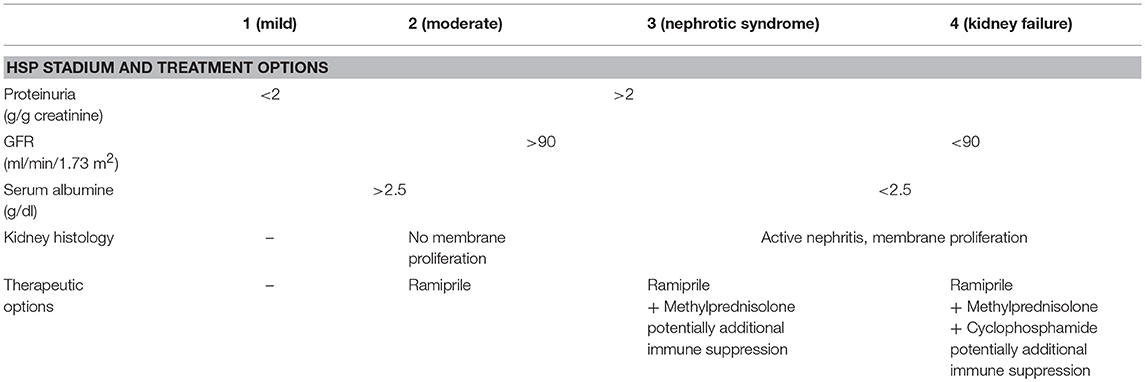
Table 7. Treatment recommendations for severe HSP nephritis (50).
The group of ANCA-associated vasculitis includes granulomatosis with polyangitis (GPA, formerly Wegener's granulomatosis), eosinophilic granulomatosis with polyangitis (EGPA, formerly Churg-Strauss syndrome), and microscopic polyangitis (MPA). All of which are characterized by destructive vasculitis of small and medium-sized arterial vessels, the presence of ANCA antibodies, and multi-organ involvement (1, 3, 4).
Granulomatosis with polyangitis (GPA) is rare in children. Its incidence is estimated to be around 1/1,000,000. The mean age at disease-onset is around 14 years. General symptoms, including fevers, weight loss, and fatigue are present in over 90% of all patients. Diagnosis is usually made based on EULAR/PRINTO/PRES classification criteria (Table 8) (3), and additional symptoms can include arthritis, erythema, ulcerations, and gastrointestinal involvement (Figure 5) (51).
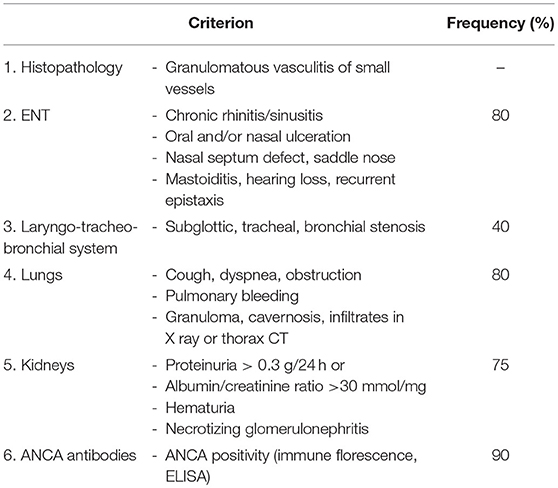
Table 8. EULAR/PRINTO/PRES criteria for GPA (3).
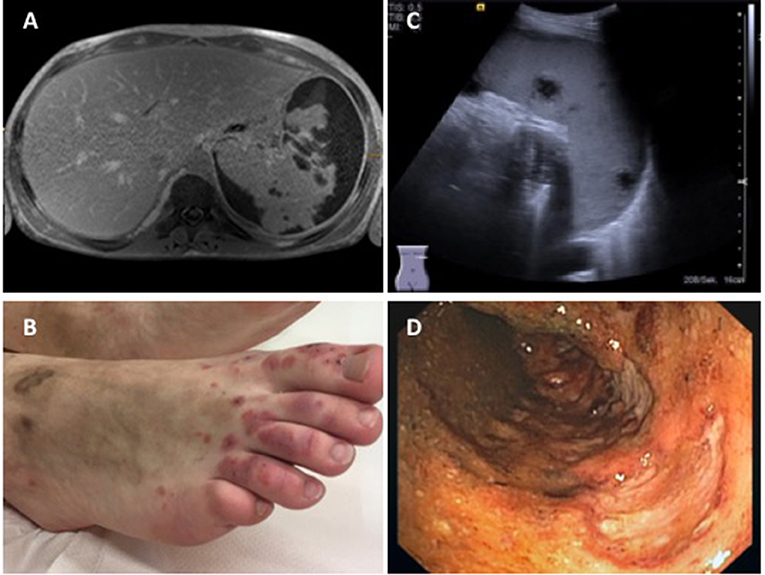
Figure 5. Findings in a 16-year-old boy with GPA. (A) Abdominal MRI showing pronounced parenchymatous necrosis of the spleen with distension of the spleen capsule (provided by Dr. Gabriele Hahn, Pädiatrische Radiologie, Universitätsklinikum Carl Gustav Carus, TU Dresden); (B) Necrotizing skin vasculitis on legs and feet; (C) Splenomegaly, spleen with increased echogenicity, subcapsular fluid accumulation, hypoechogenic necrotic areas with absence of perfusion (provided by Dr. Heike Taut, Klinik und Poliklinik für Kinder- und Jugendmedizin, Universitätsklinikum Carl Gustav Carus, TU Dresden); (D) Severe enterocolitis (shown: transversal colon) with deep ulcerations as a result of necrotizing vasculitis (provided by Dr. Martin Laass, Klinik und Poliklinik für Kinder- und Jugendmedizin, Universitätsklinikum Carl Gustav Carus, TU Dresden). Consent of the patient was obtained to publish these images.
Untreated, GPA has a mortality close to 100% within the first year, and regardless of treatment, up to 60% of patients experience subsequent disease flares. When compared to GPA in adults, children, and young people more frequently experience multi-organ involvement, kidney involvement, subglottic stenosis and nose deformities (51, 52).
Eosinophilic granulomatosis with polyangitis (EGPA) is a necrotizing vasculitis characterized by eosinophilic infiltrates in small and medium-sized vessels (53, 54). Reliable demographic information and/or classification criteria for the pediatric age-group do not exist, and diagnosis is informed by ACR criteria for adults with GPA (53). For classification as EGPA, four of six criteria must be fulfilled: (i) Asthma, (ii) Eosinophilia (peripheral blood) >10%, (iii) Mono- or polyneuropathy, (iv) pulmonary infiltrates, (v) para-nasal sinus anomalies, and (vi) biopsy with extra vascular eosinophilic infiltrates.
The exact pathophysiology of EGPA remains unclear. Inappropriate Th2 activation and IL-4, IL-5, and Il-13 expression play a central role and reflect parallels to allergic disease. Progressive immune dysregulation triggers a prodromal state that can last several years and include chronic sinusitis, allergies, and severe corticosteroid-dependent asthma. Later, sinusitis, pulmonary infiltrations and sometimes severe gastrointestinal, cardiac, skin manifestations (Livedo, painful subcutaneous lesions, infarctions, etc.), and peripheral neuropathy can be present (54, 55).
Microscopic polyangitis (MPA), in contrast to GPA and EGPA, is a non-granulomatous necrotizing vasculitis of small vessels (56). Immune complex deposition is limited or absent (pauci-immune vasculitis). Most patients develop pulmonary (but not upper respiratory tract) involvement with pulmonic hemorrhage and sometimes kidney involvement (27%). Additional organ systems, including the gastrointestinal system, CNS, skin, musculoskeletal system, and end eyes can be affected. Diagnostic or classification criteria for childhood MPA do not exist. Thus, Microscopic polyangitis (MPA) remains a diagnosis of exclusions and is informed by adult rheumatology. The presence of ANCA can be helpful (in adult cohorts: >50% pANCA, 40% cANCA positive) (56, 57).
Treatment of childhood ANCA-associated vasculitis is guided by disease severity. Generally, patients should receive more or less aggressive induction treatment to control inflammation (high-dose prednisolone or i.v. methylprednisolone pulses ± CPM) that is followed by maintenance treatment with more tolerable therapeutic agents (MTX, AZA, MMF) (58). Based on studies in adult cohorts, RTX can be discussed as alternative induction treatment in some cases and appears to be superior to CPM in GPA flares after initial induction treatment with CPM (59, 60). Trimetoprim/sufamethoxazole should be considered for pneumocystis prophylaxis. Furthermore, flare reduction has been described in patients with GPA and S. aureus colonization (61, 62). In devastating and treatment-refractory cases, autologous stem cell transplantation can be considered (63).
Several forms of vasculitis can affect vessels of variable types and diameters.
Behcet's disease (BD) is characterized by inflammatory lesions in vessel walls of all sizes, which may lead to endothelial damage, thrombosis, and aneurysms (64). Chronic recurrent oral and/or genital ulcers occur can be accompanied by additional cutaneous (erythema nodosum, cutaneous pustular vasculitis, etc.), ocular (posterior uveitis, retinal vasculitis), articular (non-erosive poly- or oligo-arthritis), gastrointestinal (abdominal pain, nausea, diarrhea, etc.), and/or central nervous symptoms (aseptic meningitis, vascular thrombosis) (1). Cases of BD can be seen across the globe and in all ethnicities (64). However, prevalence is highest in countries along the Silk Road, where it ranges between 77 and 100/100,000 individuals (0.1–15.9/100,000 in Western Europe) (65, 66). While most patients develop symptoms in young adulthood, 5–10% exhibit childhood-onset BD (67). The pathophysiology of BD is incompletely understood, but genetic associations are likely involved and may be influenced by environmental factors (13, 68, 69). HLA-B51/B5 allele carriers have considerably high risk for BD indicating a possible gene-dose effect (70). Diagnosis can be challenging, especially since children and young people frequently do not develop the full clinical picture of BD and progress over time (64, 71). More than 15 sets of classification or diagnostic criteria have been published (72). Based on clinical differences between age-groups, recently, pediatric classification criteria have been suggested (1, 73). Treatment of BD can be complex and should be informed by clinical symptoms and disease severity. Topical treatment (steroids and/or sucralfate) and systemic treatments (corticosteroids, colchicine, AZA, CsA, thalidomide, apremilast, TNF inhibitors, etc.) are discussed elsewhere (1, 64).
Cogan syndrome (CS) is characterized by predominantly large vessel vasculitis, but can affect any vessel size (1). CS is an extraordinarily rare multisystem inflammatory condition that can involve eyes (keratitis, uveitis, episcleritis) and inner ears (sensorineural deafness, vestibular dysfunction) (2, 74). Unspecific systemic symptoms occur in 50% of all patients, including arthralgia and manifestations of medium-size and small vessel vasculitis. To date, only few pediatric patients have been reported (75). Based on the rarity and lack of pathophysiological understanding of the disorder, data on effective treatments are lacking. Available reports favor DMARDs (AZA, MTX) in combination with TNF inhibitors (75).
Primary organ vasculitis covers a range of particularly rare disorders characterized by vasculitis of a single organ in the absence of signs indicative of systemic vasculitis (1). Various organ systems can be involved, including the CNS (primary large or small vessel CNS vasculitis) (76, 77), primary testicular vasculitis (78), cutaneous leukocytoclastic vasculitis (1), etc.
Autoinflammatory disorders are characterized by systemic or organ-specific inflammation that is (at least initially) caused by dysregulation of the innate immune system (79, 80). Vasculitis can be a feature seen with several autoinflammatory conditions. Indeed, in some autoinflammatory disorders, including aforementioned BD (1, 81), previously discussed DADA2 (35–37), primary type I interferonopathies STING-associated vasculopathy with onset in infancy (SAVI) (82) and Aicardi Goutières syndrome (83), and haploinsufficiency of H20 (HA20) (84), vasculitis can be the dominant feature. Autoinflammatory conditions are still relatively “new” to the field of Rheumatology and underlying pathomechanisms of systemic inflammation and/or vasculitis remain unclear in many cases. Thus, (with the exception of BD) vasculitis in the context of autoinflammatory disease is not part of currently available classifications for vasculitis, which will likely change in the years to come.
Vasculitis are rare conditions in children and young people that can be subdivided and classified based on clinical phenotypes (e.g., organ-specific vs. systemic) underlying causes (primary vs. secondary disease), histological patterns (granulomatous, non-granulomatous, necrotizing, etc.), and primarily affected vessel sizes (Chapel Hill and EULAR/PRES classifications: small/medium/large). Timely and accurate diagnosis and (where necessary) treatment initiation are essential, provided the variable severity and outcomes of individual forms of vasculitis. In light of new developments [including the identification of genetic causes, sometimes resulting in expansion of disease-associated phenotypes (e.g., DADA2)] and the identification of autoinflammatory conditions with vasculitis as key feature [including complex genetic BD, but also monogenic disease (DADA2, SAVI, HA20)], new classification tools may be justified in the near future.
AS and CH equally contributed to all stages of this manuscript, including conception and writing of the manuscript. AS and CH contributed to manuscript revision, read, and approved the submitted version.
CH's work is supported by the Fritz-Thyssen Foundation, Novartis Pharmaceuticals (research grant), the intramural MeDDrive Program of TU Dresden, LUPUS UK, and the FAIR charity.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Sag E, Batu ED, Ozen S. Childhood systemic vasculitis. Best Pract Res Clin Rheumatol. (2017) 31:558–75. doi: 10.1016/j.berh.2017.11.009
2. Jennette JC. Overview of the 2012 revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Clin Exp Nephrol. (2013) 17:603–6. doi: 10.1007/s10157-013-0869-6
3. Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, et al. EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: final classification criteria. Ann Rheum Dis. (2010) 69:798–806. doi: 10.1136/ard.2009.116657
4. Ozen S, Ruperto N, Dillon MJ, Bagga A, Barron K, Davin JC, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. (2006) 65:936–41. doi: 10.1136/ard.2005.046300
5. Ruperto N, Ozen S, Pistorio A, Dolezalova P, Brogan P, Cabral DA, et al. EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part I: overall methodology and clinical characterisation. Ann Rheum Dis. (2010) 69:790–7. doi: 10.1136/ard.2009.116624
6. Uppal SS, Verma S. Analysis of the clinical profile, autoimmune phenomena and T cell subsets (CD4 and CD8) in Takayasu's arteritis: a hospital-based study. Clin Exp Rheumatol. (2003) 21(6 Suppl. 32):S112–6. Available online at: http://www.clinexprheumatol.org/article.asp?a=2260&origin=publication_detail
7. Serra R, Butrico L, Fugetto F, Chibireva MD, Malva A, De Caridi G, et al. Updates in pathophysiology, diagnosis and management of takayasu arteritis. Ann Vasc Surg. (2016) 35:210–25. doi: 10.1016/j.avsg.2016.02.011
8. Koide K. Takayasu arteritis in Japan. Heart Vessels Suppl. (1992) 7:48–54. doi: 10.1007/BF01744544
9. Watts R, Al-Taiar A, Mooney J, Scott D, Macgregor A. The epidemiology of Takayasu arteritis in the UK. Rheumatology (2009) 48:1008–11. doi: 10.1093/rheumatology/kep153
10. Cakar N, Yalcinkaya F, Duzova A, Caliskan S, Sirin A, Oner A, et al. Takayasu arteritis in children. J Rheumatol. (2008) 35:913–9.
11. Johnston SL, Lock RJ, Gompels MM. Takayasu arteritis: a review. J Clin Pathol. (2002) 55:481–6. doi: 10.1136/jcp.55.7.481
12. Kissin EY, Merkel PA. Diagnostic imaging in Takayasu arteritis. Curr Opin Rheumatol. (2004) 16:31–7. doi: 10.1097/00002281-200401000-00007
13. Takahashi K, Oharaseki T, Yokouchi Y, Yamada H, Shibuya K, Naoe S. A half-century of autopsy results–incidence of pediatric vasculitis syndromes, especially Kawasaki disease. Circ J. (2012) 76:964–70. doi: 10.1253/circj.CJ-11-0928
14. Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, et al. EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis. (2009) 68:318–23. doi: 10.1136/ard.2008.088351
15. Barra L, Yang G, Pagnoux C, Canadian Vasculitis Network (CanVasc). Non-glucocorticoid drugs for the treatment of Takayasu's arteritis: A systematic review and meta-analysis. Autoimmun Rev. (2018) 17:683–93. doi: 10.1016/j.autrev.2018.01.019
16. Hoyer BF, Mumtaz IM, Loddenkemper K, Bruns A, Sengler C, Hermann KG, et al. Takayasu arteritis is characterised by disturbances of B cell homeostasis and responds to B cell depletion therapy with rituximab. Ann Rheum Dis. (2012) 71:75–9. doi: 10.1136/ard.2011.153007
17. Decker P, Olivier P, Risse J, Zuily S, Wahl D. Tocilizumab and refractory Takayasu disease: Four case reports and systematic review. Autoimmun Rev. (2018) 17:353–60. doi: 10.1016/j.autrev.2017.11.026
18. Tato F, Rieger J, Hoffmann U. Refractory Takayasu's arteritis successfully treated with the human, monoclonal anti-tumor necrosis factor antibody adalimumab. Int Angiol. (2005) 24:304–7. Available online at: https://pdfs.semanticscholar.org/c0fa/7aaa605fae766d354bf42d7af6f79ef98fef.pdf
19. Stern S, Clemente G, Reiff A, Ramos MP, Marzan KA, Terreri MT. Treatment of Pediatric Takayasu arteritis with infliximab and cyclophosphamide: experience from an American-Brazilian cohort study. J Clin Rheumatol. (2014) 20:183–8. doi: 10.1097/RHU.0000000000000106
20. Kawasaki T. [Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children]. Arerugi. (1967) 16:178–222.
21. Wolff AE, Hansen KE, Zakowski L. Acute Kawasaki disease: not just for kids. J Gen Intern Med. (2007) 22:681–4. doi: 10.1007/s11606-006-0100-5
22. Hedrich CM, Schnabel A, Hospach T. Kawasaki disease. Front Pediatr. (2018) 6:198. doi: 10.3389/fped.2018.00198
23. McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation (2017) 135:e927–99. doi: 10.1161/CIR.0000000000000484
24. Weiss PF. Pediatric vasculitis. Pediatr Clin North Am. (2012) 59:407–23. doi: 10.1016/j.pcl.2012.03.013
25. Yellen ES, Gauvreau K, Takahashi M, Burns JC, Shulman S, Baker AL, et al. Performance of 2004 American Heart Association recommendations for treatment of Kawasaki disease. Pediatrics (2010) 125:e234–41. doi: 10.1542/peds.2009-0606
26. Rosenfeld EA, Corydon KE, Shulman ST. Kawasaki disease in infants less than one year of age. J Pediatr. (1995) 126:524–9. doi: 10.1016/S0022-3476(95)70344-6
27. Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics (2004) 114:1708–33. doi: 10.1542/peds.2004-2182
28. Burns JC, Glode MP. Kawasaki syndrome. Lancet (2004) 364:533–44. doi: 10.1016/S0140-6736(04)16814-1
30. Newburger JW, Takahashi M, Burns JC, Beiser AS, Chung KJ, Duffy CE, et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med. (1986) 315:341–7. doi: 10.1056/NEJM198608073150601
31. Kobayashi T, Inoue Y, Otani T, Morikawa A, Kobayashi T, Takeuchi K, et al. Risk stratification in the decision to include prednisolone with intravenous immunoglobulin in primary therapy of Kawasaki disease. Pediatr Infect Dis J. (2009) 28:498–502. doi: 10.1097/INF.0b013e3181950b64
32. Kobayashi T, Inoue Y, Takeuchi K, Okada Y, Tamura K, Tomomasa T, et al. Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease. Circulation (2006) 113:2606–12. doi: 10.1161/CIRCULATIONAHA.105.592865
33. Jakob A, von Kries R, Horstmann J, Hufnagel M, Stiller B, Berner R, et al. Failure to predict high-risk Kawasaki disease patients in a population-based study cohort in Germany. Pediatr Infect Dis J. (2018) 37:850–5. doi: 10.1097/INF.0000000000001923
34. Ozen S, Anton J, Arisoy N, Bakkaloglu A, Besbas N, Brogan P, et al. Juvenile polyarteritis: results of a multicenter survey of 110 children. J Pediatr. (2004) 145:517–22. doi: 10.1016/j.jpeds.2004.06.046
35. Meyts I, Aksentijevich I. Deficiency of Adenosine Deaminase 2 (DADA2): updates on the phenotype, genetics, pathogenesis, and treatment. J Clin Immunol. (2018) 38:569–78. doi: 10.1007/s10875-018-0525-8
36. Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. (2014) 370:921–31. doi: 10.1056/NEJMoa1307362
37. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. (2014) 370:911–20. doi: 10.1056/NEJMoa1307361
38. Ozen S, Ben-Chetrit E, Bakkaloglu A, Gur H, Tinaztepe K, Calguneri M, et al. Polyarteritis nodosa in patients with Familial Mediterranean Fever (FMF): a concomitant disease or a feature of FMF? Semin Arthritis Rheum. (2001) 30:281–7. doi: 10.1053/sarh.2001.19958
39. Stanson AW, Friese JL, Johnson CM, McKusick MA, Breen JF, Sabater EA, et al. Polyarteritis nodosa: spectrum of angiographic findings. Radiographics (2001) 21:151–9. doi: 10.1148/radiographics.21.1.g01ja16151
40. Eleftheriou D, Dillon MJ, Tullus K, Marks SD, Pilkington CA, Roebuck DJ, et al. Systemic polyarteritis nodosa in the young: a single-center experience over thirty-two years. Arthritis Rheum. (2013) 65:2476–85. doi: 10.1002/art.38024
41. Pagnoux C, Quemeneur T, Ninet J, Diot E, Kyndt X, de Wazieres B, et al. Treatment of systemic necrotizing vasculitides in patients aged sixty-five years or older: results of a multicenter, open-label, randomized controlled trial of corticosteroid and cyclophosphamide-based induction therapy. Arthritis Rheumatol. (2015) 67:1117–27. doi: 10.1002/art.39011
42. Watts RA, Scott DG. Epidemiology of the vasculitides. Semin Respir Crit Care Med. (2004) 25:455–64. doi: 10.1055/s-2004-836139
43. Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schonlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet (2002) 360:1197–202. doi: 10.1016/S0140-6736(02)11279-7
44. Weiss PF, Klink AJ, Luan X, Feudtner C. Temporal association of Streptococcus, Staphylococcus, and parainfluenza pediatric hospitalizations and hospitalized cases of Henoch-Schonlein purpura. J Rheumatol. (2010) 37:2587–94. doi: 10.3899/jrheum.100364
45. McCarthy HJ, Tizard EJ. Clinical practice: diagnosis and management of Henoch-Schonlein purpura. Eur J Pediatr. (2010) 169:643–50. doi: 10.1007/s00431-009-1101-2
46. Kawasaki Y, Ono A, Ohara S, Suzuki Y, Suyama K, Suzuki J, et al. Henoch-Schonlein purpura nephritis in childhood: pathogenesis, prognostic factors and treatment. Fukushima J Med Sci. (2013) 59:15–26. doi: 10.5387/fms.59.15
47. Yavuz H, Arslan A. Henoch-Schonlein purpura-related intestinal perforation: a steroid complication? Pediatr Int. (2001) 43:423–5. doi: 10.1046/j.1442-200X.2001.01417.x
48. Ronkainen J, Koskimies O, Ala-Houhala M, Antikainen M, Merenmies J, Rajantie J, et al. Early prednisone therapy in Henoch-Schonlein purpura: a randomized, double-blind, placebo-controlled trial. J Pediatr. (2006) 149:241–7. doi: 10.1016/j.jpeds.2006.03.024
49. Ohara S, Kawasaki Y, Miyazaki K, Ono A, Suzuki Y, Suyama K, et al. Efficacy of cyclosporine A for steroid-resistant severe Henoch-Schonlein purpura nephritis. Fukushima J Med Sci. (2013) 59:102–7. doi: 10.5387/fms.59.102
50. Pohl M, Dittrich K, Ehrich JHH, Hoppe B, Kemper MJ, Klaus G, Schmitt CP, et al. Behandlung der Purpura-Schönlein-Henoch-Nephritis bei Kindern und Jugendlichen - Therapieempfehlungen der Gesellschaft für Pädiatrische Nephrologie (GPN). Monatsscrift Kinderheilkunde. (2013) 161:543–53. doi: 10.1007/s00112-013-2896-5
51. Bohm M, Gonzalez Fernandez MI, Ozen S, Pistorio A, Dolezalova P, Brogan P, et al. Clinical features of childhood granulomatosis with polyangiitis (wegener's granulomatosis). Pediatr Rheumatol Online J. (2014) 12:18. doi: 10.1186/1546-0096-12-18
52. Cabral DA, Uribe AG, Benseler S, O'Neil KM, Hashkes PJ, Higgins G, et al. Classification, presentation, and initial treatment of Wegener's granulomatosis in childhood. Arthritis Rheum. (2009) 60:3413–24. doi: 10.1002/art.24876
53. Masi AT, Hunder GG, Lie JT, Michel BA, Bloch DA, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. (1990) 33:1094–100. doi: 10.1002/art.1780330806
54. Zwerina J, Eger G, Englbrecht M, Manger B, Schett G. Churg-Strauss syndrome in childhood: a systematic literature review and clinical comparison with adult patients. Semin Arthritis Rheum. (2009) 39:108–15. doi: 10.1016/j.semarthrit.2008.05.004
55. Zwerina J, Axmann R, Jatzwauk M, Sahinbegovic E, Polzer K, Schett G. Pathogenesis of Churg-Strauss syndrome: recent insights. Autoimmunity (2009) 42:376–9. doi: 10.1080/08916930902832348
56. Sun L, Wang H, Jiang X, Mo Y, Yue Z, Huang L, et al. Clinical and pathological features of microscopic polyangiitis in 20 children. J Rheumatol. (2014) 41:1712–9. doi: 10.3899/jrheum.131300
57. Peco-Antic A, Bonaci-Nikolic B, Basta-Jovanovic G, Kostic M, Markovic-Lipkovski J, Nikolic M, et al. Childhood microscopic polyangiitis associated with MPO-ANCA. Pediatr Nephrol. (2006) 21:46–53. doi: 10.1007/s00467-005-2063-x
58. Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis. (2009) 68:310–7. doi: 10.1136/ard.2008.088096
59. Jones RB, Furuta S, Tervaert JW, Hauser T, Luqmani R, Morgan MD, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis: 2-year results of a randomised trial. Ann Rheum Dis. (2015) 74:1178–82. doi: 10.1136/annrheumdis-2014-206404
60. Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. (2010) 363:221–32. doi: 10.1056/NEJMoa0909905
61. de Groot K, Reinhold-Keller E, Tatsis E, Paulsen J, Heller M, Nolle B, et al. Therapy for the maintenance of remission in sixty-five patients with generalized Wegener's granulomatosis. Methotrexate versus trimethoprim/sulfamethoxazole. Arthritis Rheum. (1996) 39:2052–61. doi: 10.1002/art.1780391215
62. Reinhold-Keller E, De Groot K, Rudert H, Nolle B, Heller M, Gross WL. Response to trimethoprim/sulfamethoxazole in Wegener's granulomatosis depends on the phase of disease. QJM (1996) 89:15–23. doi: 10.1093/oxfordjournals.qjmed.a030133
63. Hedrich CM, Fiebig B, Bruck N, Hahn G, Bornhauser M, Gahr M, et al. Autologous stem cell transplantation in refractory paediatric Wegener's granulomatosis. Clin Exp Rheumatol. (2011) 29:897–8.
64. Kone-Paut I. Behcet's disease in children, an overview. Pediatr Rheumatol Online J. (2016) 14:10. doi: 10.1186/s12969-016-0070-z
65. Davatchi F, Shahram F, Chams-Davatchi C, Shams H, Nadji A, Akhlaghi M, et al. Behcet's disease in Iran: analysis of 6500 cases. Int J Rheum Dis. (2010) 13:367–73. doi: 10.1111/j.1756-185X.2010.01549.x
66. Olivieri I, Leccese P, Padula A, Nigro A, Palazzi C, Gilio M, et al. High prevalence of Behcet's disease in southern Italy. Clin Exp Rheumatol. (2013) 31(3 Suppl. 77):28–31. Available online at: https://www.clinexprheumatol.org/article.asp?a=6384
67. Sarica R, Azizlerli G, Kose A, Disci R, Ovul C, Kural Z. Juvenile Behcet's disease among 1784 Turkish Behcet's patients. Int J Dermatol. (1996) 35:109–11. doi: 10.1111/j.1365-4362.1996.tb03272.x
68. Muhammad JS, Ishaq M, Ahmed K. Genetics and epigenetics pathogenesis of Behcet's syndrome. Curr Rheumatol Rev. (2018) 15:7–13. doi: 10.2174/1573397114666180521090335
69. Takeuchi M, Ombrello MJ, Kirino Y, Erer B, Tugal-Tutkun I, Seyahi E, et al. A single endoplasmic reticulum aminopeptidase-1 protein allotype is a strong risk factor for Behcet's disease in HLA-B*51 carriers. Ann Rheum Dis. (2016) 75:2208–11. doi: 10.1136/annrheumdis-2015-209059
70. de Menthon M, Lavalley MP, Maldini C, Guillevin L, Mahr A. HLA-B51/B5 and the risk of Behcet's disease: a systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheum. (2009) 61:1287–96. doi: 10.1002/art.24642
71. Kotter I, Vonthein R, Muller CA, Gunaydin I, Zierhut M, Stubiger N. Behcet's disease in patients of German and Turkish origin living in Germany: a comparative analysis. J Rheumatol. (2004) 31:133–9. Available online at: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.905.1181&rep=rep1&type=pdf
72. Davatchi F, Sadeghi Abdollahi B, Chams-Davatchi C, Shahram F, Shams H, Nadji A, et al. The saga of diagnostic/classification criteria in Behcet's disease. Int J Rheum Dis. (2015) 18:594–605. doi: 10.1111/1756-185X.12520
73. Kone-Paut I, Shahram F, Darce-Bello M, Cantarini L, Cimaz R, Gattorno M, et al. Consensus classification criteria for paediatric Behcet's disease from a prospective observational cohort: PEDBD. Ann Rheum Dis. (2016) 75:958–64. doi: 10.1136/annrheumdis-2015-208491
74. Singer O. Cogan and Behcet syndromes. Rheum Dis Clin North Am. (2015) 41:75–91. doi: 10.1016/j.rdc.2014.09.007
75. Mora P, Calzetti G, Ghirardini S, Rubino P, Gandolfi S, Orsoni J. Cogan's syndrome: state of the art of systemic immunosuppressive treatment in adult and pediatric patients. Autoimmun Rev. (2017) 16:385–90. doi: 10.1016/j.autrev.2017.02.009
76. Walsh S, Knofler R, Hahn G, Lohse J, Berner R, Brenner S, et al. Childhood primary large vessel CNS vasculitis: single-centre experience and review of the literature. Clin Exp Rheumatol. (2017) 35(Suppl. 103):213–20. Available online at: https://livrepository.liverpool.ac.uk/3019142/1/10067-WALSH.pdf
77. Twilt M, Benseler SM. CNS vasculitis in children. Mult Scler Relat Disord. (2013) 2:162–71. doi: 10.1016/j.msard.2012.11.002
78. Dotan ZA, Laufer M, Heldenberg E, Langevitz P, Fridman E, Duvdevani M, et al. Isolated testicular polyarteritis nodosa mimicking testicular neoplasm-long-term follow-up. Urology (2003) 62:352. doi: 10.1016/S0090-4295(03)00388-1
79. Hedrich CM. Shaping the spectrum - From autoinflammation to autoimmunity. Clin Immunol. (2016) 165:21–8. doi: 10.1016/j.clim.2016.03.002
80. Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol. (2009) 27:621–68. doi: 10.1146/annurev.immunol.25.022106.141627
81. Ozen S, Eroglu FK. Pediatric-onset Behcet disease. Curr Opin Rheumatol. (2013) 25:636–42. doi: 10.1097/BOR.0b013e328363ea8b
82. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
83. Yan N. Immune diseases associated with TREX1 and STING dysfunction. J Interferon Cytokine Res. (2017) 37:198–206. doi: 10.1089/jir.2016.0086
Keywords: vasculitis, inflammation, systemic disease, kawasaki disease, granulomatosis with polyangitis, purpura, classification, paediatric
Citation: Schnabel A and Hedrich CM (2019) Childhood Vasculitis. Front. Pediatr. 6:421. doi: 10.3389/fped.2018.00421
Received: 15 September 2018; Accepted: 19 December 2018;
Published: 10 January 2019.
Edited by:
Rita Consolini, University of Pisa, ItalyReviewed by:
Toni Hospach, Klinikum Stuttgart, GermanyCopyright © 2019 Schnabel and Hedrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anja Schnabel, YW5qYS5zY2huYWJlbEB1bmlrbGluaWt1bS1kcmVzZGVuLmRl
Christian M. Hedrich, Y2hyaXN0aWFuLmhlZHJpY2hAbGl2ZXJwb29sLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.